 ,1,*, 张孟臣,2,*
,1,*, 张孟臣,2,*Mapping Main-effect and Epistatic QTL for Hard Seededness in Soybean
AI Li-Juan1,2,**, CHEN Qiang2,**, YANG Chun-Yan2, YAN Long2, WANG Feng-Min2, GE Rong-Chao,1,*, ZHANG Meng-Chen,2,*通讯作者:
第一联系人:
收稿日期:2017-09-5接受日期:2018-01-8网络出版日期:2018-06-12
| 基金资助: |
Received:2017-09-5Accepted:2018-01-8Online:2018-06-12
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (439KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
艾丽娟, 陈强, 杨春燕, 闫龙, 王凤敏, 葛荣朝, 张孟臣. 大豆籽粒硬实加性和上位性QTL定位[J]. 作物学报, 2018, 44(6): 852-858. doi:10.3724/SP.J.1006.2018.00852
AI Li-Juan, CHEN Qiang, YANG Chun-Yan, YAN Long, WANG Feng-Min, GE Rong-Chao, ZHANG Meng-Chen.
硬实是种子休眠类型之一, 普遍存在于豆科作物中[1]。硬实主要是由于种皮的不渗透性吸水导致, 而种皮迅速均匀的渗透性吸水是许多豆科作物驯化的关键特征之一[2,3,4,5]。在农业生产中, 硬实严重影响种子的发芽率和出苗率[6,7]; 在大豆食品加工过程中, 硬实对大豆食品的感观品质及碾磨品质产生影响[8,9]; 在品种改良中, 硬实严重阻碍种质资源的利用。硬实对种子的休眠萌发和延长种子寿命起着重要作用, 因此有利于种质保存[1]; 硬实也被认为是野生物种长期生存所需的重要特性[10,11]。此外, 硬实种子与种皮中的钙含量相关[12,13], 可以潜在增强大豆食品的营养价值。目前, 关于种子硬实遗传控制的信息较少, 因此有必要对大豆籽粒硬实特性的遗传机制进一步解析。硬实特性是受多基因控制的复杂数量性状[1], 通过QTL (quantitative trait locus)定位分析, 检测与种子硬实相关的QTL, 对解析硬实形成相关分子机制及分子标记辅助选择(marker-assisted selection, MAS)育种具有重要的指导意义。
前人利用经典遗传学方法研究硬实性状, 发现大豆籽粒硬实性状受少数基因控制, 但在显隐性关系和基因数目上具有争议[14,15,16]。近年来, 随着分子标记技术的快速发展, 国内外****对大豆籽粒硬实性状QTL定位分析发现, 籽粒硬实QTL主要分布在第2、第3、第6、第8、第10、第19和第20等[9,17-18]连锁群上。Sun等[19]在第2染色体上定位到控制野生大豆籽粒种皮渗透性的QTL (GmHs1-1), 通过精细定位及基因功能分析发现, 在编码钙调磷酸跨膜蛋白与种皮钙含量有关的基因Glyma02g43700上的点突变导致了硬实, 而在该基因附近, Jang等[20]通过精细定位、基因表达和测序分析发现, Glyma02g43680同样可导致籽粒硬实, 它编码内切- 1,4-β-葡聚糖酶可在栅栏细胞的外层上产生1,4-β-葡聚糖衍生物使得种皮更硬, 导致大豆种皮不渗透性吸水从而产生硬实。随着研究的深入, QTL间的上位性互作逐渐受到重视, 前人相继研究证明上位性效应在复杂数量性状的遗传中起着重要的作用[21]。上位性效应的机制分析和作用效果对于品种改良至关重要, 2017年Soyk等[22]在番茄研究中发现, 负向的上位性效应会阻碍优势性状的表现, 该研究利用基因编辑手段控制负向上位性效应, 从而培育出高产番茄品种。与进化和自然选择相比, 强烈的人工选择可能会推动更频繁的上位性发生, 上位性效应普遍存在。前人研究大豆籽粒硬实性状多分析加性效应影响, 虽已有籽粒硬实相关基因报道, 对上位性效应检测研究涉及较少, 仅Liu等[23]通过双因素方差分析检测到位于第2和第6染色体上的籽粒硬实QTL间存在上位性互作效应, 但上位效应值尚不明确, 因此解析QTL之间的上位性互作并估算其效应, 明确现有QTL间的上位性互作关系及效应, 发掘上位性互作QTL新位点可为进一步解析大豆籽粒硬实遗传基础及辅助育种提供依据。
本研究利用高世代重组自交系群体及WinQTL Cartographer V. 2.5和IciMapping 4.1软件对不同年份的大豆籽粒硬实性状进行加性检测。并使用IciMapping 4.1软件对籽粒硬实性状进行上位性QTL分析, 探明加性和上位性互作效应对籽粒硬实性状的影响, 为QTL的精细定位及基因克隆奠定基础, 为分子标记辅助育种提供依据。
1 材料与方法
1.1 供试材料
以育成品种冀豆12和农家种黑豆(ZDD03651)杂交通过单粒传法构建的包含186个家系的F6:8和F6:9重组自交系为材料。其中冀豆12是河北省农林科学院粮油作物研究所育成的百粒重为22 g的大粒品种, 黑豆(ZDD03651)为百粒重7 g的小粒农家种。将该群体F6:8(2011年)和F6:9(2013年)重组自交系及其亲本播种于石家庄田间, 采用3 m行长, 3行区, 随机区组实验设计, 行距0.5 m, 株距0.1 m。分别从自然成熟的亲本和重组自交系群体每个株系随机收获10株, 研究其籽粒。1.2 实验方法
分别从亲本和重组自交系每个株系中随机选取60粒正常籽粒, 设每份材料3个重复, 将20粒籽粒浸泡在盛有20 mL蒸馏水的培养皿中, 于室温下浸泡7 d。每天统计籽粒硬实数, 硬实率Y = Yi/20× 100%, 其中Yi为籽粒硬实数。1.3 数据分析
采用SPSS 17.0软件完成籽粒硬实性状的均值、偏度、峰度及正态分布检验等分析。1.4 QTL定位
本研究所用分子遗传连锁图谱由本实验室前人[24,25]构建完成, 包含SSR标记154个, 总长度为1516.1 cM, 标记间的平均遗传距离为9.3 cM。采用Windows QTL Cartographer 2.5软件中的复合区间作图法(CIM)检测不同年份下大豆籽粒硬实性状相关的主效QTL; 以IciMapping 4.1软件中的完备区间作图法(ICIM)进行籽粒硬实性状的加性及上位性QTL定位分析。显著主效QTL的LOD阈值为2.5, 显著上位性QTL的LOD阈值为5.0。采取McCouch等[26]的方式命名QTL。2 结果与分析
2.1 籽粒硬实测定的种子浸泡时间
浸泡1 d后群体籽粒大部分表现为非硬实状态, 随着浸泡天数的增加, 硬实率的变化差异不显著(P<0.05)。不同年份的RIL (recombinant inbred lines)群体籽粒硬实率平均值随浸泡天数的变化略有差异, 但总体趋势基本一致(图1)。因此以浸泡1 d后群体的籽粒硬实率进行后续分析及QTL定位。图1
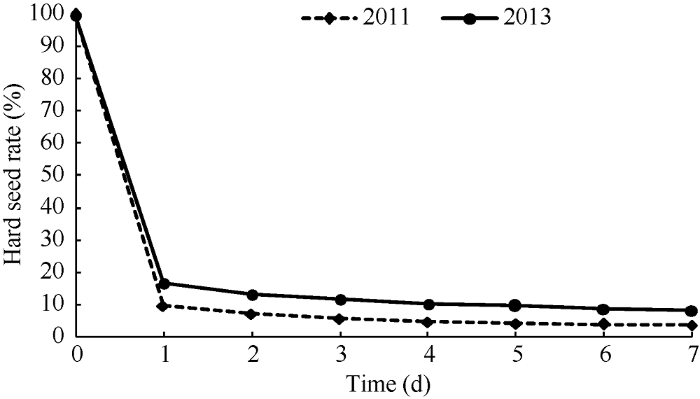 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1籽粒硬实率变化情况
Fig. 1Change of hard seededness rate
2.2 表型数据分析
由表1可知, 浸泡1 d后, 亲本冀豆12的硬实率为0, 黑豆的硬实率在71.65%~83.33%左右, 两亲本间籽粒硬实率存在明显差异, 为QTL定位分析提供了较好的遗传背景。RIL群体各家系籽粒硬实率的分离程度较大, 变异范围为0~96.67%, 变异系数介于56%~75%之间, 且RIL群体的籽粒硬实性状存在超高亲分离。由其频率分布图可知籽粒硬实性状的表型值呈连续性变异(图2)。Table 1
表1
表1不同环境下大豆亲本及RIL群体的籽粒硬实率的表型值
Table 1
| 年份 Year | 亲本Parents | 重组自交系群体RILs population | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 冀豆12 Jidou 12 (%) | 黑豆 Heidou (%) | 平均值 Mean (%) | 最大值 Maximum (%) | 最小值 Minimum (%) | 标准差 SD | 变异系数 CV (%) | 偏度 Skewness | 峰度 Kurtosis | ||
| 2011 | 0 | 71.65 | 9.78 | 90.00 | 0 | 17.52 | 56 | 2.07 | 4.27 | |
| 2013 | 0 | 83.33 | 16.85 | 96.67 | 0 | 22.45 | 75 | 1.42 | 1.29 | |
新窗口打开|下载CSV
图2
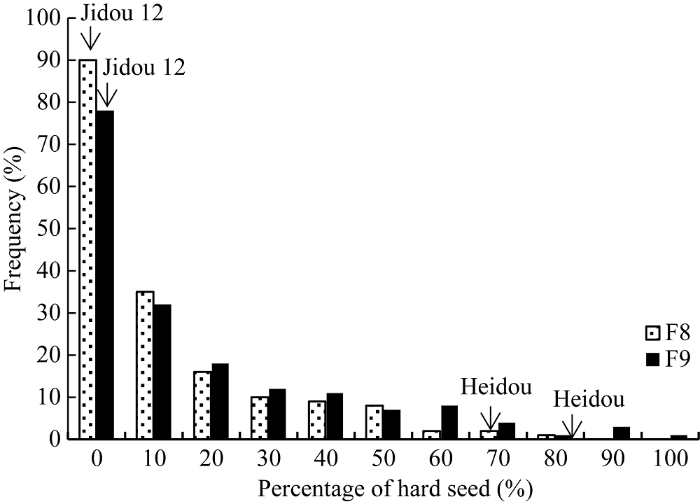 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2大豆籽粒硬实性状频率分布图
Fig. 2Frequency distributions for hard seededness in 186 soybean RILs
2.3 籽粒硬实加性QTL分析
由表2可知, 两年中共检测到3个籽粒硬实性状相关的加性QTL, 分别位于第2 (D1b)、第6 (C2)和第14 (B2)染色体, 可解释的表型变异率在5.54%~ 12.94%之间。其中位于第2染色体的Sat_069- Sat_183标记之间的qHS-2-1和位于第6染色体的Sat_402-Satt460标记之间的qHS-6-1用两种方法均能被检测到, 且qHS-2-1和qHS-6-1在CIM法中两年环境下均能被检测到, 为稳定的QTL位点, 贡献率最大, 分别为6.82%和12.94%, 加性效应最大, 分别为5.16和7.92, 增效基因来自黑豆。qHS-14-1用两种方法均仅在2013年检测到, 贡献率分别为8.25%和9.39%, 加性效应分别为-6.26和-7.47, 位 于第14染色体的Satt577-Sat_287标记之间, 其增效基因来源于冀豆12。Table 2
表2
表2用不同方法检测到的不同环境下大豆籽粒硬实加性QTL
Table 2
| QTL名称 Name of QTL | 染色体/连锁群 Chr./LG | 标记区间 Marker interval | WinQTL Cart 2.5 | IciMapping 4.1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 年份 Year | 阈值 LOD | 贡献率 R2 (%) | 加性效应 Additive effect | 年份 Year | 阈值 LOD | 贡献率 R2 (%) | 加性效应 Additive effect | ||||
| qHS-2-1 | Chr.02/D1b | Sat_069-Sat_183 | 2011 | 2.94 | 6.82 | 4.62 | 2011 | 2.99 | 5.57 | 4.57 | |
| 2013 | 2.51 | 5.54 | 5.16 | ||||||||
| qHS-6-1 | Chr.06/C2 | Sat_402-Satt460 | 2011 | 6.02 | 11.99 | 6.09 | 2011 | 5.87 | 11.04 | 6.47 | |
| 2013 | 6.64 | 12.94 | 7.92 | 2013 | 2.77 | 6.52 | 6.29 | ||||
| qHS-14-1 | Chr.14/B2 | Satt577-Sat_287 | 2013 | 3.34 | 8.25 | -6.26 | 2013 | 3.31 | 9.39 | -7.47 | |
新窗口打开|下载CSV
2.4 籽粒硬实上位性QTL分析
对于两年环境下的大豆籽粒硬实性状共检测到4对上位性互作QTL (表3和图3)。上位性效应值在-10.08~11.64之间, 贡献率在2.53%~3.47%之间。其中第6染色体上的qHS-6-1和第2染色体上的qHS-2-1、第9染色体上的qHS-9-1以及第12染色体上的qHS-12-1均存在上位性互作。qHS-6-1和qHS-2-1之间的互作仅在2011年中被检测到, 贡献率为2.69%, 上位性效应为8.66, 为亲本型大于重组型互作。qHS-6-1和qHS-9-1以及qHS-12-1间的互作在两年中均能被检测到, 贡献率最大, 分别为3.36%和3.47%, 上位性效应最大, 分别为11.64和-8.15。另外, 第14染色体上qHS-14-1和第12染色体上的qHS-12-1也存在上位性互作, 仅在2013年被检测到, 贡献率为3.13%, 上位性效应为10.10, 表现为亲本型大于重组型。Table 3
表3
表3不同环境下大豆籽粒硬实性状的上位性效应
Table 3
| QTL名称 Name of QTL | 标记区间 Marker interval | QTL名称 Name of QTL | 标记区间 Marker interval | 年份 Years | 阈值 LOD | 贡献率 R2 (%) | 上位性效应 Add by add |
|---|---|---|---|---|---|---|---|
| qHS-6-1 | Sat_402-Satt460 | qHS-2-1 | Sat_069-Sat_183 | 2011 | 10.41 | 2.69 | 8.66 |
| qHS-9-1 | Sat_399-Satt273 | 2011 | 10.93 | 2.74 | 8.82 | ||
| 2013 | 7.24 | 3.36 | 11.64 | ||||
| qHS-12-1 | Barcsoyssr_12_1-Satt353 | 2011 | 6.22 | 2.53 | -8.15 | ||
| 2013 | 7.50 | 3.47 | -10.08 | ||||
| qHS-14-1 | Satt577-Sat_287 | qHS-12-1 | Barcsoyssr_12_1-Satt353 | 2013 | 5.87 | 3.13 | 10.10 |
新窗口打开|下载CSV
图3
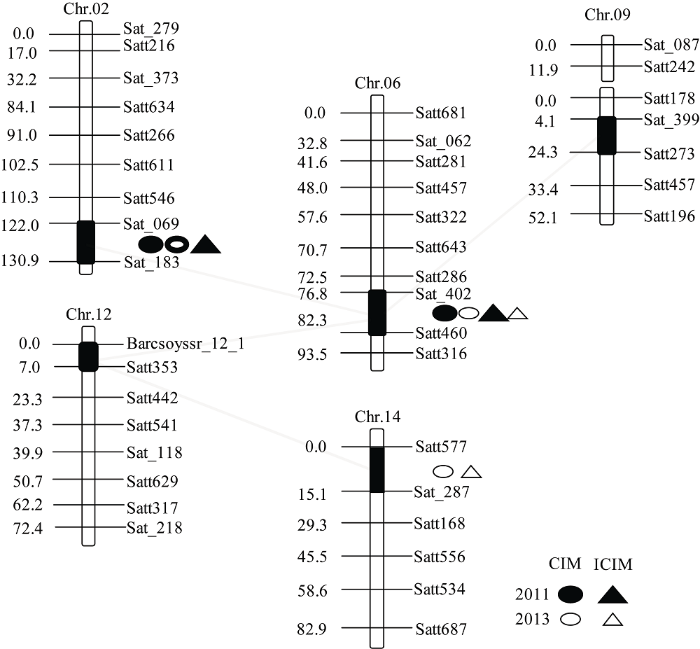 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3检测到的加性QTL及上位性互作QTL在连锁群上的分布
黑色区段为QTL所在区域, 虚线代表上位性互作QTL位点。
Fig. 3Location of additive QTLs and epistatic effects QTLs on linkage groups
The black bars show the support interval of QTL position, the dotted line represents epistatic QTLs.
3 讨论
3.1 与前人QTL定位结果的比较
本研究检测到籽粒硬实性状相关的加性QTL 3个, 分别位于第2、第6和第14染色体。与QTL qHS-2-1和qHS-6-1与前人所报道标记区间相同或相近, 这两个位点在前人研究中多次被检测到, 且在本研究中的不同年份间稳定表达, 表明qHS-2-1和qHS-6-1为籽粒硬实的稳定主效QTL。Liu等[23]研究中检测到第2染色体上的硬实相关QTL位于标记Satt456附近, 与本研究得到的qHS-2-1位点的标记区间(Sat_096-Sat_183)区域相近, 且Jang等[20]和Sun等[19]分别在该区域附近发现与大豆种皮渗透性相关的基因, 分别为Glyma02g43680和Glyma02g43700。本研究所检测到的qHS-6-1位于第6染色体的Sat_402-Satt460标记区间, 与Watanabe等[17]检测到的籽粒硬实QTL区间Satt489-Satt100部分重叠。本研究所检测到的qHS-14-1位于第14染色体的Satt577-Sat_287标记区间, 前人研究中均没检测到该位点, 因只在一年中检测到, 该位点是否为新位点需进行进一步的研究。此外, Keim等[18]和Watanabe等[17]在第3、第8、第19和第20染色体上也检测到籽粒硬实相关的加性QTL, 而在本研究与Liu等[23]研究中均没检测到, 可能是由于定位群体遗传背景或环境差异造成。QTL间的上位性互作是数量性状遗传基础的重要组成部分, 上位性互作对解析复杂性状的遗传基础具有十分重要的意义[27,28,29,30]。本研究检测到4对籽粒硬实性状相关的上位性互作QTL, 其中位于第6染色体上的qHS-6-1分别与第2染色体上的qHS-2-1, 第9染色体上的qHS-9-1及第12染色体上的qHS-12-1间均存在上位性互作。qHS-2-1与qHS-6-1间的互作发生在加性QTL间, Liu等[23]同样检测到位于第2和第6染色体上控制籽粒硬实性状相关的加性QTL间发生互作。qHS-6-1与qHS-9-1及qHS-12-1间的互作发生在加性效应QTL与非加性效应QTL之间, 且在不同年份间均被检测到。另外, qHS-12-1还与qHS-14-1存在上位性互作。说明除加性QTL起作用外, QTL之间的上位性互作也是影响性状的重要因素。
3.2 籽粒硬实上位性互作的遗传机制
在遗传学领域, 上位性效应(epistasis)是指某基因的表达受到另一非等位基因的作用, 这种非等位基因间的抑制或遮掩作用叫上位效应。在不同的生物体中各种各样的上位相互作用的分子机制被研究发现[31]。最近Cho等[32]研究表明, 控制大豆种皮色的I位点和K1位点间的互作主要是由于K1位点的功能基因AG05编码Argonaute类蛋白, 该蛋白可以结合到小RNA上, 进而通过剪切mRNA、抑制翻译、促进DNA甲基化等各种形式实现RNAi干扰的效果, 影响了小RNA的分布从而调节I位点的CHS基因的表达, 最终控制大豆种皮颜色的调控过程。因此, 小RNA在QTL间的上位性互作中起着非常重要的作用。另外QTL间的上位性互作可能与基因的调控网络有关, 即QTL间主要是通过基因间的正向诱导表达或负向反馈抑制的调控网络来实现上位性互作[33]。本研究检测到4对上位性互作QTL, 其中第2染色体上的qHS-2-1和第6染色体上的qHS-6-1间存在上位性互作, 且第2染色体上已报道了2个与大豆种皮渗透性相关的基因, 分别为Glyma02g43680和Glyma02g43700。若上位性互作QTL的遗传机制可能与上述遗传机制相一致, 即与Glyma02g43680和Glyma02g43700两个基因的表达有关。因此可以这2个已报道的基因为出发点, 寻找与其表达过程中相关的酶或调控因子, 进而确定第6染色体上QTL位点区域内与其互作的基因位点。同样另外3对上位性互作位点也可通过以上方法进一步研究, 从而确定互作的基因。因此我们可以将基因的表达及调控网络作为解析籽粒硬实遗传机制的突破口, 研究这4对互作QTL之间的遗传机制, 探究籽粒硬实形成机制, 挖掘出与籽粒硬实相关的基因。4 结论
首次报道了籽粒硬实相关的4对上位性互作QTL。确定了加性效应及QTL间的上位性互作效应在大豆籽粒硬实性状的遗传基础中均具有重要的作用。因此, 在大豆籽粒硬实性状分子标记辅助育种中, 既要考虑具有加性效应的QTL, 也要考虑具有上位性互作的QTL。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.3969/j.issn.1674-3547.2014.03.007URL [本文引用: 3]

硬实是种子休眠类型之一,在豆科作物中最为常见。种皮缺乏透性是硬实大豆形成的主要原因。种子硬实的影响因素包括遗传因素、大豆生长后期的环境条件以及贮藏条件等。硬实的处理方法机械划破种皮、温度处理、干湿交错处理和化学处理等。在种质保存过程中还可对种子硬实特性加以利用,发挥坚硬种皮的保水作用。
DOI:10.3969/j.issn.1674-3547.2014.03.007URL [本文引用: 3]
硬实是种子休眠类型之一,在豆科作物中最为常见。种皮缺乏透性是硬实大豆形成的主要原因。种子硬实的影响因素包括遗传因素、大豆生长后期的环境条件以及贮藏条件等。硬实的处理方法机械划破种皮、温度处理、干湿交错处理和化学处理等。在种质保存过程中还可对种子硬实特性加以利用,发挥坚硬种皮的保水作用。
DOI:10.1007/BF00029189URL [本文引用: 1]
Following hybridization experiments and cytogenetic analysis of interspecific hybrids three chromosome interchanges were found between the cultivated lentil L. culinaris and L. nigricans , and only one between the cultivated species and L. orientalis . This indicates that the latter species is more likely to be wild progenitor of lentil. The partial fertility of the interspecific hybrids indicate further that both L. nigricans and L. orientalis should be included in the wild genepool of lentil, and their variation can be exploited by relatively simple hybridization techniques. The wild lentils L. orientalis and L. nigricans are morphologically very similar but reproductively strongly isolated from one another by the albino seedling of their hybrids. It has been suggested that the populations of L. orientalis that gave rise to the cultivated lentil still possess a similar chromosome arrangement as in L. culinaris and are also capable of forming normal hybrids with L. nigricans . According to these considerations it is unlikely that lentil originated from populations at the south western corner of the distribution area of L. orientalis .
[本文引用: 1]
DOI:10.1109/55.215086URLPMID:17660515 [本文引用: 1]
Background and Aims The changes that occur during the domestication of crops such as maize and common bean appear to be controlled by relatively few genes. This study investigates the genetic basis of domestication in pea (Pisum sativum) and compares the genes involved with those determined to be important in common bean domestication. Methods Quantitative trait loci and classical genetic analysis are used to investigate and identify the genes modified at three stages of the domestication process. Five recombinant inbred populations involving crosses between different lines representing different stages are examined. Key Results A minimum of 15 known genes, in addition to a relatively few major quantitative trait loci, are identified as being critical to the domestication process. These genes control traits such as pod dehiscence, seed dormancy, seed size and other seed quality characters, stem height, root mass, and harvest index. Several of the genes have pleiotropic effects that in species possessing a more rudimentary genetic characterization might have been interpreted as clusters of genes. Very little evidence for gene clustering was found in pea. When compared with common bean, pea has used a different set of genes to produce the same or similar phenotypic changes. Conclusions Similar to results for common bean, relatively few genes appear to have been modified during the domestication of pea. However, the genes involved are different, and there does not appear to be a common genetic basis to 'domestication syndrome' in the Fabaceae.
[本文引用: 1]
DOI:10.2135/cropsci1978.0011183X001800020006xURL [本文引用: 1]
The effects of the impermeable seed coat characteristic in soybeans (Glycine max (L.) Merr.) on the seed’s sponse to field environment, artificial drying, and storage potential were evaluated. Comparisons were made be. tween seed of ‘Dare’ and those of an experimental hardseeded line, D67-5677-1 which is similar to Dare in growth type and maturity. Hardseededness was beneficial in maintaining the viability of seed remaining in the field for up to 9 weeks after seed moisture initially declined to 20%. Resistance to moisture reabsorption by seed on unharvested plants of the hardseeded line was clearly superior to that of Dare, indicating a possible solution to seed viability problems encountered in areas where mature seed are exposed to extended periods of warm, humid weather. Drying hand harvested seed had no effect on viability but increased the number of hard seed. Seed from the hardseeded line displayed a substantially greater storage potential than those of the normal, Dare.
[本文引用: 1]
[本文引用: 1]
DOI:10.2135/cropsci2007.10.0544URL [本文引用: 2]
DOI:10.1007/BF02957854URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/BF02591040URL [本文引用: 1]
Book Review: Real Lives: Personal and Photographic Perspectives on Albinism Archie W. N. Roy and Robin Mackenzie Spinks Glasgow: Albinism Fellowship, 2005: ISBN 0 9550344 0 X. 00 20.00 (pbk) - PubliCAT
DOI:10.2134/agronj1946.00021962003800050001xURL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1270/jsbbs.54.399URL [本文引用: 3]
[本文引用: 2]
[本文引用: 2]
DOI:10.1371/journal.pone.0128527URLPMID:4454576 [本文引用: 2]
Physical dormancy, a structural feature of the seed coat known as hard seededness, is an important characteristic for adaptation of plants against unstable and unpredictable environments. To dissect the molecular basis of qHS1, a quantitative trait locus for hard seededness in soybean (Glycine max (L) Merr.), we developed a near-isogenic line (NIL) of a permeable (soft-seeded) cultivar, Tachinagaha, containing a hard-seed allele from wild soybean (G. soja) introduced by successive backcrossings. The hard-seed allele made the seed coat of Tachinagaha more rigid by increasing the amount of -1,4-glucans in the outer layer of palisade cells of the seed coat on the dorsal side of seeds, known to be a point of entrance of water. Fine-mapping and subsequent expression and sequencing analyses revealed that qHS1 encodes an endo-1,4- -glucanase. A single-nucleotide polymorphism (SNP) introduced an amino acid substitution in a substrate-binding cleft of the enzyme, possibly reducing or eliminating its affinity for substrates in permeable cultivars. Introduction of the genomic region of qHS1 from the impermeable (hard-seeded) NIL into the permeable cultivar Kariyutaka resulted in accumulation of -1,4-glucan in the outer layer of palisade cells and production of hard seeds. The SNP allele found in the NIL was further associated with the occurrence of hard seeds in soybean cultivars of various origins. The findings of this and previous studies may indicate that qHS1 is involved in the accumulation of -1,4-glucan derivatives such as xyloglucan and/or -(1,3)(1,4)-glucan that reinforce the impermeability of seed coats in soybean.
DOI:10.3321/j.issn:1001-7216.2008.03.006URL [本文引用: 1]
利用由181个家系组成的Lemont/特青籼粳交重组自交系群 体,以及由161个RFLP、SSR标记和3个形态标记构建的全长为1916.5 cM、覆盖水稻基因组12 条染色体的连锁图,采用线性模型的复合区间作图方法(QTLMapper V1 0),对粒长、粒宽、长宽比和垩白度等4个稻米品质性状的数量性状座位(QTL)进行了分析.在水稻的所有12 条染色体上共定位到7个加性主效QTL和19对上位性QTL,其中控制粒长、粒宽、长宽比的主效QTL各2个,控制垩白度的QTL 1个,分别解释12.8%、40.0%、26.0%和42.1%的表型变异;共检测到6对影响垩白度、6对影响粒长、7对影响长宽比的上位性QTL,分别 解释52.2%、31.3%和38.2% 的表型变异.结果表明,上位性QTL和主效QTL一样在稻米粒形和垩白度的遗传中起着重要的作用.
DOI:10.3321/j.issn:1001-7216.2008.03.006URL [本文引用: 1]
利用由181个家系组成的Lemont/特青籼粳交重组自交系群 体,以及由161个RFLP、SSR标记和3个形态标记构建的全长为1916.5 cM、覆盖水稻基因组12 条染色体的连锁图,采用线性模型的复合区间作图方法(QTLMapper V1 0),对粒长、粒宽、长宽比和垩白度等4个稻米品质性状的数量性状座位(QTL)进行了分析.在水稻的所有12 条染色体上共定位到7个加性主效QTL和19对上位性QTL,其中控制粒长、粒宽、长宽比的主效QTL各2个,控制垩白度的QTL 1个,分别解释12.8%、40.0%、26.0%和42.1%的表型变异;共检测到6对影响垩白度、6对影响粒长、7对影响长宽比的上位性QTL,分别 解释52.2%、31.3%和38.2% 的表型变异.结果表明,上位性QTL和主效QTL一样在稻米粒形和垩白度的遗传中起着重要的作用.
DOI:10.1016/j.cell.2017.04.032URL [本文引用: 1]
[本文引用: 4]
.
DOI:10.7666/d.Y2762071URL [本文引用: 1]
大豆是重要的粮油作物,目前我国大豆产量仅能满足国内1/3左右的消费量,因此大豆高产育种成为我国首要的育种目标。影响大豆产量重要因素之一是粒重。随着农业现代化的发展,机械化程度逐渐提高,粒形(长宽比、长厚比和宽厚比等)影响机械播种及其出苗的均一度,从而影响大豆产量;同时,粒形性状(长、宽、厚等)是构成籽粒大小的重要指标。籽粒性状为多基因控制的数量性状,通过研究其相关QTL位点,实现产量性状的分子标记辅助育种,对我国大豆产业具有重大应用意义。 本研究以216个冀豆12×黑豆(ZDD03651)的重组自交系(RILs)为材料,分别在石家庄和三亚两地三个年次获得F6:8和F6:9代家系粒重和粒型表型数据,利用在全基因组均匀分布的163对SSR多态引物构建大豆分子遗传图谱。定位在多个环境下的粒重和粒形相关QTL位点,并挖掘粒重相关上位性效应互作QTL。进一步以自然群体为材料挖掘粒重相关的优异等位变异,为该QTL位点在多环境下的广泛应用提供依据,为分子辅助育种奠定基础。 本研究结果如下: 1.构建完成一张包含20个连锁群163对SSR标记的大豆遗传连锁图谱,覆盖大豆基因组的1516cM,标记间平均距离为9.3cM。 2.共检测到控制百粒重的QTL位点5个,分别为qSDW-A2-1、qSDW-A2-2、qSDW-C2-1、qSDW-D1b-1和qSDW-D2-1。其中,qSDW-C2-1在石家庄和三亚环境下均可被检测到,贡献率最大为11.85%;qSDW-D2-1在石家庄多年环境下均可被检测到,贡献率最大为13.01%。此外,本研究还检测到对粒重具有上位性互作效应的QTL位点3对,分别位于A2、C2、D1b、D2、N和E连锁群上,其贡献率在7.80-16.67%之间。 3.检测到29个与粒形相关的QTL,其中qSL-D2-1、qSW-C2-1、qSW-D2-1、qST-A2-2、qST-C2-1,qST-D2-1,qSLW-C1-1、qSLW-D1b-3、qSLT-A2-2、qSLT-F-1和qSWT-D1b-1在多环境均可被检测到,其贡献率范围在7.71-14.76%。粒长、粒宽和粒厚的QTL增效基因均来自于冀豆12;长宽比、长厚比和宽厚比的QTL增效基因均来自于黑豆。 4.本研究中在5个粒重QTL位点上共检测到140个等位变异,进一步筛选到11个优异等位变异,分别为:sat_406-212、sat_406-214、sat_406-216、satt119-124、satt281-227、sat_279-259、B-2-304-245、satt301-199、satt301-259。
.
DOI:10.7666/d.Y2762071URL [本文引用: 1]
大豆是重要的粮油作物,目前我国大豆产量仅能满足国内1/3左右的消费量,因此大豆高产育种成为我国首要的育种目标。影响大豆产量重要因素之一是粒重。随着农业现代化的发展,机械化程度逐渐提高,粒形(长宽比、长厚比和宽厚比等)影响机械播种及其出苗的均一度,从而影响大豆产量;同时,粒形性状(长、宽、厚等)是构成籽粒大小的重要指标。籽粒性状为多基因控制的数量性状,通过研究其相关QTL位点,实现产量性状的分子标记辅助育种,对我国大豆产业具有重大应用意义。 本研究以216个冀豆12×黑豆(ZDD03651)的重组自交系(RILs)为材料,分别在石家庄和三亚两地三个年次获得F6:8和F6:9代家系粒重和粒型表型数据,利用在全基因组均匀分布的163对SSR多态引物构建大豆分子遗传图谱。定位在多个环境下的粒重和粒形相关QTL位点,并挖掘粒重相关上位性效应互作QTL。进一步以自然群体为材料挖掘粒重相关的优异等位变异,为该QTL位点在多环境下的广泛应用提供依据,为分子辅助育种奠定基础。 本研究结果如下: 1.构建完成一张包含20个连锁群163对SSR标记的大豆遗传连锁图谱,覆盖大豆基因组的1516cM,标记间平均距离为9.3cM。 2.共检测到控制百粒重的QTL位点5个,分别为qSDW-A2-1、qSDW-A2-2、qSDW-C2-1、qSDW-D1b-1和qSDW-D2-1。其中,qSDW-C2-1在石家庄和三亚环境下均可被检测到,贡献率最大为11.85%;qSDW-D2-1在石家庄多年环境下均可被检测到,贡献率最大为13.01%。此外,本研究还检测到对粒重具有上位性互作效应的QTL位点3对,分别位于A2、C2、D1b、D2、N和E连锁群上,其贡献率在7.80-16.67%之间。 3.检测到29个与粒形相关的QTL,其中qSL-D2-1、qSW-C2-1、qSW-D2-1、qST-A2-2、qST-C2-1,qST-D2-1,qSLW-C1-1、qSLW-D1b-3、qSLT-A2-2、qSLT-F-1和qSWT-D1b-1在多环境均可被检测到,其贡献率范围在7.71-14.76%。粒长、粒宽和粒厚的QTL增效基因均来自于冀豆12;长宽比、长厚比和宽厚比的QTL增效基因均来自于黑豆。 4.本研究中在5个粒重QTL位点上共检测到140个等位变异,进一步筛选到11个优异等位变异,分别为:sat_406-212、sat_406-214、sat_406-216、satt119-124、satt281-227、sat_279-259、B-2-304-245、satt301-199、satt301-259。
DOI:10.3969/j.issn.1000-7091.2012.06.002URL [本文引用: 1]
为进一步饱和大豆公共图谱SSR标记,以大豆育成品种冀豆12×地方品种ZDD03651组合的211个F6株系为作图群体,以Kosambi作图函数构建SSR标记遗传连锁图谱。结果表明,栽培大豆冀豆12与大豆地方品种ZDD03651间SSR标记多态率为44.6%,遗传图谱包含21个连锁群,117个SSR标记,遗传距离总长度1 501 cM,标记间平均距离15.6 cM,其中包含8个偏分离标记。与公共遗传图谱相比,位点间排列顺序、遗传距离和偏分离位点比例基本相同。将SSR新标记Barcsoyssr_4_1181、Barcsoyssr_4_1201、Barcsoyssr_4_1235和Barcsoyssr_5_1266整合到C1连锁群上,填补了国际大豆公共遗传图谱中C1连锁群94.62~120.12 cM之间的SSR标记空白区段。
DOI:10.3969/j.issn.1000-7091.2012.06.002URL [本文引用: 1]
为进一步饱和大豆公共图谱SSR标记,以大豆育成品种冀豆12×地方品种ZDD03651组合的211个F6株系为作图群体,以Kosambi作图函数构建SSR标记遗传连锁图谱。结果表明,栽培大豆冀豆12与大豆地方品种ZDD03651间SSR标记多态率为44.6%,遗传图谱包含21个连锁群,117个SSR标记,遗传距离总长度1 501 cM,标记间平均距离15.6 cM,其中包含8个偏分离标记。与公共遗传图谱相比,位点间排列顺序、遗传距离和偏分离位点比例基本相同。将SSR新标记Barcsoyssr_4_1181、Barcsoyssr_4_1201、Barcsoyssr_4_1235和Barcsoyssr_5_1266整合到C1连锁群上,填补了国际大豆公共遗传图谱中C1连锁群94.62~120.12 cM之间的SSR标记空白区段。
DOI:10.1023/A:1005711431474URLPMID:9291963 [本文引用: 1]
Microsatellites are simple, tandemly repeated di- to tetra-nucleotide sequence motifs flanked by unique sequences. They are valuable as genetic markers because they are co-dominant, detect high levels of allelic diversity, and are easily and economically assayed by the polymerase chain reaction (PCR). Results from screening a rice genomic library suggest that there are an estimated 5700-10 000 microsatellites in rice, with the relative frequency of different repeats decreasing with increasing size of the motif. A map consisting of 120 microsatellite markers demonstrates that they are well distributed throughout the 12 chromosomes of rice. Five multiple copy primer sequences have been identified that could be mapped to independent chromosomal locations. The current level of genome coverage provided by these simple sequence length polymorphisms (SSLPs) in rice is sufficient to be useful for genotype identification, gene and quantitative trait locus (QTL) analysis, screening of large insert libraries, and marker-assisted selection in breeding. Studies of allelic diversity have documented up to 25 alleles at a single locus in cultivated rice germplasm and provide evidence that amplification in wild relatives of Oryza sativa is generally reliable. The availability of increasing numbers of mapped SSLP markers can be expected to complement existing RFLP and AFLP maps, increasing the power and resolution of genome analysis in rice.
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s10681-006-9085-8URL [本文引用: 1]
It has been theoretically proposed that multiple linked quantitative trait loci (QTLs) play a role in the accumulation of hidden variation within and between populations. In this study, the genetic bases for grain characteristics were examined by comparing two accessions representing the two rice subspecies by QTL analysis. Grain dimensions are known to be quantitative traits and to be diagnostic between these two subspecies. To enhance the power to detect QTL with small effects, after transferring a segment of chromosome 6 from an Indica type into a Japonica type of rice by repeated backcrosses, the introgressed segment was dissected by making recombinant inbred lines (RILs) which were expected to have different sizes of the introgressed segment in the same genetic background. The resulting RILs showed distinct transgression of the grain characteristics examined. Multiple QTLs controlling each of the length and breadth of seeds were detected on the introgressed segment, and showed positive and negative additive effects as well as epistatic interactions. The present study confirmed that transgressive segregation resulted from a breakdown of linkage and that the detection of QTLs was highly dependent upon the genetic effects of the neighboring QTLs, indicating the need for caution in interpreting QTL effects.
.
DOI:10.1016/j.fcr.2004.06.004URL [本文引用: 1]
Seedling-vigor is important for optimum stand establishment and increasing weed competitive ability in rice cropping systems. In the current study, three seedling-vigor-related traits, seed germination rate, seedling shoot length and dry matter weight, were investigated by the paper-roll tests with rice recombinant inbred lines derived from a cross between Lemont ( japonica ) and Teqing ( indica ). The phenotype data, together with a linkage map consisting of 198 marker loci, was used to conduct composite interval mapping by QTLMapper 1.0 to simultaneously map both main-effect and epistatic QTLs for seedling-vigor in rice. Totally, 13 putative main-effect QTLs and 19 pairs of epistatic loci with R 2 ≥ 5% were identified. Almost all of these QTLs or interactions individually explained only around 5–10% of the phenotypic variation. The majority (68%) of these main-effect and epistatic loci were clustered in seven chromosome regions, each spanning 12–2802cM (centi-Morgan) and containing three or more detectable loci. When detectable for the multiple seedling-vigor-related traits, either the main-effect QTLs or the epistatic interactions sharing the same map location had their additive or epistatic effects in the same direction, which agreed well with the positive correlations among the traits. The results demonstrated that seedling-vigor in rice could be controlled by many loci, most of which had small effects, but, relatively, epistasis as a genetic factor was much more important than main-effects of QTLs. Along with the results reported previously, this study revealed the extensive genetic diversity for seedling-vigor in rice. In addition, the QTL q SV-7 on chromosome 7 was found to have the largest main-effects on multiple seedling-vigor-related traits and therefore could be used as a potential target to be genetically manipulated by marker-assisted selection in rice seedling-vigor breeding programs.
DOI:10.1007/s001220050628URL [本文引用: 1]
An F 2 and two equivalent F 3 populations of an indica-indica cross of rice, Tesanai 2/CB, were constructed and grown in different environments. The identification of quantitative trait loci (QTL) for yield components and plant height and an analysis of QTL脳environment interaction were conducted for three trials. Interval mapping of QTL for eight traits was employed with a threshold of LOD=2 using the computer package MAPMAKER/QTL . A total of 44 QTL were detected in 18 intervals of nine chromosomes, including 3 for the number of panicles (NP), 5 for the number of filled grains (NFG), 6 for total number of spikelets (TNS), 3 for spikelet fertility (SF), 7 for 1000-grain weight (TGWT), 5 for grain weight per plant (GWT), 8 for plant height (PH) and 7 for panicle length (PL). The numbers of QTL detected in two or three trials were 1 for NP, 1 for NFG, 1 for TNS, none for SF, 4 for TGWT, 3 for GWT, 2 for PH and 5 for PL, making a total of 17. When a QTL was detected in more than one trial the direction and magnitude of its additive effect, the dominance effect and the degree of dominance were generally in good agreement. In all three trials, QTL were frequently detected for related traits in the same intervals. The directions of additive effect of QTL for related traits in a given interval were in agreement with few exceptions, no matter whether they were detected in the same trial or not. This result suggested that pleiotropism rather than close linkage of different QTL was the major reason why QTL for different traits were frequently detected in the same intervals. When gene pleiotropism was considered, 23 of the 29 QTL for yield and its components and 9 of the 15 QTL for plant stature were detected in more than one trial. This indicated that the detection of chromosomal segments harboring QTL was hardly affected by environmental factors.
DOI:10.1016/j.tig.2011.05.007URL [本文引用: 1]
DOI:10.1105/tpc.17.00162URLPMID:28351993 [本文引用: 1]
Abstract The soybean (Glycine max) seed coat has distinctive, genetically programmed patterns of pigmentation and the recessive k1 mutation can epistatically overcome the dominant I and ii alleles, which inhibit seed color by producing small interfering RNAs (siRNAs) targeting chalcone synthase (CHS) mRNAs. Small RNA sequencing of dissected regions of immature seed coats demonstrated that CHS siRNA levels cause the patterns produced by the ii and ik alleles of the I locus, which restrict pigment to the hilum or saddle region of the seed coat, respectively. To identify the K1 locus, we compared RNA-Seq data from dissected regions of two Clark isolines having similar saddle phenotypes mediated by CHS siRNAs but different genotypes (homozygous ik K1 versus homozygous ii k1). By examining differentially expressed genes, mapping information, and genome resequencing, we identified a 129-bp deletion in Glyma.11G190900 encoding Argonaute5 (AGO5), a member of the Argonaute family. Amplicon sequencing of several independent saddle pattern mutants from different genetic backgrounds revealed independent lesions affecting AGO5, thus establishing Glyma.11G190900 as the K1 locus. Non-functional AGO5 from k1 alleles leads to altered distributions of CHS siRNAs, thus explaining how the k1 mutation reverses the phenotype of the seed coat regions from yellow to pigmented, even in the presence of the normally dominant I or ii alleles. {copyright, serif} 2017 American Society of Plant Biologists. All rights reserved.
DOI:10.1038/nrg1407URL [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}