全文HTML
--> --> -->单层CrBr3作为二维铁磁材料CrX3 (X = I, Br, Cl)中的一员, 已经在实验上被成功制备[23,24]. 单层CrBr3具有本征铁磁性和半导体性质, 是自旋电子器件的潜在候选者. 研究发现, 石墨烯/CrBr3范德瓦耳斯异质结由于其界面处磁场交换的可调控性, 有利于存储器件的应用[25]. 此外, 单层MoSe2与CrBr3接触后, 层间的电荷转移导致了磁光响应, 可用于新型光学探针的研发[26]. 但是, 单层CrBr3的居里温度TC仅有41 K[27], 远低于室温, 这极大地限制了其在自旋电子学领域的应用. 因此, 如何提高CrBr3的居里温度, 增强其铁磁稳定性是至关重要的问题.
掺杂是调控二维材料电学和磁学性能的最常用方法之一. 例如, Cheng等[28]基于第一性原理方法预测了Mn, Fe, Co和Zn原子掺杂可以使单层MoS2产生磁性. Li等[29]采用化学气相输运法成功制备了铁掺杂的SnS2单晶, 利用振动样品磁强计观察到了铁磁性, 测得居里温度为31 K. 同样地, 第一性原理方法预测铜掺杂可以将单层ZnO转变为半金属铁磁体[30]. 碱金属掺杂显著增加了单层Cr2Ge2Te6的磁各向异性和居里温度, 实现了铁磁稳定性的增强[31]. 锂原子的掺杂可以提升CrI3体系的磁矩和居里温度, 从而实现CrI3铁磁稳定性的增强[32].
本文基于密度泛函理论, 研究了3d过渡金属(TM)掺杂对单层CrBr3电学和磁学性能的影响. 研究发现, 过渡金属掺杂可以显著增强单层CrBr3的铁磁稳定性, 这主要归因于直接交换和超交换相互作用之间的竞争. 此外, 依赖于不同的TM原子掺杂, TM-CrBr3体系表现出半金属性和自旋零带隙半导体(spin gapless semiconductor, SGS)性质. 这些结果表明, 过渡金属掺杂是调控单层CrBr3电学和磁学性能的有效方法, 有助于推动单层CrBr3在纳米电子和自旋电子器件方面的应用.



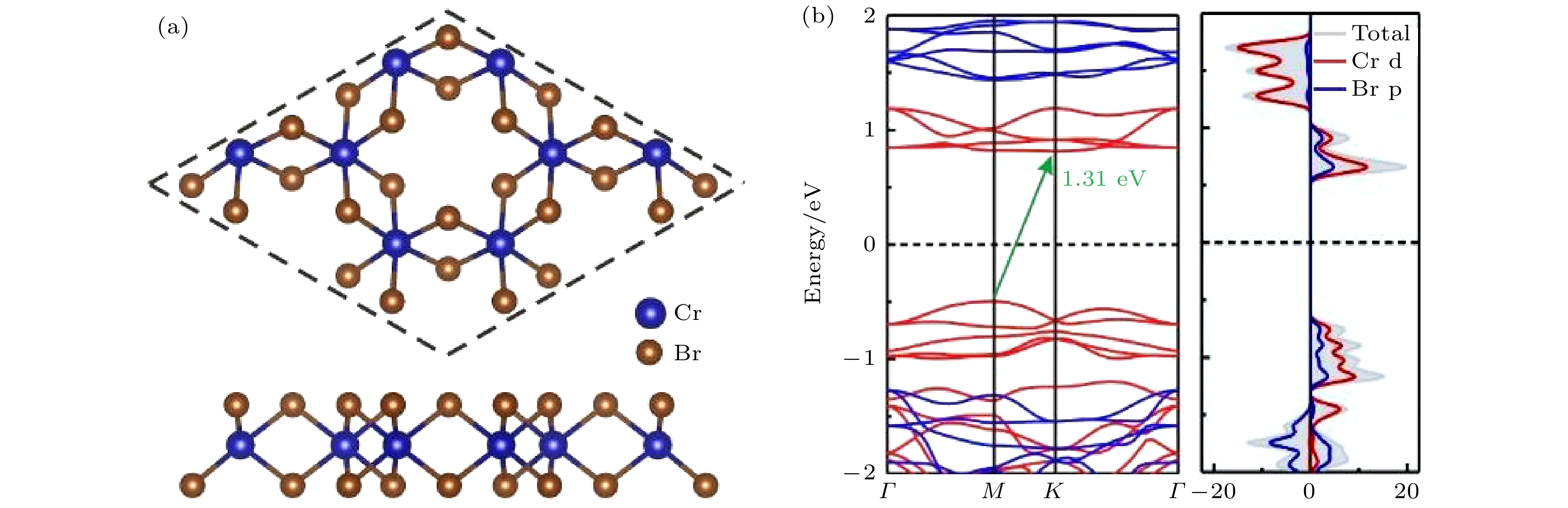 图 1 (a) 单层CrBr3的结构示意图; (b) 单层CrBr3的能带结构和态密度. 能带结构中自旋向上和自旋向下分别用红色实线和蓝色实线表示
图 1 (a) 单层CrBr3的结构示意图; (b) 单层CrBr3的能带结构和态密度. 能带结构中自旋向上和自旋向下分别用红色实线和蓝色实线表示Figure1. (a) Structural diagram of CrBr3 monolayer; (b) band structure and density of states of CrBr3 monolayer. The red and blue solid lines indicate spin-up and spin-down channels in the band structures, respectively.
首先, 选取了2 × 2 × 1的CrBr3超胞来构建3d过渡金属 (TM = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu和Zn) 掺杂单层CrBr3的模型结构. 根据CrBr3的结构特点, 考虑了3种可能的过渡金属原子掺杂位点: Br原子构成的六元环中心(H), Cr原子的上方(Cr-Top) 和Br原子的上方(Br-Top), 如图2(a)—(c)所示. 通过比较不同掺杂位点对应的TM-CrBr3结构的形成能, 确定最稳定的模型结构. 形成能定义为
 图 2 (a)?(c) TM原子分别掺杂在H, Cr-Top和Br-Top位点时TM-CrBr3晶体结构的俯视图和侧视图; (d) TM-CrBr3的形成能; (e) 在H构型中, TM原子到CrBr3表层Br原子的高度以及TM原子与最邻近Br原子共价键的键长
图 2 (a)?(c) TM原子分别掺杂在H, Cr-Top和Br-Top位点时TM-CrBr3晶体结构的俯视图和侧视图; (d) TM-CrBr3的形成能; (e) 在H构型中, TM原子到CrBr3表层Br原子的高度以及TM原子与最邻近Br原子共价键的键长Figure2. Top and side views of the crystalline structure of three different doped positions of TM atoms labeled as (a) H, (b) Cr-Top and (c) Br-Top; (d) the formation energy of TM-CrBr3; (e) the height of the TM to Br on the surface of CrBr3 and the length of covalent bond between TM and nearest Br atom.



根据形成能的定义, 掺杂体系的形成能越低, 稳定性越高. 图2(d)是不同掺杂位点对应的TM-CrBr3的形成能, 发现相比Cr-Top和Br-Top位点, H位点对应的TM-CrBr3的形成能最低, 这意味着3d TM原子掺杂在CrBr3的H位点时体系更稳定. 因此, 在后续的讨论中, 将研究重点放在H构型的TM-CrBr3体系. 从图2(d)看出TM-CrBr3 (Zn除外)的形成能较低(–1.19 至 –5.32 eV), 表明TM-CrBr3的稳定性较强. 其中, Sc-CrBr3具有最低的形成能(–5.32 eV), 表明Sc-CrBr3的结构最稳定. 而对于满壳层的Zn原子而言, 最外层的12个电子完全填满了3d和4s轨道, 因此, Zn与单层CrBr3的结合较弱, 形成能最高(–0.47 eV), 体系结构的稳定性较差. 进一步, 基于DFT的分子动力学模拟(DFT-MD), 研究了TM-CrBr3掺杂体系H构型在300 K的热稳定性. 结果表明, H构型在5 ps后晶体结构保持稳定(图3). 此外, 在H构型中, TM原子与最邻近的6个Br原子以共价键的形式连接, TM—Br的键长为2.59—2.68 ?, TM原子到CrBr3表面的高度h在1.45—1.54 ?的范围内 (如图2(e)所示).
 图 3 在300 K下, TM-CrBr3掺杂体系的H构型在5 ps后分子动力学模拟的结构示意图
图 3 在300 K下, TM-CrBr3掺杂体系的H构型在5 ps后分子动力学模拟的结构示意图Figure3. Snapshots of TM-CrBr3 on the H site taken after 5 ps of DFT-MD simulations at 300 K.
接下来, 研究了TM-CrBr3的磁学性能. 为了便于描述, 将Cr原子的磁矩、TM原子的磁矩和TM-CrBr3的总磁矩分别命名为MCr, MTM和Mtotal. 与本征CrBr3的MCr (3.00 μB) 相比, TM-CrBr3中与TM原子最邻近Cr原子的MCr有所增加(3.08 μB—3.31 μB), 如图4(a)所示. 产生这种现象的原因是Cr原子获得了来自于TM原子的部分电荷(图4(b)). 不同的TM原子之间存在电负性的差异, 即TM原子在化合物中吸引电子的能力不同, 导致了TM-CrBr3中电荷转移的差异. 在所有TM-CrBr3中, Sc-CrBr3中Cr原子获得的电荷最多, 约为0.06 e, 因而对应的MCr最大 (3.31 μB). 与Sc-CrBr3相反, Cu-CrBr3中Cr原子获得的电荷最少, 约为0.02 e, 所以MCr最小 (3.08 μB).
 图 4 (a) H构型的TM-CrBr3中TM原子的磁矩以及与TM原子最近邻的Cr原子的磁矩; (b) TM-CrBr3中Cr和TM原子的电荷转移; (c) TM-CrBr3体系的总磁矩(Mtotal)
图 4 (a) H构型的TM-CrBr3中TM原子的磁矩以及与TM原子最近邻的Cr原子的磁矩; (b) TM-CrBr3中Cr和TM原子的电荷转移; (c) TM-CrBr3体系的总磁矩(Mtotal)Figure4. (a) Magnetic moments of TM atom and Cr atom nearest to TM atom in TM-CrBr3 of H configuration; (b) charge transfer between Cr and TM atoms in TM-CrBr3; (c) the total magnetic moments (Mtotal) of TM-CrBr3.
在TM-CrBr3体系中, 磁性原子为Cr和TM原子, 总磁矩(Mtotal)主要由Cr原子贡献, 部分来源于TM原子. 根据Mtotal的变化趋势(图4(c)), 掺杂体系可分为3类. 1) Sc, Ti, V, Cr, Mn掺杂的CrBr3体系, 其Mtotal大于本征CrBr3的Mtotal (24 μB), 随着掺杂元素原子序数的增加 (从Sc到Mn), 其Mtotal是线性增加的. 进一步的观察发现, 掺杂体系中增加的磁矩与掺杂TM原子的最外层电子数有关, 如图4(c)所示. 例如, Sc最外层的4s和3d轨道的电子共有3个, 因此, Sc-CrBr3体系的Mtotal (27 μB) 比本征CrBr3的总磁矩 (24 μB) 增加了3 μB. 随着TM原子序数的增加, 到Mn原子时, 最外层的4s和3d轨道的电子数线性增加到7, 此时掺杂体系的Mtotal从27 μB线性增加到31 μB (从Sc到Mn). 2) Fe-CrBr3, Co-CrBr3和Ni-CrBr3体系的Mtotal为分数, 整体大致呈现为线性下降趋势, Mtotal分别为29.08 μB, 27.43 μB和26.69 μB. 3) Cu-CrBr3和Zn-CrBr3体系, Mtotal分别为25 μB和26 μB. 在Cu-CrBr3体系中, Cu原子最外层共有11个电子, 其中10个电子填满了所有的3d轨道[41], 剩下的一个未配对的游离电子贡献了磁矩, 使Mtotal比本征CrBr3的总磁矩增加了1 μB. 对于Zn-CrBr3体系, Zn原子最外层3d和4s轨道被12个电子完全填充, 导致Zn原子的净磁矩为零, 因此Zn-CrBr3体系的Mtotal几乎完全来自于Cr原子. 总的来说, 随着TM原子序数的增加, 掺杂体系的Mtotal大致呈现先增加, 再减小的趋势. 在3d过渡金属掺杂单层CrI3, MoSe2和磷烯的研究中, 也发现了类似的变化趋势[15,42,43].
接下来, 进一步研究了TM-CrBr3体系的铁磁稳定性. 采用了基于Ising模型的蒙特卡罗模拟方法, 对TM-CrBr3的居里温度(TC)进行了计算, 以此来反映TM-CrBr3体系铁磁耦合的稳定性. 本征CrBr3和TM-CrBr3中的交换耦合参数(J )的具体数值见表1. 计算得到本征CrBr3的J值为2.39 meV, 与之前的研究结果一致[27]. 通过计算得到的J值, 计算了单层本征CrBr3的居里温度, 计算结果表明, 单层CrBr3的TC为41 K, 与之前的计算结果一致[27,44]. TM-CrBr3体系的铁磁(FM)构型与反铁磁(AFM)构型的能量差 (EAFM – EFM), 以及TM-CrBr3的TC, 如图5(a)所示. 从图5(a)可以看出, 除Co-CrBr3以外, 其他掺杂体系的能量差为144—322 meV, TC为46—106 K, 与本征CrBr3的TC相比, 均有所增加. 其中, Sc, V和Zn掺杂CrBr3的TC显著增加. 特别地, ScCrBr3的TC增加了159%. 这些结果表明, 3d过渡金属 (Co除外) 掺杂能够增强单层CrBr3的铁磁稳定性.
| CrBr3 | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | |
| J/meV | 2.39 | 5.95 | 3.95 | 5.10 | 2.66 | 3.96 | 2.80 | 1.25 | 2.97 | 3.27 | 4.73 |
表1本征CrBr3和TM-CrBr3体系中的交换耦合参数 (J )
Table1.Exchange coupling parameter (J ) of pristine CrBr3 and TM-CrBr3.
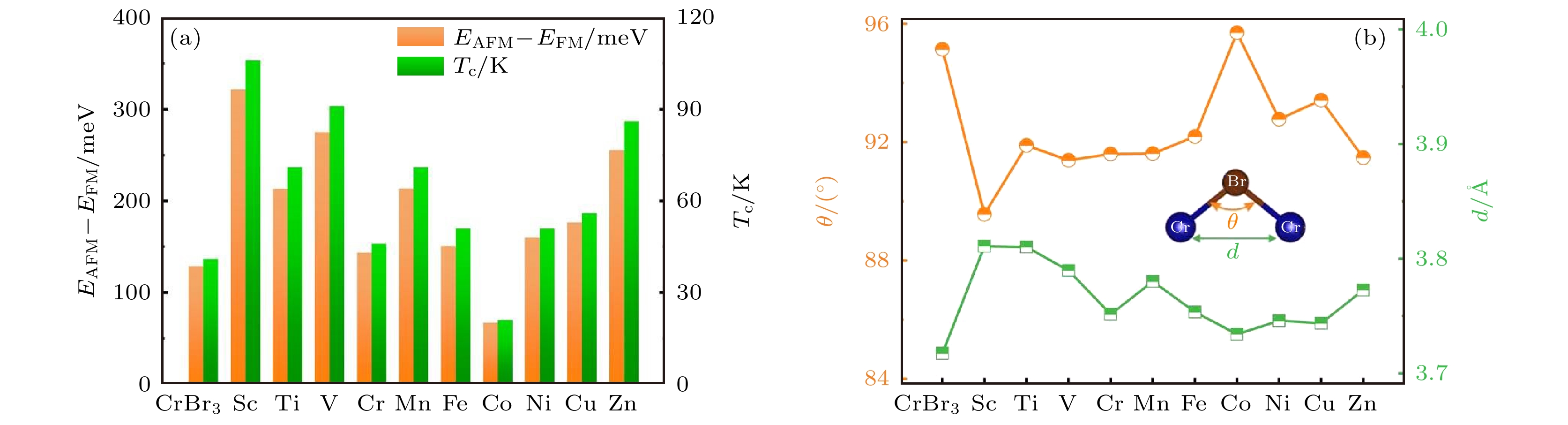 图 5 (a) 本征CrBr3和TM-CrBr3体系的AFM构型与FM构型的能量差EAFM – EFM和居里温度TC; (b) 本征CrBr3和TM-CrBr3的Cr—Cr距离和Cr—I—Cr键角
图 5 (a) 本征CrBr3和TM-CrBr3体系的AFM构型与FM构型的能量差EAFM – EFM和居里温度TC; (b) 本征CrBr3和TM-CrBr3的Cr—Cr距离和Cr—I—Cr键角Figure5. (a) The EAFM – EFM and Curie temperature of pristine CrBr3 and TM-CrBr3; (b) the Cr—Cr distance and Cr—I—Cr bond angle in pristine CrBr3 and TM-CrBr3.
单层CrBr3的铁磁耦合可以根据Goodenough-Kanamori-Anderson规则[45-47]来解释. 直接交换相互作用会受到相邻Cr原子之间的直接电子跃迁的影响, 与Cr原子之间距离d相关, 反铁磁耦合通常随着距离d的减小而增强. 超交换相互作用主要源于Cr的d轨道和Br的p轨道重叠, 由Cr—Br—Cr键角θ决定, 键角θ越接近90°, 则铁磁耦合越强. 体系的磁基态由直接交换和超交换相互作用之间的竞争决定. 图5(b)给出了TM-CrBr3体系中的Cr—Cr距离和Cr—Br—Cr键角的变化. 相比于本征CrBr3, TM原子(除Co以外)掺杂后, TM-CrBr3体系的Cr—Br—Cr键角更接近90°, Cr原子之间的距离d增大, 导致影响铁磁耦合的超交换相互作用增强, 影响反铁磁耦合的直接交换相互作用减弱. 因此, TM原子 (除Co以外) 掺杂CrBr3的铁磁稳定性显著增强. 对于Co-CrBr3, 相较于本征CrBr3, Cr原子之间距离d几乎不变, 然而Cr—Br—Cr键角增大了1°, 导致Co-CrBr3中影响铁磁耦合的超交换相互作用减弱. 因此, 与本征CrBr3相比, Co-CrBr3的铁磁稳定性减弱.
最后, 研究了过渡金属原子掺杂对单层CrBr3电学性质的影响, TM-CrBr3体系的能带结构, 如图6所示. 在Cr-CrBr3体系中, 自旋向上和自旋向下的通道分别表现出金属性和半导体性, 因此, Cr原子掺杂使CrBr3从半导体转变为半金属, 能够实现100%自旋极化的电流. 在Sc-CrBr3掺杂体系中, 其自旋向上和自旋向下的带隙值分别为0.06 eV和2.37 eV. 根据Wang[48]的定义, SGS是指能带结构中的一个自旋方向表现为零带隙, 另一个自旋方向表现出半导体性质; 或者两个自旋方向的通道都有带隙, 但是其中一个自旋通道的导带与另一个自旋通道的价带之间是零带隙. 这里的“零带隙”是指能带间隙等于或小于0.1 eV. 在Sc-CrBr3体系的能带结构中, 自旋向上通道的带隙值为2.37 eV, 自旋向下通道的带隙值为0.06 eV, 因此, Sc-CrBr3可以称为SGS. 在SGS材料中, 电子无需能量就可以从价带跃迁到导带, 从而在费米能级处产生100%的自旋极化, 因此, 这种材料可以作为理想的自旋电子器件候选材料. 同样地, 在除Cr-CrBr3以外的其他TM-CrBr3 (Sc-, Ti-, V-, Mn-, Fe-, Co-, Ni-, Cu-和Zn-CrBr3)体系中, 发现其中一个自旋通道的带隙小于0.1 eV, 表现为SGS性质. 上述结果表明, 依赖于不同的TM原子掺杂, TM-CrBr3体系表现出半金属性或SGS性质, 3d过渡金属原子的掺杂可以使CrBr3在纳米电子器件中具有潜在的应用价值.
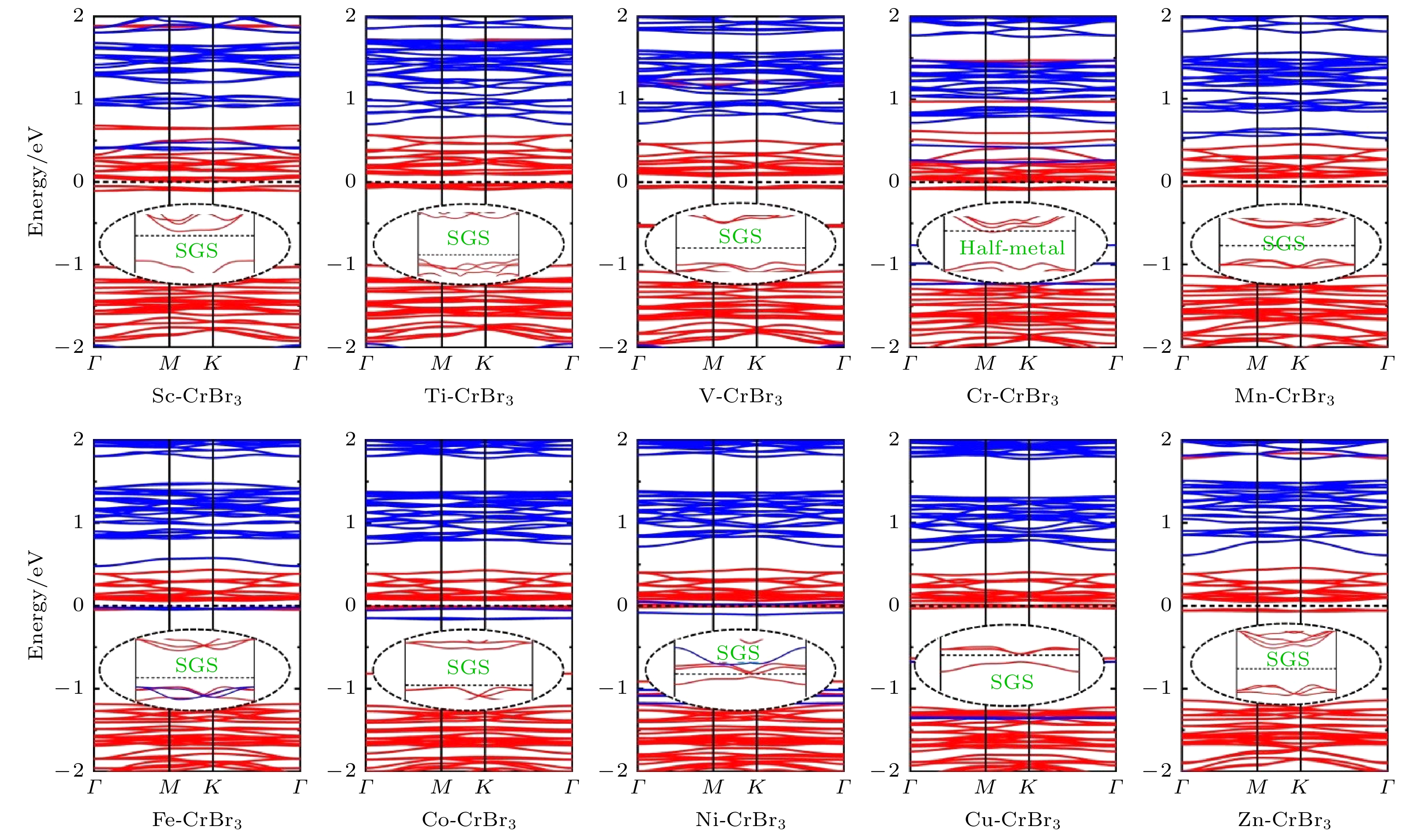 图 6 3d TM原子掺杂单层CrBr3的自旋极化能带结构, 插图是费米能级附近能带结构的放大图. 自旋向上和自旋向下分别用红色实线和蓝色实线表示
图 6 3d TM原子掺杂单层CrBr3的自旋极化能带结构, 插图是费米能级附近能带结构的放大图. 自旋向上和自旋向下分别用红色实线和蓝色实线表示Figure6. Spin-polarized band structures of 3d TM atoms doped CrBr3 monolayer. The illustration is an enlarged picture of the band structures near the Fermi level. The red and blue solid lines indicate spin-up and spin-down channels in the band structures, respectively.
感谢南京工业大学高性能计算中心的计算支持.
