摘要选用Xushu 18 (6 x)和Nancy Hall (6 x)、2个二倍体 I. trifida (2 x)品系(4597-10和4597-21)、四倍体 I. trifida (4 x)、六倍体 I. trifida (6 x)、二倍体 I. temussima (2 x)和 I. littorallis (2 x)以简化基因组测序技术SLAF-seq测序, 获得724 589个SLAF标签, 其中多态性SLAF标签35 310个。通过序列分析, 获得40 765个有效单核苷酸多态(SNP), 并用这些SNP分析了8个种质的群体结构和系统发生树。结果表明, 利用简化基因组测序技术SLAF-seq能高效、低成本地开发出大量可用于群体遗传分析的SNP标记; 通过构建进化树发现甘薯栽培种和野生种 I. trifida的亲缘关系比较近。这些分析结果为进一步研究甘薯栽培种的起源提供了基础数据。
关键词:甘薯; SLAF-seq; 分子标记; SNP; 进化分析 Analysis of Interspecific SNPs in Sweetpotato Using a Reduced-Representation Genotyping Technology SHI Xuan1, WANG Ru-Yuan1, TANG Jun2, LI Zong-Yun1,*, LUO Yong-Hai1,* 1 School of Life Science, Jiangsu Normal University, Xuzhou 221116, China
2 Sweetpotato Research Institute, Chinese Academy of Agricultural Sciences / National Sweetpotato Improvement Centre, Xuzhou 221121, China
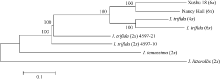
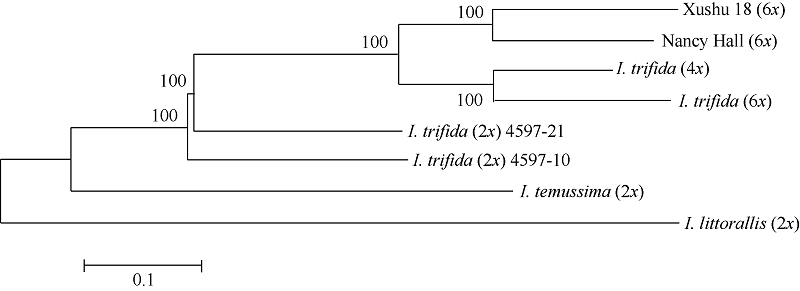
Fund:This study was supported by the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, the Project on Crop Conservation Funded by the Ministry of Agriculture of China (2015NWB006), the General Projects Funded by Natural Science Foundation of Jiangsu Province (BK2012579, BK20141146), and the Major Project Funded by Natural Science Foundation of Jiangsu Higher Education Institutions (12KJA180001) AbstractXushu 18 (6 x), Nancy Hall (6 x), I. trifida (2 x) 4597-10, I. trifida (2 x) 4597-21, I. trifida (4 x), I. trifida (6x), I. temussima (2 x), and I. littorallis (2 x) were used as experimental materials for sequencing by specific-locus amplified fragment sequencing (SLAF-seq), a high-throughput reduced-representation genotyping technology. In total, 724 589 SLAF tags were obtained and 40 765 SNPs were identified out of 35 310 polymorphic SLAF tags. A total of 40 765 single nucleotide polymorphisms (SNPs) were obtained by sequence analysis. Population structure and phylogenetic relationship of eight germplasm were analyzed using the SNP dataset, which suggests that SLAF-seq can be used to develop large-scale SNPs for population genetic analysis, effectively and economically. Our analysis revealed that the relationship between sweet potato cultivars and the wild species I. trifida is closer . These results provide empirical data for further study of the origin of sweet potato.
Keyword:Sweetpotato; SLAF-seq; Molecular marker; SNP; Phylogenetic analysis Show Figures Show Figures
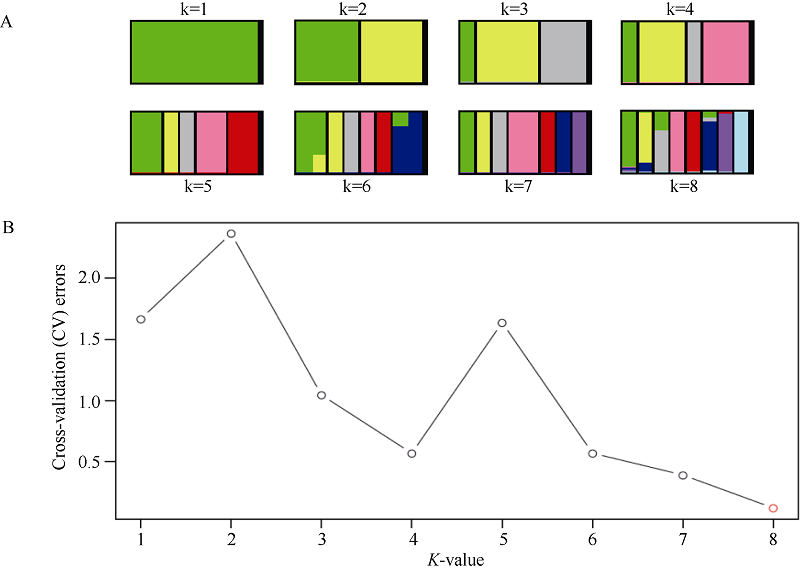
图1 基于所鉴定SNP分析群体结构A: 不同分群数(K值)的聚类结果显示无明显群体结构存在; B: 不同K值对应的交叉验证错误率显示k为8的时候交叉验证错误率最小。Fig. 1 Analysis of population structure by using identified SNPsA: cluster results of different structuring number (K-values) show no obvious population structure; B: cross validation error rates corresponding to different K-values show k = 8 is the best.
Land eR. Neutral theory of quantitative genetic variance in an island model with local extinction and colonization. Evolution, 1992, 46: 381-389[本文引用:1]
[2]
Huang JC, SunM. Genetic diversity and relationships of sweetpotato and its wild relatives in Ipomoea series Batatas (Convolvulaceae) as revealed by inter-simple sequence repeat (ISSR) and restriction analysis of chloroplast DNA. Theor Appl Genet, 2000, 100: 1050-1060[本文引用:4]
[3]
Jarret RL, GawelN, Whittemore A. Phylogenetic relationships of the sweetpotato [Ipomoea batatas (L. ) Lam. ]. J Am Soc Hort Sci, 1992, 117: 633-637[本文引用:3]
[4]
WrightS. The genetical structure of populations. Ann Eugen, 1951, 15: 323-354[本文引用:1]
[5]
Buteler MI, Jarret RL, LaBonteD R. Sequence characterization of microsatellites in diploid and polyploid Ipomoea. Theor Appl Genet, 1999, 99: 123-132[本文引用:1]
[6]
Huang JC, CorkeH, SunM. Highly polymorphic AFLP markers as a complementary tool to ITS sequences in assessing genetic diversity and phylogenetic relationships of sweetpotato (Ipomoea batatas (L. ) Lam. ) and its wild relatives. Genet Resour Crop Evol, 2000, 49: 541-550[本文引用:1]
[7]
RajapakseS, Nilmalgoda SD, MolnarM, Ballard RE, Austin DF, Bohac JR. Phylogenetic relationships of the sweetpotato in Ipomoea series Batatas (Convolvulaceae) based on nuclear beta-amylase gene sequences. Mol Phylogenet Evol, 2004, 30: 623-632[本文引用:1]
[8]
SrisuwanS, SihachakrD, Siljak-YakovlevS. The origin and evolution of sweet potato (Ipomoea batatas (L. ) Lam. ) and its wild relatives through the cytogenetic approaches. Plant Sci, 2006, 171: 424-433[本文引用:1]
[9]
Jarret RL, Austin DF. Genetic diversity and systematic relationship in sweetpotato (Ipomoea batatas (L. ) Lam. ) and related species as revealed by RAPD analysis. Gen Resour Crop Evol, 1994, 41: 165-173[本文引用:2]
[10]
贺学勤, 刘庆昌, 翟红, 王玉萍. 用RAPD、ISSR和AFLP标记分析系谱关系明确的甘薯品种的亲缘关系. 作物学报, 2005, 10: 1300-1304He XQ, Liu QC, ZhaiH, Wang YP. The use of RAPD, ISSR and AFLP markers for analyzing genetic relationships among sweetpotato cultivars with known origin. Acta Agron Sin, 2005, 10: 1300-1304 (in Chinese with English abstract)[本文引用:2]
[11]
KobayashiM. The Ipomoea trifida complex closely related to sweet potato. In: Shideler S F, Rincon H, eds. Proceedings of the 6th Symposium of the International Society of Tropical Root Crop. Lima, Peru: CIP. 1984. pp 561-568[本文引用:3]
[12]
RoullierC, KambouoR, PaofaJ, MckeyD, LebotV. On the origin of sweet potato (Ipomoea batatas (L. ) Lam. ) genetic diversity in New Guinea, a secondary center of diversity. Heredity, 2013, 1-11[本文引用:3]
[13]
HeffelfingerC, Fragoso CA, Moreno MA, Overton JD, Mottinger JP, Zhao HY, TohmeJ, Dellaporta SL. Flexible and scalable genotyping-by-sequencing strategies for population studies. BMC Genom, 2014, 15: 979-1001[本文引用:1]
[14]
王洋坤, 胡艳, 张天真. RAD-seq技术在基因组研究中的现状及展望. 遗传, 2014, 36: 41-49Wang YK, HuY, Zhang TZ. Current status and perspective of RAD-seq in genomic research. Hereditas (Beijing), 2014, 36: 41-49 (in Chinese with English abstract)[本文引用:1]
[15]
Crow JF, KimuraM. Population genetics. (Book Reviews: An introduction to population genetics theory). Science, 1971, 171: 666--667[本文引用:]
[16]
SunX, Liu DY, Zhang XF, Li WB, LiuH, Hong WG, Jiang CB, GuanN, Ma CX, Zeng HP, Xu CH, SongJ, HuangL, Wang CM, Shi JJ, WangR, Zheng XH, Lu CY, Wang XW, Zheng HK. SLAF-seq: an efficient method of large-scale De novo SNP discovery and genotyping using high-throughput sequencing. PloS One, 2013, 8: e58700[本文引用:1]
[17]
Davey JW, CezardT, Fuentes-UtrilaP, Eland C, GharbiK, Blaxter ML. Special features of RAD Sequencing data: implications for genotyping. Mol Ecol, 2013, 22: 3151-3164[本文引用:2]
Alexand er DH, NovembreJ, LangeK. Fast model-based estimation of ancestry in unrelated individuals. Genome Res, 2009, 19: 1655-1664[本文引用:2]
[20]
HoonM J L D, ImotoS, NolanJ, MiyanoS. Open source clustering software. Bioinformatics, 2004, 20: 1453-1454[本文引用:1]
[21]
TamuraK, PetersonD, PetersonN, StecherG, NeiM, KumarS. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol, 2011, 28: 2731-2739[本文引用:1]
[22]
SaitouN, NeiM. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol, 1987, 4: 406-425[本文引用:1]
[23]
KriegnerA, Cervantes JC, BurgK, MwangaR O M, ZhangD P. A genetic linkage map of sweetpotato [Ipomoea batatas (L. ) Lam. ] based on AFLP markers. Mol Breed, 2003, 11: 169-185[本文引用:2]
[24]
Cervantes JC, Yencho GC, KriegnerA, Pecota KV, Faulk MA, MwangaR O M, SosinskiB R. Development of a genetic linkage map and identification of homologous linkage groups in sweetpotato using multiple-dose AFLP markers. Mol Breed, 2008, 21: 511-532[本文引用:1]
[25]
吴洁, 谭文芳, 何俊蓉, 蒲志刚, 王大一, 阎文昭. 甘薯SRAP连锁图构建淀粉含量QTL检测. 植物分子育种, 2005, 6: 841-845WuJ, Tan WF, He JR, Pu ZG, Wang DY, Yan WZ. Construct on of SRAP linkage map and qtl mapping for starch content in sweet potato. Mol Plant Breed, 2005, 6: 841-845 (in Chinese with English abstract)[本文引用:1]
[26]
唐茜, 何凤发, 王季春, 王瑞娜. 甘薯SRAP遗传图谱构建及淀粉含量QTL初步定位. 西南大学学报(自然科学版), 2010, 32(6): 40-45TangQ, He FF, Wang JC, Wang RN. Construct on of SRAP genetic map and qtl mapping for starch content in sweet potato. J Southwest Univ (Nat Sci Edn), 2010, 32(6): 40-45 (in Chinese)[本文引用:1]
[27]
蒲志刚, 王大一, 谭文芳, 吴洁, 阎文昭. 利用AFLP构建甘薯连锁图及淀粉含量QTL定位. 西南农业学报, 2010, 23: 1047-1050Pu ZG, Wang DY, Tan WF, WuJ, Yan WZ. AFLP maps and QTL analysis of starch content of sweet potato. Southwest China J Agric Sci, 2010, 23: 1047-1050 (in Chinese with English abstract)[本文引用:1]
[28]
李爱贤, 刘庆昌, 王庆美, 张立明, 翟红, 刘树震. 利用SRAP标记构建甘薯分子连锁图谱. 作物学报, 2010, 36: 1286-1295Li AX, Liu QC, Wang QM, Zhang LM, ZhaiH, Liu SZ. Establishment of molecular linkage maps using SRAP markers in sweet potato. Acta Agron Sin, 2010, 36: 1286-1295 (in Chinese with English abstract)[本文引用:1]
[29]
ZhaoN, Yu XX, JieQ, LiH, LiH, HuJ, ZhaiH, He SZ, Liu QC. A genetic linkage map based on AFLP and SSR markers and mapping of QTL for dry-matter content in sweetpotato. Mol Breed, 2013, 32: 807-820[本文引用:1]
[30]
LiH, VikramP, Singh RP, KilianA, CarlingJ, SongJ, Burgueno-Ferreira JA, BhavaniS, Huerta-EspinoJ, PayneT, SehgalD, WenzlP, SinghS. A high density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genomics, 2015, 16: 1-15[本文引用:1]
[31]
SrisuwanS, SihachakrD, Siljak-YakovlevS. The origin and evolution of sweet potato (Ipomoea batatas (L. ) Lam. ) and its wild relatives through the cytogenetic approaches. Plant Sci, 2006, 171: 424-433[本文引用:2]
[32]
NishiyamaI. Evaluation and domestication of the sweet potato. Bot Mag, 1971, 84: 377-387[本文引用:1]
[33]
Austin DF. The taxonomy, evolution and genetic diversity of sweet potatoes and related wild species. In: Gregory P ed. Exploration, Maintenance, and Utilization of Sweet Potato Genetic Resources. 1988, pp 27-60[本文引用:2]
[34]
RoullierC, DuputiéA, WennekesP, BenoitL, Fernández BringasV M, RosselG, TayD, McKeyD, LebotV. Disentangling the origins of cultivated sweet potato (Ipomoea batatas (L. ) Lam. ). PLoS One, 2013, 8: e62707[本文引用:2]
[35]
Otto SP. The evolutionary consequences of polyploidy. Cell, 2007, 131: 452-462[本文引用:2]
[36]
ZoharyD. Unconscious selection and the evolution of domesticated plants. Econ Bot, 2004, 58: 5-10[本文引用:1]
[37]
Allaby RG, Fuller DQ, Brown TA. The genetic expectations of a protracted model for the origins of domesticated crops. Proc Natl Acad Sci USA, 2008, 105: 13982-13986[本文引用:1]
 , 罗永海
, 罗永海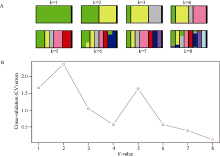

{kind=link}
{kind=link}