关键词:玉米; 穗行数; 全基因组关联分析; 候选基因 Genome-wide Association Analysis of Kernel Row Number in Maize ZHANG Huan-Xin, WENG Jian-Feng*, ZHANG Xiao-Cong, LIU Chang-Lin, YONG Hong-Jun, HAO Zhuan-Fang, LI Xin-Hai* Institute of Crop Science, Chinese Academy of Agricultural Sciences / National Engineer Laboratory of Crop Molecular Breeding, Beijing 100081, China
AbstractKernel row number (KRN) is one of grain yield components in maize (Zea mays L.). Investigation of its genetic architecture will help develop high-yield varieties in maize. In this study, the KRN in a panel of 203 maize inbred lines was detected in Urumqi of Xinjiang, Gongzhuling of Jilin, and Sanya of Hainan in 2007, and used to perform the genome-wide analysis for KRN using MaizeSNP50 BeadChip. A total of nine SNPs were found to be significantly associated with KRN at a threshold ofP< 0.0001, which were on chromosome Bins 1.02, 1.10, 7.03, 8.02, 9.06, and 10.03, respectively. Eight of these SNPs were located in the QTL intervals reported previously. Meanwhile, four candidate genes were scanned, encoding auxin signaling F-box containing protein, kn1 protein, AP2 domain containing protein and leucine-rich repeat transmembrane protein kinase respectively. In summary, these identified genes and SNPs will offer essential information for cloning yield-related genes in maize.
Keyword:Maize; Kernel row number; Genome-wide association analysis; Candidate gene Show Figures Show Figures
图1 穗行数全基因组关联分析XJ、JL和HN分别代表新疆、吉林和海南。黑色水平线代表全基因组关联分析的显著阈值。Fig. 1 Genome-wide association study of kernel row number with mixed linear modelXJ, JL, and HN indicate experiments at Xinjiang, Jilin and Hainan, respectively. Black horizontal line indicates the genome-wide significance threshold.
表2 Table 2 表2(Table 2)
表2 与穗行数显著关联的SNP位点( P< 0.0001) Table 2 SNPs identified to be associated with kernel row number ( P< 0.0001)
中的QTL与本文检测到的SNP具有一致性。 MAF: minor allele frequencies. There exists consistency between SNP and QTL interval reported in the references. XJ, JL, and HN indicate experiments at Xinjiang, Jilin and Hainan, respectively.
表2 与穗行数显著关联的SNP位点( P< 0.0001) Table 2 SNPs identified to be associated with kernel row number ( P< 0.0001)
KerstetterR A, Laudencia-ChingcuancoD, SmithL G, HakeS. Loss-of-function mutations in the maize homeobox gene, knotted1, are defective in shoot meristem maintenance. Development, 1997, 124: 3045-3054[本文引用:2][CJCR: 0.1123]
[2]
DhillonB S, SinghJ. Estimation and inheritance of stability parameters of grain yield in maize. J Agric Sci, 1977, 88: 257-265[本文引用:1][JCR: 2.041]
[3]
LimaM L A, SouzaC L, BentoD A V, SouzaA P, Carlini-GarciaL A. Mapping QTL for grain yield and plant traits in a tropical maize population. Mol Breed, 2006, 17: 227-239[本文引用:1][JCR: 2.852]
[4]
MaX Q, TangJ H, TengW T, YanJ B, MengY J, LiJ S. Epistatic interaction is an important genetic basis of grain yield and its components in maize. Mol Breed, 2007, 20: 41-51[本文引用:2][JCR: 2.852]
[5]
LuM, XieC X, LiX H, HaoZ F, LiM S, WengJ F, ZhangD G, BaiL, ZhangS H. Mapping of quantitative trait loci for kernel row number in maize across seven environments. Mol Breed, 2010, 28: 143-152[本文引用:4][JCR: 2.852]
[6]
GuoJ J, ChenZ L, LiuZ P, WangB B, SongW B, LiW, ChenJ, DaiJ G, LaiJ S. Identification of genetic factors affecting plant density response through QTL mapping of yield component traits in maize (Zea mays L. ). Euphytica, 2011, 182: 409-422[本文引用:2][JCR: 1.554]
[7]
谭巍巍, 李永祥, 王阳, 刘成, 刘志斋, 彭勃, 王迪, 张岩, 孙宝成, 石云素, 宋燕春, 杨德光, 王天宇, 黎裕. 在干旱和正常水分条件下玉米穗部性状QTL分析. 作物学报, 2011, 37: 235-248TanW W, LiY X, WangY, LiuC, LiuZ Z, PengB, WangD, ZhangY, SunB C, ShiY S, SongY C, YangD G, WangT Y, LiY. QTL mapping of ear traits of maize under different water regimes. Acta Agron Sin, 2011, 37: 235-248 (in Chinese with English abstract)[本文引用:2][CJCR: 1.8267]
[8]
江培顺, 张焕欣, 李博, 郝转芳, 吕香玲, 李明顺, 王宏伟, 慈晓科, 张世煌, 李新海, 翁建峰, 史振声. 玉米产量相关性状Meta-QTL及候选基因分析. 作物学报, 2013, 39: 969-978JiangP S, ZhangH X, LiB, HaoZ F, LüX L, LiM S, WangH W, CiX K, ZhangS H, LiX H, WengJ F, ShiZ S. Analysis of Meta-QTL and cand idate genes related to yield components in maize. Acta Agron Sin, 2013, 39: 969-978 (in Chinese with English abstract)[本文引用:1][CJCR: 1.8267]
[9]
YuJ M, BucklerE S. Genetic association mapping and genome organization of maize. Curr Opin Biotechnol, 2006, 17: 155-160[本文引用:2][JCR: 7.711]
[10]
ZhuC S, GoreM, BucklerE S, YuJ M. Status and prospects of association mapping in plants. Plant Genome, 2008, 1: 5-20[本文引用:2]
[11]
HuangX H, ZhaoY, WeiX H, LiC Y, WangA H, ZhaoQ, LiW J, GuoY L, DengL W, ZhuC R, FanD L, LuY Q, WengQ J, LiuK Y, ZhouT Y, JingY F, SiL Z, DongG J, HuangT, LuT T, FengQ, QianQ, LiJ Y, HanB. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet, 2012, 44: 32-39[本文引用:2][JCR: 35.532]
[12]
WengJ F, XieC X, HaoZ F, WangJ J, LiuC L, LiM S, ZhangD G, BaiL, ZhangS H, LiX H. Genome-wide association study identifies cand idate genes that affect plant height in Chinese elite maize (Zea mays L. ) inbred lines. PLoS One, 2011, 6: e29229[本文引用:5][JCR: 4.092]
[13]
WengJ F, LiuX J, WangZ H, WangJ J, ZhangL, HaoZ F, XieC X, LiM S, ZhangD G, BaiL, LiuC L, ZhangS H, LiX H. Molecular mapping of the major resistance quantitative trait locus qHS2. 09 with simple sequence repeat and single nucleotide polymorphism markers in maize. Phytopathology, 2012, 102: 692-699[本文引用:1][JCR: 2.799]
[14]
TianF, BradburyP J, BrownP J, HungH, SunQ, Flint-GarciaS, RochefordT R, McMullenM D, Holland J B, BucklerE S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet, 2011, 43: 159-162[本文引用:2][JCR: 35.532]
[15]
BucklerE S, Holland J B, BradburyP J, AcharyaC B, BrownP J, BrowneC, ErsozE, Flint-GarciaS, GarciaA, GlaubitzJ C, GoodmanM M, HarjesC, GuillK, KroonD E, LarssonS, LepakN K, LiH H, MitchellS E, PressoirG, PeifferJ A, RosasM O, RochefordT R, RomayM C, RomeroS, SalvoS, VilledaH S, SunQ, TianF, UpadyayulaN, WareD, YatesH, YuJ M, ZhangZ W, KresovichS, McMullenM D. The genetic architecture of maize flowering time. Science, 2009, 325: 714-718[本文引用:2]
[16]
BrownP J, UpadyayulaN, MahoneG S, TianF, BradburyP J, MylesS, Holland J B, Flint-GarciaS, McMullenM D, BucklerE S, RochefordT R. Distinct genetic architectures for male and female inflorescence traits of maize. PLoS Genet, 2011, 7: e1002383[本文引用:2][JCR: 8.694]
[17]
MurrayM G, ThompsonW F. Rapid isolation of high molecular weight plant DNA. Nucl Acids Res, 1980, 8: 4321-4326[本文引用:1]
[18]
KnappS J, StroupW W, RossW M. Exact confidence intervals for heritability on a progeny mean basis. Crop Sci, 1985, 25: 192-194[本文引用:1][JCR: 1.641]
[19]
PritchardJ K, StephensM, DonnellyP. Inference of population structure using multilocus genotype data. Genetics, 2000, 155: 945-959[本文引用:1][JCR: 4.007]
[20]
HardyO J, VekemansX. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes, 2002, 2: 618-620[本文引用:1][JCR: 2.384]
[21]
BradburyP J, ZhangZ W, KroonD E, CasstevensT M, RamdossY, BucklerE S. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 2007, 23: 2633-2635[本文引用:1][JCR: 5.468]
[22]
刘宗华, 汤继华, 卫晓轶, 王春丽, 田国伟, 胡彦民, 陈伟程. 氮胁迫和正常条件下玉米穗部性状的QTL分析. 中国农业科学, 2007, 40: 2409-2417LiuZ H, TangJ H, WeiX Y, WangC L, TianG W, HuY M, ChenW C. QTL mapping of ear traits under low and high nitrogen conditions in maize. Sci Agric Sin, 2007, 40: 2409-2417 (in Chinese with English abstract)[本文引用:1][CJCR: 1.4522]
[23]
SmithL G, GreeneB, VeitB, HakeS. A dominant mutation in the maize homeobox gene, knotted-1, causes its ectopic expression in leaf cells with altered fates. Development, 1992, 116: 21-30[本文引用:1][CJCR: 0.1123]
[24]
KumpK L, BradburyP J, WisserR J, BucklerE S, BelcherA R, Oropeza-RosasM A, ZwonitzerJ C, KresovichS, McMullenM D, WareD, Balint-KurtiP J, Holland J B. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat Genet, 2011, 43: 163-168[本文引用:1][JCR: 35.532]
[25]
Poland J A, BradburyP J, BucklerE S, NelsonR J. Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc Natl Acad Sci USA, 2011, 108: 6893-6898[本文引用:1][JCR: 9.737]
[26]
LiY, HuangY, BergelsonJ, NordborgM, BorevitzJ O. Association mapping of local climate-sensitive quantitative trait loci in Arabidopsis thaliana. Proc Natl Acad Sci USA, 2010, 107: 21199-21204[本文引用:1][JCR: 9.737]
[27]
MassmanJ, CooperB, HorsleyR, NeateS, Dill-MackyR, ChaoS, DongY, SchwarzP, MuehlbauerG J, SmithK P. Genome-wide association mapping of fusarium head blight resistance in contemporary barley breeding germplasm. Mol Breed, 2011, 27: 439-454[本文引用:2][JCR: 2.852]
[28]
Flint-GarciaS A, ThornsberryJ M, BucklerE S. Structure of linkage disequilibrium in plants. Annu Rev Plant Biol, 2003, 54: 357-374[本文引用:1][JCR: 25.962]
[29]
BarrettJ C, FryB, MallerJ, DalyM J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics, 2005, 21: 263-265[本文引用:1][JCR: 5.468]
[30]
杨小红, 严建兵, 郑艳萍, 余建明, 李建生. 植物数量性状关联分析研究进展. 作物学报, 2007, 33: 523-530YangX H, YanJ B, ZhengY P, YuJ M, LiJ S. Reviews of association analysis for quantitative traits in plants. Acta Agron Sin, 2007, 33: 523-530 (in Chinese with English abstract)[本文引用:1][CJCR: 1.8267]
[31]
AranzanaM J, KimS, ZhaoK Y, BakkerE, HortonM, JakobK, ListerC, MolitorJ, ShindoC, TangC L, ToomajianC, TrawB, ZhengH G, BergelsonJ, DeanC, MarjoramP, NordborgM. Genome-wide association mapping in Arabidopsis thaliana identifies previously known genes responsible for variation in flowering time and pathogen resistance. PLoS Genet, 2005, 1: 531-539[本文引用:1][JCR: 8.694]
[32]
HuangX H, WeiX H, SangT, ZhaoQ, FengQ, ZhaoY, LiC Y, ZhuC R, LuT T, ZhangZ W, LiM, FanD L, GuoY L, WangA H, WangL, DengL W, LiW J, LuY Q, WengQ J, LiuK Y, HuangT, ZhouT Y, JingY F, LiW, LinZ, BucklerE S, QianQ, ZhangQ F, LiJ Y, HanB. Genome-wide association studies of 14 agronomic traits in rice land races. Nat Genet, 2010, 42: 961-967[本文引用:1][JCR: 35.532]
[33]
KepinskiS, LeyserO. The Arabidopsis F-box protein TIR1 is an auxin receptor. Nature, 2005, 435: 446-451[本文引用:1][JCR: 36.28]
[34]
VollbrechtE, ReiserL, HakeS. Shoot meristem size is dependent on inbred background and presence of the maize homeobox gene, knotted1. Development, 2000, 127: 3161-3172[本文引用:1][CJCR: 0.1123]
[35]
ChuckG, MeeleyR B, HakeS. The control of maize spikelet meristem fate by the APETALA2-like gene indeterminate spikelet1. Genes Dev, 1998, 12: 1145-1154[本文引用:1][JCR: 11.659]
[36]
ChuckG, MeeleyR B, HakeS. Floral meristem initiation and meristem cell fate are regulated by the maize AP2 genes ids1 and sid1. Development, 2008, 135: 3013-3019[本文引用:1][CJCR: 0.1123]
[37]
LeeD Y, AnG. Two AP2 family genes, supernumerary bract (SNB) and osindeterminate spikelet 1 (OsIDS1), synergistically control inflorescence architecture and floral meristem establishment in rice. Plant J, 2012, 69: 445-461[本文引用:1][JCR: 6.16]
[38]
BommertP, LundeC, NardmannJ, VollbrechtE, RunningM, JacksonD, HakeS, WerrW. thick tassel dwarf1 encodes a putative maize ortholog of the Arabidopsis CLAVATA1 leucine- rich repeat receptor-like kinase. Development, 2005, 132: 1235-1245[本文引用:1][CJCR: 0.1123]
[39]
Taguchi-ShiobaraF, YuanZ, HakeS, JacksonD. The fasciated ear2 gene encodes a leucine-rich repeat receptor-like protein that regulates shoot meristem proliferation in maize. Genes Dev, 2001, 15: 2755-2766[本文引用:1][JCR: 11.659]
[40]
BommertP, NagasawaN S, JacksonD. Quantitative variation in maize kernel row number is controlled by the FASCIATED EAR2 locus. Nat Genet, 2013, 45: 334-337[本文引用:1][JCR: 35.532]
 , 翁建峰
, 翁建峰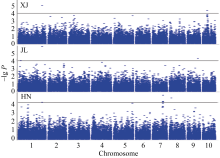


{kind=link}
{kind=link}