 ,1, 王新涛1, 杨青1, 王艳1, 张莹莹1, 席章营2, 李保全,1,*
,1, 王新涛1, 杨青1, 王艳1, 张莹莹1, 席章营2, 李保全,1,*Major Quantitative Trait Loci Mapping for Tassel Branch Number and Construction of qTBN5 Near-isogenic Lines in Maize (Zea mays L.)
DAI Zi-Ju,1, WANG Xin-Tao1, YANG Qing1, WANG Yan1, ZHANG Ying-Ying1, XI Zhang-Ying2, LI Bao-Quan,1,*通讯作者:
第一联系人:
收稿日期:2017-12-20接受日期:2018-06-9网络出版日期:2018-06-11
| 基金资助: |
Received:2017-12-20Accepted:2018-06-9Online:2018-06-11
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (5081KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
代资举, 王新涛, 杨青, 王艳, 张莹莹, 席章营, 李保全. 玉米雄穗分枝数主效QTL定位及qTBN5近等基因系构建[J]. 作物学报, 2018, 44(8): 1127-1135. doi:10.3724/SP.J.1006.2018.01127
DAI Zi-Ju, WANG Xin-Tao, YANG Qing, WANG Yan, ZHANG Ying-Ying, XI Zhang-Ying, LI Bao-Quan.
玉米雄穗分枝数(tassel branch number, TBN)作为衡量雄穗大小的重要指标, 是玉米育种与种子生产过程中被研究的与产量形成有关的重要农艺性状之一。Geraldi等[1]的研究表明雄穗分枝数与产量呈负相关。然而在玉米杂交种种子生产过程中却要求作父本的亲本植株具有较为发达的雄穗, 以利于提高制种质量与降低成本[2]。同时较小适中的雄穗又是理想株型的构成要素之一, 过度发达的雄穗影响株型的冠层结构[3]。因此在育种实践中选育适度减少雄穗分枝数的杂交种是目前玉米育种的一个趋势, 而研究玉米雄穗分枝数基因的遗传效应, 挖掘玉米雄穗分枝数基因资源可以为玉米产量和株型相关性状的遗传改良提供依据。
随着现代分子生物技术的发展, 一些控制玉米雄穗分枝数QTL被鉴定出来。Berke和Rocheford[4]利用F2群体定位到3个雄穗分枝数目QTL, 分布于第2、第4、第7染色体上, 其中第7染色体上的QTL贡献率达35.3%。Mickolson等[5]利用重组自交系群体, 定位到6个雄穗分枝数目QTL, 分布于第1、第2、第3、第4染色体上, 其中第2染色体QTL贡献率达19.2%。汤华等[6]利用N87-1×综3构建F2:3群体, 定位到9个雄穗分枝数目QTL, 分布于第1、第3、第4、第5、第9、第10染色体上, 贡献率为6.11%~12.01%。Upadyayula等[7]利用BC1S1家系群体, 定位到2个雄穗分枝数目QTL, 分布于第4、第7染色体, 贡献率分别为7.9%和11.7%。高世斌等[8]利用N87-1×9526构建F2:3群体, 定位到6个雄穗分枝数目QTL, 分布于第2、第4、第7、第10染色体上, 贡献率为5.6%~28.4%。王迪等[9]利用齐319×黄早四和披478×黄早四分别构建了2套F2:3群体, 定位到40个雄穗分枝数目QTL, 10条染色体上均有分布, 贡献率为2.93%~23.78%。Brown等[10]利用5000份重组自交系群体组成的(nested association mapping, NAM)群体, 检测到39个雄穗分枝数目QTL。杨钊钊等[11]以黄早四为共同亲本构建了11个不同组合的重组自交系群体, 定位到11个雄穗分枝数目QTL, 分布于第2、第3、第4、第5、第7、第8染色体, 贡献率为9.7%~20.9%。Chen等[12]利用F2群体和高密度遗传图谱检测到7个控制雄穗分枝数QTL, 分布于第1、第3、第4、第5、第7、第8、第9染色体上。Wu等[13]利用NAM群体和全基因组关联分析检测到63个控制雄穗分枝数QTL。而Xu等[14]利用栽培玉米和类蜀黍(teosinte)发展作图群体, 利用SNP标记全基因组扫描检测到14个控制玉米雄穗分枝数QTL。以上研究表明玉米雄穗分枝数是多基因控制数量性状, 控制该性状基因在多数染色体上都是存在的, 且存在主效QTL。但由于不同的研究所用到的F2:3、RIL、BC1S1等群体类型易受到遗传背景的影响及试验环境和标记密度等因素干扰, 使得彼此定位结果存在差异, 具有一致性且贡献率较大的QTL比较少, 因此难以通过精细定位克隆雄穗分枝数基因。
玉米雄穗分枝发育涉及众多关键基因, 基于突变体的遗传分析, 已鉴定分离了多个调控玉米雄穗分枝数发育基因, 如参与玉米花序分枝分生组织调控的ramosa基因突变体系列ramosa1 (ra1)、ramosa2 (ra2)和ramosa3 (ra3), 主要表现为雄穗基部分枝数增多, 其中ra1和ra3基因分别位于第7染色体的两个不同区域, ra1编码含有一个Cys2-His2锌指结构域且具植物特异性的类EPF蛋白, 而ra3基因编码6-磷酸海藻糖酶用以修饰进入花序分生组织的糖信号, 进而调控ra1的转录活性, ra2基因位于第3染色体, 是一个含有LOB-domain的转录因子, 在茎顶端分生组织和花序分生组织细胞中表达较高, 其中ra1起关键作用, 调节ra2和ra3表达[15,16,17]。barren stalk1 (ba1)突变体雄穗只有一个主轴而没有分枝、小穗和小花, 导致突变的原因是位于第3染色体ba1基因编码起始位点上游存在Helitron转座子插入, 该基因编码一个碱性螺旋-环-螺旋(helix-loop-helix)结构蛋白, 是一个植物特有的bHLH 转录因子[18]。liguleless2 (lg2)主要控制玉米叶舌和叶耳的发育, 也参与花序建成的调控, 雄穗下部长分枝不能启始, 该基因位于第3染色体上, 编码一个bZIP结构蛋白[19]。barren inflorescence2 (bif2)基因是位于第1染色体上的一个编码丝氨酸/苏氨酸激酶, 参与生长素极性运输, bif2突变体表现为雄穗分枝难以分化、小穗分生组织减少[20]。而unbranched2 (ub2)和unbranched2 (ub3)基因分别位于第1和第4染色体, 是一类SBP-box转录因子, 增强其表达可以增加玉米雄穗分枝和花药数[21]。尽管这些突变体导致了雄穗分枝数的改变, 为雄穗分枝数相关基因的克隆及功能研究提供了参考, 但部分突变体表现出其他性状(如雌穗性状、植株形态性状等)的改变, 影响了优异种质材料的育种利用。
本研究首先利用郑单958的骨干亲本郑58和昌7-2构建的重组自交系群体对控制玉米雄穗分枝数主效QTL进行鉴定, 然后构建主效QTL的近等基因系验证基因遗传效应, 开发连锁标记, 利用重组株系缩短定位区间, 为玉米雄穗分枝数主效基因的精细定位和克隆及分子育种实践奠定基础。
1 材料与方法
1.1 供试材料与田间试验
以生产上大量应用玉米品种郑单958的2个优良自交系亲本郑58和昌7-2杂交, 采用单粒传法连续自交至F7:8构建188个重组自交系群体。分别于2015年和2016年夏季在河南省农业科学院原阳试验基地种植188个重组自交系及亲本, 采用随机区组设计, 不设重复, 单行区, 行长2 m, 每行10穴, 每穴1株, 植株授粉15 d后, 调查雄穗分枝数, 每行从第2株开始, 连续调查5株完整雄穗, 统计各家系的雄穗分枝数平均值用于进一步分析。qTBN5-NILs构建试验材料于2008—2017年夏季种植于河南郑州, 冬季种植于海南三亚, 春季在河南省农业科学院原阳试验基地温室加代。1.2 分子标记检测
按CTAB法从玉米幼苗叶片组织提取分离基因组DNA。引物合成的序列来自于玉米基因组数据库(MaizeGDB,1.3 玉米雄穗分枝数QTL定位
从1928个分子标记中筛选到在郑58与昌7-2之间有多态性的分子标记300个, 用于植株的基因型鉴定。经χ2检验后, 选择扩增位点符合孟德尔分离定律(1:1)的引物288个, 利用MapMaker/EXP 3.0 软件构建群体的遗传图谱[22], 采用Haldane函数, 图距单位为cM (centiMorgan)。根据引物序列比对B73 RefGen-v3基因组序列, 找出标记在图谱上的物理位置, 利用MapChart 2.1绘制物理图谱。利用188个家系的雄穗分枝数平均数进行QTL定位, 选用QTL IciMapping Version 4.0软件的复合区间作图法[23]分析QTL效应。设定QTL检测的LOD阈值为3.0, PIN值为0.001。1.4 主效QTL-qTBN5近等基因系构建
利用分子标记辅助选择(marker-assisted selection, MAS)及表型选择创制qTBN5-NILs流程(图1)。在qTBN5-NILs创制过程中, 子代群体只保留经分子标记选择基因型正确、雄穗分枝数与受体亲本植株表型具有显著差异、而其他性状与轮回亲本相近的单株, 并严格进行杂交或自交授粉。以昌7-2为供体亲本, 郑58为受体材料并作为轮回亲本(recurrent parent)回交5代后自交, 用140个多态性SSR进行全基因组背景选择, 构建以郑58为受体的染色体片段代换系(chromosome segment substitution line, CSSL)。然后利用qTBN5连锁标记umc1853选择基因型为qTBN5且雄穗分枝数与受体亲本存在显著差异的CSSL, 用均匀分布于全基因组的288个多态性标记进行背景分析, 选出片段较少且携带目标基因片段的CSSL与郑58杂交、回交, 最后自交, 选择qTBN5基因型纯合且除雄穗分枝数外其他性状没有明显差异的植株, 表型稳定遗传后创制qTBN5- NILs。图1
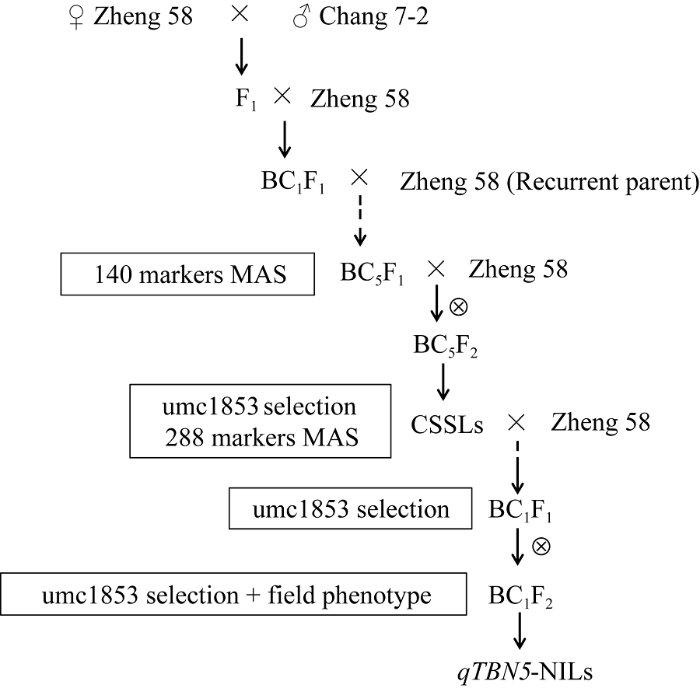 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1qTBN5-NILs的构建流程
Fig. 1Flow-process diagram of construction of qTBN5-NILs
2 结果与分析
2.1 亲本与RIL家系雄穗分枝数表型分析
对亲本郑58和昌7-2及188个RIL家系的两年雄穗分枝数调查结果(表1)表明, 188个RIL家系雄穗分枝数的总平均数分别为10.7个和11.2个, 家系间从最少的2.4个和2.8个到最多的22.6个和21.4个, 变异系数分别为43.60和38.83, 分布范围较广。而且广义遗传率(H2)较高, 为89.74%, 表明RIL家系群体大部分表型变异受遗传控制。Table 1
表1
表1亲本及RIL家系在两年环境下的雄穗分枝数的表型分析
Table 1
| 年份 Year | 郑58 Zheng 58 | 昌7-2 Chang 7-2 | 均值 Average | 范围 Range | 变异系数 Variation coefficient (%) |
|---|---|---|---|---|---|
| 2015 | 4.6 | 17.8 | 10.7 | 2.4-22.6 | 43.60 |
| 2016 | 4.4 | 17.2 | 11.2 | 2.8-21.4 | 38.83 |
新窗口打开|下载CSV
分别以188个RIL家系雄穗分枝数平均值的两年数据制作频次分布, 呈现为一种连续的近似正态分布(图2), 表明玉米雄穗分枝数是典型的多基因控制的数量性状。亲本郑58雄穗分枝数的平均个数2年分别为4.6个和4.4个, 而昌7-2两年分别为17.8个和17.2个, 多数家系雄穗分枝数的平均值都介于两个亲本之间, 但也有部分家系雄穗分枝数在两个亲本区间之外, 说明控制雄穗分枝数基因存在双向重组超亲分离, 双亲均含有影响雄穗分枝数性状的增效、减效基因。
图2
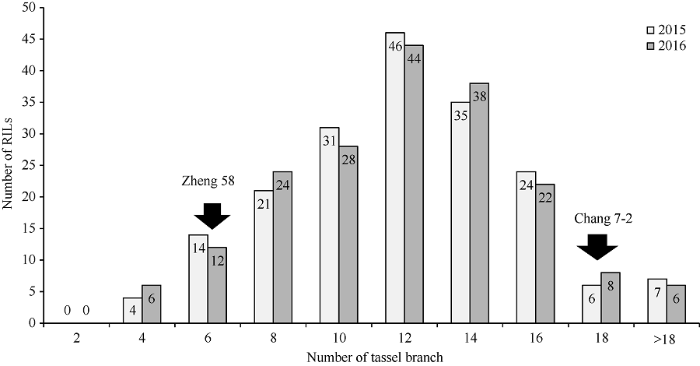 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2RIL家系雄穗分枝数频次分布
Fig. 2Frequency distribution of average values for tassel branch number of parents and 188 RILs populations from cross between Zheng 58 and Chang 7-2
2.2 QTL检测与遗传效应分析
连锁图谱共包含288个在郑58和昌7-2之间存在多态性的分子标记, 覆盖玉米整个10条染色体的基因组, 除第4和第7染色体存在2个较大gap外, 其他标记分布较为均匀。根据引物序列参考比对B73 RefGen-v3基因组序列, 明确了分子标记在图谱上的物理位置(图3)。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3雄穗分枝数性状主效QTL在标记连锁图上的位置
连锁图上左边数字表示标记在B73 RefGen-v3基因组序列图谱上的物理位置。
Fig. 3Position of major QTLs for tassel branch number detected in RILs population of Zheng 58 × Chang 7-2 within linkage map
Physical map distances (Mb) referring B73 RefGen-v3 are shown on the left of linkage map.
利用188个RIL家系两年的雄穗分枝数表型数据, 经复合区间作图法在包含288个分子标记的连锁图谱上一共检测到了5个两年均可重复检测的主效QTL, 分别位于第3、第4、第5、第7和第10染色体上, 即qTBN3、qTBN4、qTBN5、qTBN7和qTBN10 (图3), 各QTL的贡献率变异范围在5.81%~ 19.92%之间(表2), 在5个QTL中贡献率超过10%的有3个, 最高的是位于第5染色体bin 5.05上的qTBN5。在被检测到的5个QTL中, 有3个加性效应值为负值, 表明来自于郑58的等位基因可以使雄穗分枝数减少; 而加性效应值为正值、位于第3和第5染色体上的qTBN3和qTBN5的贡献率较高, 分别达到了13.67%和19.92%, 表明来自于亲本昌7-2的增效等位基因可以使雄穗分枝数显著增加, 这与作为父本的昌7-2自身具有相对较多的雄穗分枝数有关。
Table 2
表2
表2玉米雄穗分枝数主效QTL及其遗传效应
Table 2
| QTL | 染色体 Chromosome | 物理区间 Position (Mb) | 标记区间 Marker interval | LOD | 加性效应 Additive effect | 贡献率 R2 (%) |
|---|---|---|---|---|---|---|
| qTBN3 | 3 | 146.7-175.5 | umc1954-bnlg1798 | 5.06 | 1.40 | 13.67 |
| qTBN4 | 4 | 169.2-186.7 | bnlg2291-umc2041 | 3.15 | -1.01 | 5.81 |
| qTBN5 | 5 | 170.1-200.2 | umc2111-bnlg278 | 9.58 | 1.88 | 19.92 |
| qTBN7 | 7 | 29.4-83.2 | bnlg2233-bnlg1380 | 4.65 | -0.82 | 16.53 |
| qTBN10 | 10 | 127.3-145.5 | umc2003-phi323152 | 6.11 | -1.73 | 9.06 |
新窗口打开|下载CSV
2.3 qTBN5-NILs的表型分析
根据雄穗分枝数主效QTL-qTBN5定位(图4-a), 利用回交、自交和分子标记前景及背景选择, 成功创制了qTBN5-NILs。图4-b是qTBN5-NILs基因型, 除位于第5染色体上qTBN5基因所在片段来自昌7-2之外, 其他背景均来自郑58。对创制成功的qTBN5-NILs及其受体亲本郑58和供体亲本昌7-2的雄穗分枝数、株高、穗位高、抽雄期和雄穗主轴长进行调查, t检验分析表明: 受体亲本郑58和供体亲本昌7-2的雄穗分枝数与qTBN5-NILs之间均存在显著性差异(图4-c), 而其他主要性状如株高、穗位高、抽雄期和雄穗主轴长在郑58与qTBN5-NILs之间差异不显著(图4-d~g)。由此表明, qTBN5基因的导入可以使郑58雄穗分枝数显著增加, 但是达不到昌7-2水平, 而且并没有造成郑58其他主要性状的改变。图4
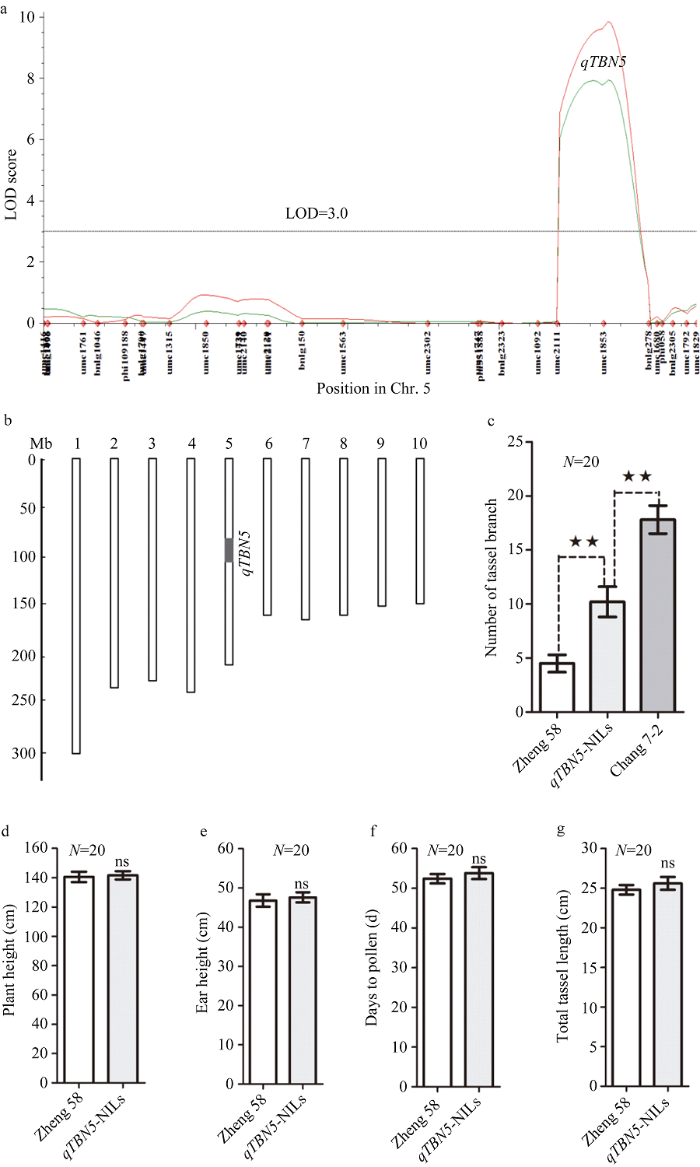 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4qTBN5-NILs的基因型和表型效应
a: 雄穗分枝数主效基因qTBN5的定位; b: qTBN5-NILs的基因型, 白色为郑58背景, 黑色部分为来自昌7-2的qTBN5基因所在片段; c: 雄穗分枝数; d: 株高; e: 穗位高; f: 抽雄期; g: 雄穗主轴长。**表示在 0.01水平差异显著, ns表示差异不显著。
Fig. 4Genotypes and phenotypes of qTBN5-NILs
a: physical locus of qTBN5 identified in RILs population. b: genotypes of qTBN5-NILs. Black part is segment from Chang 7-2 indicating gene position and white parts are genetic background from Zheng 58. c: tassel branch number. d: plant height. e: ear height. f: days to pollen. g: total tassel length. Error bars represent SD. “**” and“ns” show significance at P<0.01 level and on significant difference, respectively as determined by t-test.
2.4 雄穗分枝数主效QTL-qTBN5的进一步定位及连锁标记开发
根据qTBN5在第5染色体bin 5.05的定位区间, 利用CSSL和郑58发展的BC1F2作图群体, 提取DNA筛选交换单株, 种植重组单株自交发展株系, 纯合株系用于表型考察和进一步定位(图5-a), 从网上公布的引物数据库及相关序列, 开发筛选到3个与qTBN5紧密连锁的分子标记, 即引物phi333597、e2040、e2042 (图5-b), 结合重组株表型将qTBN5进一步定位在phi333597和e2042的13.2 Mb区域之内, 为qTBN5的精细定位和分子标记辅助育种奠定基础。图5
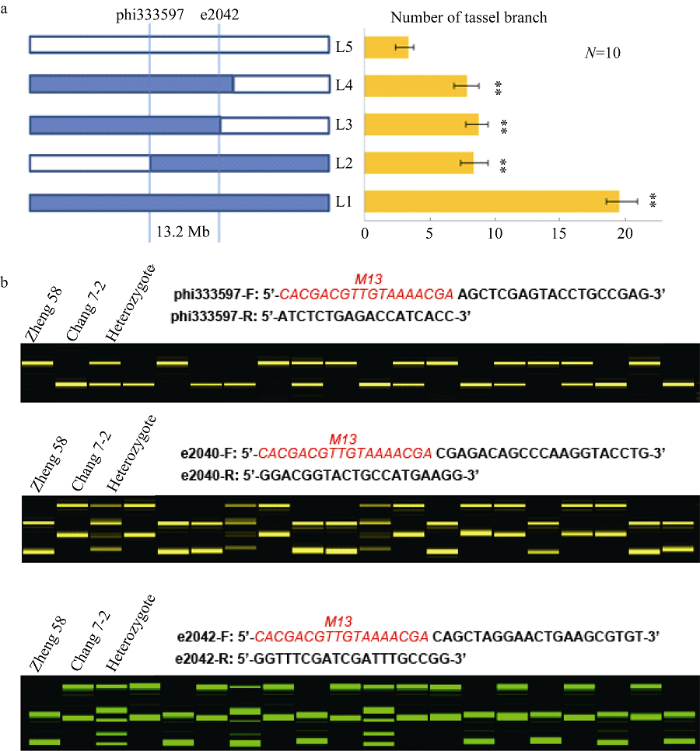 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5qTBN5进一步定位及连锁标记开发
a: 交换株代换作图定位qTBN5, L1为昌7-2, L5为郑58, L2~L4为重组株, **表示在 0.01水平差异显著; b: 标记检测郑58和昌7-2及BC1F2世代部分单株基因型, 红色M13为通用引物序列。
Fig. 5Further mapping and linked markers development of qTBN5
a: qTBN5 detected on the substituted segments in recombinant lines; L1: Chang 7-2; L5: Zheng 58. L2-L4: recombinant lines; “**” shows significance at P<0.01 as determined by t-test. b: genotypes identification of qTBN5 in BC1F2 backcross population. M13: Red indicates universal primer sequences.
3 讨论
玉米雄穗分枝数是复杂的数量性状, 玉米雄穗分枝数QTL定位易受遗传背景、群体类型、标记密度和试验环境等因素影响, 迄今对控制玉米雄穗分枝数的基因研究报道相对较少且存在不同的看法[24]。不同的研究者利用不同研究材料、不同的标记密度, 在玉米10条染色体上都定位出了玉米雄穗分枝数的QTL[4,5,6,7,8,9,10,11,12,13,14]。本研究共检测到5个均可重复检测的主效QTL, 分别位于第3、第4、第5、第7、第10染色体上(图3), 各QTL座位的表型贡献率变异范围在5.81%~19.92%之间(表2)。其中第3染色体qTBN3定位区间位于ba1[18]和lg2[19]基因附近, 第7染色体qTBN7定位区间附近有ra1[15]突变体基因, 而qTBN4定位的区间与Chen等[12]报道区间有少部分重合, 且该区间附近存在ub3[21]突变体基因。对于第5染色体上雄穗分枝数QTL定位的研究, 由于受群体类型及标记密度的影响, 不同的研究中没有相对一致的定位区间, 且效应都比较小[6,9-10,12,14,25], 本研究中定位到的qTBN5贡献率在5个主效QTL中最大, 达到19.92%。而位于第10染色体的qTBN10为一个新的QTL, 在此区域未见有QTL报道。因此, 尽管定位到的部分QTL与前人报道的具有重合定位区间, 但由于效应大小及稳定性未知, 仍然需要进行遗传效应验证。近等基因系和染色体片段代换系是在受体的遗传背景中代换某个或某些供体亲本的染色体片段, 而基因组的其余部分均与受体亲本相同[26], 可对复杂数量性状进行有效的遗传剖析, 因此在基因(QTL)的鉴定与定位, 等位基因变异分析等方面具有重要的利用价值而受到许多研究者的重视。玉米雄穗分枝数是受多基因控制的数量性状, 且存在主效QTL, 然而前人研究中所用到的F2、RIL、BC1S1等群体类型易受遗传背景的影响, 使得彼此定位结果存在差异, 难以通过精细定位克隆雄穗分枝数基因。而本研究根据定位到的控制玉米雄穗分枝数主效QTL, 构建主效基因qTBN5近等基因系, 有效解决了遗传背景的干扰, 且通过发展近等基因系对该基因遗传效应进行了验证(图4), 开发了紧密连锁标记, 并将qTBN5进一步定位在13.2 Mb区间之内(图5), 为玉米雄穗分枝数基因精细定位和分子标记辅助育种提供参考。
玉米雄穗分枝数作为衡量雄穗大小的重要指标, 是玉米育种与种子生产过程中被研究的与产量形成有关的重要农艺性状之一。育种实践中常常要求母本具有较小雄穗或者较少雄穗分枝数, 这样可以减少对雌穗能量的竞争, 父本一般要求较为发达的雄穗或者较多雄穗分枝数, 这样可以保证充足的粉量以便提高制种产量, 而二者组配的杂交种往往需要具有适度较少雄穗分枝数, 从而达到最优组合[27]。郑单958作为黄淮海地区的主推品种, 其亲本郑58和昌7-2两个优良自交系也一直是研究的热点, 二者之所以在制种过程中获得较高产量的杂交种, 重要原因之一是亲本之间雄穗的合理搭配。本研究通过1928个分子标记筛选, 最终找到288个在郑58和昌7-2之间具有多态性的分子标记, 并对标记的物理位置进行了注释, 建立了一张可以借助ABI 3500XL遗传分析仪高通量检测郑单958品种的指纹图谱, 为后续开展郑58和昌7-2之间包括雄穗分枝数在内的优良性状基因挖掘, 进而解析郑单958品种之所以能够成为主推品种的奥秘奠定基础。
4 结论
定位到5个玉米雄穗分枝数主效QTL, 分别位于玉米5条染色体上, 3个QTL表型贡献率超过了10%。构建了玉米雄穗分枝数主效QTL qTBN5近等基因系, 验证了基因遗传效应, 并将qTBN5进一步定位在13.2 Mb区间之内, 为玉米雄穗分枝数主效基因的精细定位和分子育种实践奠定了基础。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
DOI:10.1007/s00122-005-0133-xURLPMID:16395569 [本文引用: 1]

Maize ( Zea mays L.) ear inflorescence architecture is directly relevant to grain yield components, and tassel architecture is relevant to hybrid seed production. The objectives of this study were to (1) determine heritabilities and correlations of a comprehensive set of tassel and ear inflorescence architecture traits in a set of (Illinois Low Protein B73) B73 S 1 families, (2) identify chromosomal positions of QTL affecting tassel and ear architecture, and (3) identify possible candidate genes associated with these QTL. For tassel traits, the number of detected QTL ranged from one to five, and explained between 6.5 and 35.9% of phenotypic variation. For ear traits, the number of detected QTL ranged from one to nine and phenotypic variation explained by those QTL varied between 7.9 and 53.0%. We detected QTL for tassel architecture traits that required calculation of ratios from measured traits. Some of these calculated traits QTL were detected in regions that did not show QTL for the measured traits, suggesting that calculation of ratios may reveal developmentally relevant patterns of tassel architecture. We detected a QTL on chromosome 7 for tassel branch number near the gene ramosa1 ( ra1 ), which is known to control tassel branch number, making ra1 a candidate gene for tassel branch number. We detected QTL for several traits on chromosomes 6, 8, and 9, where no inflorescence architecture genes have been mapped, thus providing initial information towards new gene discovery for control of inflorescence architecture.
[本文引用: 1]
DOI:10.2135/cropsci1999.3951439xURL [本文引用: 2]
The inheritance of tassel traits in maize (Zea mays L.) is not well understood. This study was conducted to estimate the chromosomal location and magnitude of effect of major quantitative trait loci (QTL) affecting tassel branch angle (ANGLE), branches per tassel (BRANCH#), and tassel weight (TASSWT) in 200 [S.sub.1] lines derived from a single [F.sub.1] plant from a cross of Illinois High Oil (IHO) by Illinois Low Oil (Early Maturity) [ILO(EM)]. The restriction fragment length polymorphism (RFLP) genotypes of the 200 [S.sub.1] lines were determined at 80 polymorphic loci. These lines were measured for ANGLE, BRANCH#, and TASSWT in replicated field trials during 1992 and 1993 at Urbana, IL. Composite interval mapping using selected markers as cofactors detected several QTL for each tassel trait. Six QTL located on chromosomes 2, 3, 5, 6, and 10 were significantly associated with ANGLE. They showed primarily dominant gene action and explained 43.1 and 50.7% of the phenotypic and genetic variance, respectively. Three QTL located on chromosomes 2, 4, and 7 were significantly associated with BRANCH#. They showed primarily additive gene action and explained 44.3 and 49.3% of the phenotypic and genetic variance, respectively. The QTL for BRANCH# on chromosome 7 explained 35.3% ([R.sup.2]) of the phenotypic variance. Seven QTL located on chromosomes 1, 2, 3, 4, and 7 were significantly associated with TASSWT. They showed primarily negative dominant gene action and explained 35.1 and 43.4% of the phenotypic and genetic variance, respectively. Most of the major QTL detected for these tassel traits were not associated with anthesis date (ANTH) in this population.
DOI:10.2135/cropsci2002.1902URL [本文引用: 2]
URL [本文引用: 3]
利用“豫玉 2 2”构建的 2 6 6个玉米F2∶3家系为材料 ,通过一年两点的随机区组田间试验和分子标记分析 ,研究了玉米穗位高、雄穗分支数、茎粗、抽雄期、吐丝期 5个重要农艺性状。相关分析表明 ,穗位高、雄穗分支数、茎粗与单株产量显著正相关 ,抽雄期与吐丝期高度正相关 ,雄穗分支数与茎粗显著正相关。采用复合区间作图法 ,通过5 0 0次排列测验分别确定各性状的LOD阈值 ,在武汉和襄樊两地共定位了 7个穗位高QTL、9个雄穗分支数QTL、8个茎粗QTL、9个抽雄期QTL和 7个吐丝期QTL ;这些QTL在染色体上分布不均匀 ,具有集中分布的特点。研究表明 ,数量性状间的表型相关可能源于控制数量性状的QTL位点间的相关
URL [本文引用: 3]
利用“豫玉 2 2”构建的 2 6 6个玉米F2∶3家系为材料 ,通过一年两点的随机区组田间试验和分子标记分析 ,研究了玉米穗位高、雄穗分支数、茎粗、抽雄期、吐丝期 5个重要农艺性状。相关分析表明 ,穗位高、雄穗分支数、茎粗与单株产量显著正相关 ,抽雄期与吐丝期高度正相关 ,雄穗分支数与茎粗显著正相关。采用复合区间作图法 ,通过5 0 0次排列测验分别确定各性状的LOD阈值 ,在武汉和襄樊两地共定位了 7个穗位高QTL、9个雄穗分支数QTL、8个茎粗QTL、9个抽雄期QTL和 7个吐丝期QTL ;这些QTL在染色体上分布不均匀 ,具有集中分布的特点。研究表明 ,数量性状间的表型相关可能源于控制数量性状的QTL位点间的相关
DOI:10.1007/s00122-005-0133-xURLPMID:16395569 [本文引用: 2]
Maize ( Zea mays L.) ear inflorescence architecture is directly relevant to grain yield components, and tassel architecture is relevant to hybrid seed production. The objectives of this study were to (1) determine heritabilities and correlations of a comprehensive set of tassel and ear inflorescence architecture traits in a set of (Illinois Low Protein B73) B73 S 1 families, (2) identify chromosomal positions of QTL affecting tassel and ear architecture, and (3) identify possible candidate genes associated with these QTL. For tassel traits, the number of detected QTL ranged from one to five, and explained between 6.5 and 35.9% of phenotypic variation. For ear traits, the number of detected QTL ranged from one to nine and phenotypic variation explained by those QTL varied between 7.9 and 53.0%. We detected QTL for tassel architecture traits that required calculation of ratios from measured traits. Some of these calculated traits QTL were detected in regions that did not show QTL for the measured traits, suggesting that calculation of ratios may reveal developmentally relevant patterns of tassel architecture. We detected a QTL on chromosome 7 for tassel branch number near the gene ramosa1 ( ra1 ), which is known to control tassel branch number, making ra1 a candidate gene for tassel branch number. We detected QTL for several traits on chromosomes 6, 8, and 9, where no inflorescence architecture genes have been mapped, thus providing initial information towards new gene discovery for control of inflorescence architecture.
DOI:10.3321/j.issn:0253-9772.2007.08.019URL [本文引用: 2]
在包含103个SSR标记的连锁图谱基础上,运用复合区间作图法检测玉米组合 (N87-1×9526)F3家系在正常与干旱胁迫环境下的雄穗分枝数与主轴长性状QTL.雄穗分枝数在正常环境下被检测到2个QTL座位,分别位于第5 和7连锁群上;在胁迫环境下被检测到4个QTL座位分别位于2、5、7和10连锁群上,其中位于第5和7连锁群上的QTL不仅具有一致性而且与本作图群体 中曾检测到的耐旱相关性状QTL存在连锁.雄穗主轴长在正常环境下被检测到2个QTL位于第2和第6连锁群上,在干旱胁迫环境下被检测到了3个QTL分别 于第2、4和10连锁群上,其中位于第2染色体上的QTL是两种环境下所共同检测到的QTL.分析QTL的遗传作用方式表明,雄穗分枝数以部分加性效应为 主,而雄主轴长全部表现为显性和超显性.
DOI:10.3321/j.issn:0253-9772.2007.08.019URL [本文引用: 2]
在包含103个SSR标记的连锁图谱基础上,运用复合区间作图法检测玉米组合 (N87-1×9526)F3家系在正常与干旱胁迫环境下的雄穗分枝数与主轴长性状QTL.雄穗分枝数在正常环境下被检测到2个QTL座位,分别位于第5 和7连锁群上;在胁迫环境下被检测到4个QTL座位分别位于2、5、7和10连锁群上,其中位于第5和7连锁群上的QTL不仅具有一致性而且与本作图群体 中曾检测到的耐旱相关性状QTL存在连锁.雄穗主轴长在正常环境下被检测到2个QTL位于第2和第6连锁群上,在干旱胁迫环境下被检测到了3个QTL分别 于第2、4和10连锁群上,其中位于第2染色体上的QTL是两种环境下所共同检测到的QTL.分析QTL的遗传作用方式表明,雄穗分枝数以部分加性效应为 主,而雄主轴长全部表现为显性和超显性.
DOI:10.3724/SP.J.1259.2011.00011URL [本文引用: 3]
利用2套具有共同亲本黄早四且分别含有230个及235个家系的F2:3群体,结合2年多点的表型鉴定,运用完备复合区间作图方法对不同生态环境下(2007-北京、2008-北京、2007-河南、2008-河南、2007-新疆以及2008-新疆)的玉米雄穗分枝数和雄穗重进行QTL定位。同时,利用基于混合线性模型的QTLNetwork-2.0软件进行基因×环境互作及上位性分析。6个环境下2个群体共检测到51个与雄穗分枝数和雄穗重相关的QTL(Q/H群体32个,Y/H群体19个),其中包括7个主效QTL,并在Q/H群体中确定了2个重要的QTL,即位于7.01bin的Qqtpbn7-1和位于7.02bin的Qqtw7-2。对比2个群体的定位结果,共挖掘到3个在不同遗传背景下的"一致性"QTL,这些在不同环境及不同遗传背景下能够稳定存在的QTL可为玉米雄穗相关性状的生产应用以及精细定位提供有价值的参考。
DOI:10.3724/SP.J.1259.2011.00011URL [本文引用: 3]
利用2套具有共同亲本黄早四且分别含有230个及235个家系的F2:3群体,结合2年多点的表型鉴定,运用完备复合区间作图方法对不同生态环境下(2007-北京、2008-北京、2007-河南、2008-河南、2007-新疆以及2008-新疆)的玉米雄穗分枝数和雄穗重进行QTL定位。同时,利用基于混合线性模型的QTLNetwork-2.0软件进行基因×环境互作及上位性分析。6个环境下2个群体共检测到51个与雄穗分枝数和雄穗重相关的QTL(Q/H群体32个,Y/H群体19个),其中包括7个主效QTL,并在Q/H群体中确定了2个重要的QTL,即位于7.01bin的Qqtpbn7-1和位于7.02bin的Qqtw7-2。对比2个群体的定位结果,共挖掘到3个在不同遗传背景下的"一致性"QTL,这些在不同环境及不同遗传背景下能够稳定存在的QTL可为玉米雄穗相关性状的生产应用以及精细定位提供有价值的参考。
DOI:10.1371/journal.pgen.1002383URLPMID:3219606 [本文引用: 3]
We compared the genetic architecture of thirteen maize morphological traits in a large population of recombinant inbred lines. Four traits from the male inflorescence (tassel) and three traits from the female inflorescence (ear) were measured and studied using linkage and genome-wide association analyses and compared to three flowering and three leaf traits previously studied in the same population. Inflorescence loci have larger effects than flowering and leaf loci, and ear effects are larger than tassel effects. Ear trait models also have lower predictive ability than tassel, flowering, or leaf trait models. Pleiotropic loci were identified that control elongation of ear and tassel, consistent with their common developmental origin. For these pleiotropic loci, the ear effects are larger than tassel effects even though the same causal polymorphisms are likely involved. This implies that the observed differences in genetic architecture are not due to distinct features of the underlying polymorphisms. Our results support the hypothesis that genetic architecture is a function of trait stability over evolutionary time, since the traits that changed most during the relatively recent domestication of maize have the largest effects.
[本文引用: 2]
[本文引用: 2]
DOI:10.1186/1471-2164-15-433URL [本文引用: 4]
DOI:10.1111/pbi.2016.14.issue-7URL [本文引用: 2]
DOI:10.1111/nph.14400URLPMID:28067953 [本文引用: 3]
Abstract Maize (Zea mays) tassels underwent profound morphological changes during maize domestication and improvement. Although a number of genes affecting maize inflorescence development have been identified, the genetic basis of the morphological changes in maize tassels since domestication is not well understood. Here, using a large population of 866 maize-teosinte BC2 S3 recombinant inbred lines genotyped using 19 838 single nucleotide polymorphism (SNP) markers, we performed high-resolution quantitative trait locus (QTL) mapping for five tassel morphological traits. We showed that the five tassel traits were associated with different genetic architecture features. Known genes for maize inflorescence development identified by mutagenesis were significantly enriched in the tassel trait QTLs, and many of these genes, including ramosa1 (ra1), barren inflorescence2 (bif2), unbranched2 (ub2), zea floricaula leafy2 (zfl2) and barren stalk fastigiate1 (baf1), showed evidence of selection. An in-depth nucleotide diversity analysis at the bif2 locus identified strong selection signatures in the 5'-regulatory region. We also found that several known flowering time genes co-localized with tassel trait QTLs. A further association analysis indicated that the maize photoperiod gene ZmCCT was significantly associated with tassel size variation. Using near-isogenic lines, we narrowed down a major-effect QTL for tassel length, qTL9-1, to a 513-kb physical region. These results provide important insights into the genetic architecture that controls maize tassel evolution.
DOI:10.1038/nature03892URLPMID:16041362 [本文引用: 2]
Abstract The external appearance of flowering plants is determined to a large extent by the forms of flower-bearing branch systems, known as inflorescences, and their position in the overall structure of the plant. Branches and branching patterns are produced by tissues called shoot apical meristems. Thus, inflorescence architecture reflects meristem number, arrangement and activity, and the duration of meristem activity correlates with branch length. The inflorescences of maize, unlike those of related grasses such as rice and sorghum, predominantly lack long branches, giving rise to the tassel and familiar corncob. Here we report the isolation of the maize ramosa1 gene and show that it controls inflorescence architecture. Through its expression in a boundary domain near the nascent meristem base, ramosa1 imposes short branch identity as branch meristems are initiated. A second gene, ramosa2, acts through ramosa1 by regulating ramosa1 gene expression levels. ramosa1 encodes a transcription factor that appears to be absent in rice, is heterochronically expressed in sorghum, and may have played an important role in maize domestication and grass evolution.
DOI:10.1038/nature04725URLPMID:16688177 [本文引用: 1]
Abstract Inflorescence branching is a major yield trait in crop plants controlled by the developmental fate of axillary shoot meristems. Variations in branching patterns lead to diversity in flower-bearing architectures (inflorescences) and affect crop yield by influencing seed number or harvesting ability. Several growth regulators such as auxins, cytokinins and carotenoid derivatives regulate branching architectures. Inflorescence branching in maize is regulated by three RAMOSA genes. Here we show that one of these genes, RAMOSA3 (RA3), encodes a trehalose-6-phosphate phosphatase expressed in discrete domains subtending axillary inflorescence meristems. Genetic and molecular data indicate that RA3 functions through the predicted transcriptional regulator RAMOSA1 (RA1). We propose that RA3 regulates inflorescence branching by modification of a sugar signal that moves into axillary meristems. Alternatively, the fact that RA3 acts upstream of RA1 supports a hypothesis that RA3 itself may have a transcriptional regulatory function.
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1093/pcp/pcp006URL [本文引用: 1]
[本文引用: 2]
DOI:10.1016/0888-7543(87)90010-3URL [本文引用: 1]
DOI:10.1016/j.cj.2015.05.001URL [本文引用: 1]
Breeding to Optimize Chinese Agriculture (OPTICHINA) was a three-year EU–China project launched in June of 2011. As designed, the project acted as a new strategic model to reinforce systematic cooperation on agricultural research between Europe and China. The OPTICHINA International Conference“Breeding to Optimize Agriculture in a Changing World”was held in Beijing, May 26–29, 2014. The conference included six thematic areas: (1) defining and protecting the yield potential of traits and genes;(2) high-throughput precision phenotyping in the field;(3) molecular technologies in modern breeding;(4) plant ideotype;(5) data analysis, data management, and bioinformatics; and (6) national challenges and opportunities for China. The 10 articles collected in this special issue represent key contributions and topics of this conference. This editorial provides a brief introduction to the OPTICHINA project, followed by the main scientific points of articles published in this special issue. Finally, outcomes from a brainstorming discussion at the end of the conference are summarized, representing the authors' opinions on trends in breeding for a changing world.
DOI:10.2135/cropsci2014.03.0248URL [本文引用: 1]
ABSTRACT The diversity and genetics of tassel branch numbers (TBN) in maize (Zea mays L.) were evaluated in a series of five studies. Diversity was assessed in races and inbreds and genetics assessed in diallels, generation mean analyses (GMAs), nearisogenic lines (NILs), and recombinant inbred lines (RILs). High diversity characterized 215 tropical maize races that averaged 27.0 +/- 3.27 TBN and 80 modern inbreds that averaged 14.8 +/- 1.45 TBN. There was a close correlation between TBN values and husk numbers (r = 0.674). Both were much higher in tropical lowlands than in highlands. Correlations of the native elevations of races were r = -0.465 for TBN and r = -0.732 for husk numbers. Reduced tassel size and branch numbers have been viewed as advantages for grain yield, but this could also be attributed to the correlation with husk numbers. The related genetic controls of meristematic activity in tassel and ear are implicated. Heritability studies included an eight-entry diallel, five sets of progenies for GMA analysis, three sets of RILs, and evaluation of 169 NILs of inbred Hi27. A new mutant Brta (branched tassel) on Chromosome 2 was identified that doubled branch numbers. Inbred TBNs were highly correlated with means of their hybrids (r = 0.918); mean square ratios of general combining ability (GCA) to specific combining ability (SCA) averaged 11.2; and heritability values were high (h = 67%) with negligible dominance and epistasis effects. Midparent heterosis for TBN was high (32%). Variances and means were highly correlated (r = 0.677) and CVs low (average 10.4%). The RIL segregations were interpreted on the basis of three or four quantitative trait loci (QTLs) biased by highvariance -mean correlations. Inheritance of TBN is concluded to involve additive polygenic systems that could involve many loci in the genus Zea. Reducing TBN in tropical maize offers little challenge to breeders, but seems of doubtful value.
DOI:10.1534/genetics.107.076497URLPMID:17947434 [本文引用: 1]
Abstract An ultimate objective of QTL mapping is cloning genes responsible for quantitative traits. However, projects seldom go beyond segments <5 cM without subsequent breeding and genotyping lines to identify additional crossovers in a genomic region of interest. We report on a QTL analysis performed as a preliminary step in the development of a resource for map-based cloning of domestication and improvement genes in corn. A large backcross (BC)1 population derived from a cross between maize (Zea mays ssp. mays) and teosinte (ssp. parviglumis) was grown for the analysis. A total of 1749 progenies were genotyped for 304 markers and measured for 22 morphological traits. The results are in agreement with earlier studies showing a small number of genomic regions having greater impact on the morphological traits distinguishing maize and teosinte. Despite considerable power to detect epistasis, few QTL interactions were identified. To create a permanent resource, seed of BC1 plants was archived and 1000 BC2S6 BC1-derived lines are in development for fine mapping and cloning. The identification of four BC1 progeny with crossovers in a single gene, tb1, indicated that enough derived lines already exist to clone many QTL without the need to generate and identify additional crossovers.
[本文引用: 1]
DOI:10.1016/S2095-3119(16)61538-1URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}