Fund Project:Project supported by the National Natural Science Foundation of China (Grant Nos. 12064014, 12064015), the Natural Science Foundation of Jiangxi Province, China (Grant No. 20192BAB202004), and the Open fund of Fujian Provincial Innovation Laboratory of Energy Devices, China (Grant No. 21C-OP-202005)
Received Date:30 January 2021
Accepted Date:06 March 2021
Available Online:07 June 2021
Published Online:05 August 2021
Abstract:Doping is one of the most important methods to improve the electronic conductivity and modify its electrochemical performance of LiFePO4. Rare earth elements have become an effective selection for doping modification due to their high electronic charges, large ion radii and strong self-polarization ability. In this work, we study the structural, electronic and ionic diffusion properties of LiFePO4 with rare earth (RE) doping (La, Ce, Pr) by using first-principles calculation based on density functional theory. The calculated results show that the lattice constant and cell volume of LiFePO4 increase to a different degree after RE doping. In the delithiation process, the volume change rate of the material after RE doping is significantly reduced, indicating the cycle performance of the material is improved, on the other hand, the energy density is reduced. The calculated density of states suggests that RE-doped LiFePO4 exhibits metallic characteristics, which is different from the undoped one with semiconductor characteristics. As a result, the RE-doping can increase the electronic conductivity of the material. The calculation of elastic modulus demonstrates the increase of ductility for RE-doped LiFePO4, and it can be predicted that the cycle performance and the rate performance of the RE-doped battery have great improvement. In addition, La and Ce doped LiFePO4 materials exhibit that the complex energy barrier can change during the Li ion migration, and the migration barriers vary considerably, depending on different paths, which is related to the variation of potential energy surface caused by the doping of rare-earth elements. The Li-ions are far from the RE ions, the migration barriers are obviously lower than the undoped one, while the Li-ions are closest to RE ions, the migration barriers increase essentially. Compared with Ce doping, the change of the Li-ion migration barrier caused by La doping is great, indicating that RE ion doping has a greater influence on the local structure of the system. Keywords:first-principles calculations/ rare-earth doped/ lithium-ion battery/ LiFePO4
表2未掺杂LiFePO4中的Fe—O键长和稀土掺杂结构中RE—O键长 Table2.Bond lengths between Fe atoms and O atoms, rare-earth atoms and O atoms in LiFePO4 without doping and with rare-earth doping structure, respectively.
表3LiFePO4未掺杂及稀土掺杂(RE = La, Ce, Pr)的弹性常数(单位: GPa) Table3.Elastic constants (in GPa) of LiFePO4 without doping and with rare earth (RE = La, Ce, Pr) doping.
表4未掺杂与稀土掺杂(RE = La, Ce, Pr)的LiFePO4的体模量(B)、剪切模量(G)、B/G、杨氏模量(E)、泊松比(v) Table4.Bulk modulus (B), shear modulus (G), B/G, Young’s modulus (E), Poisson’s ratio (v) for LiFePO4 without doping and with rare earth (RE = La, Ce, Pr) doping.
7.稀土掺杂的LiFePO4中的Li离子迁移动力学从所周知, Li离子在电极材料中的迁移能垒对锂离子电池倍率性能的影响至关重要[43]. 根据表1和表2, 由于Ce和Pr掺杂后的LiFePO4的晶格常数以及Ce—O和Pr—O键长都非常接近, 说明Ce和Pr掺杂对LiFePO4结构产生的影响非常相似. 因此, 为了简化分析过程和降低计算量, 选取具有代表性的La和Ce元素掺杂的LiFePO4体系, 研究其中的Li离子迁移动力学. 为了更全面地研究稀土离子掺杂对LiFePO4中Li离子迁移的影响, 选择在1 × 3 × 1的LiFePO4超胞中把其中1个Fe分别替换成稀土原子La或Ce (如图4所示), 此时掺杂浓度为1/12. 考虑到实验和理论上已证实在LiFePO4化合物中, Li离子被约束在沿b方向的一维通道内迁移[44,45], 为简化起见, 本文只研究掺杂体系中Li离子在b方向的迁移通道. 根据超胞中b轴迁移通道上Li离子与稀土离子的距离, 以及超胞中的周期性边界条件, 发现Li离子存在4条不同的迁移路径, 分别是1→2, 2→3, 3→4, 4→5, 而5→6与3→4是等价路径, 如图4所示. 图 4 稀土元素(La, Ce)掺杂后的LiFePO4中不同的Li离子迁移路径 Figure4. Different Li ion migration paths of LiFePO4 with rare-earth (La, Ce) doping.
为了比较稀土掺杂对LiFePO4中Li离子迁移的影响, 首先计算了Li离子在未掺杂的1 × 3 × 1的LiFePO4超胞中的迁移能垒. 很明显, 在未掺杂的LiFePO4超胞中1→5包括的4条路径是完全等价的, 因此, 只计算了1→2路径上Li离子的迁移能垒, 为0.493 eV, 如图5所示, 比Ouyang等[45]的第一性原理计算结果小了0.1 eV左右. 图 5 未稀土掺杂的LiFePO4中Li离子迁移的能量分布 Figure5. Energy profile of the Li ion migration in LiFePO4 without rare-earth doping.
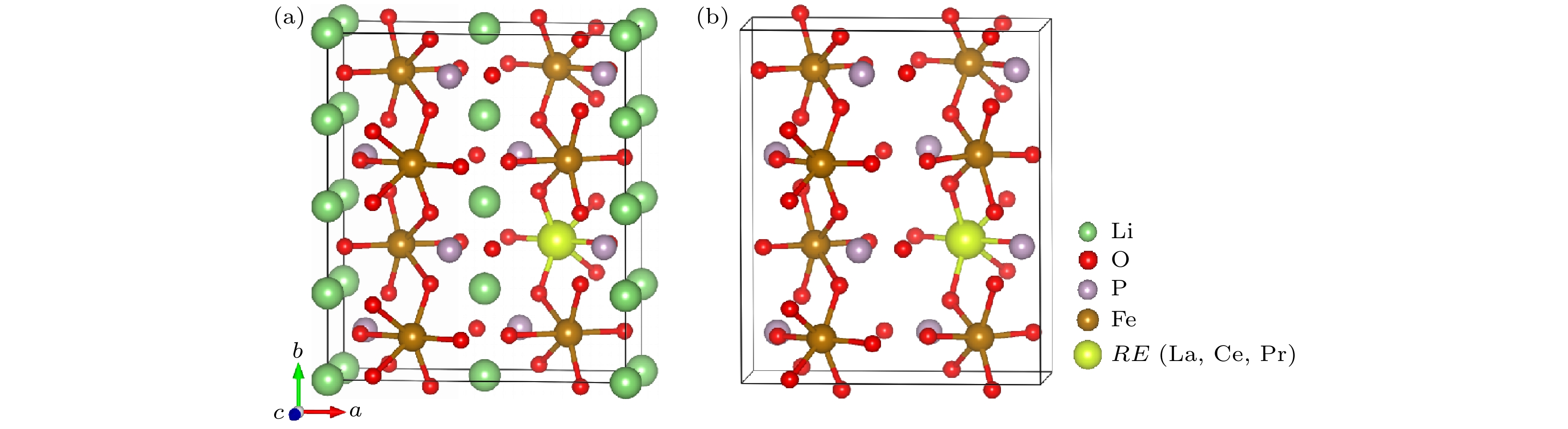 图 1 (a) LiFePO4和(b) FePO4中稀土掺杂位置示意图
图 1 (a) LiFePO4和(b) FePO4中稀土掺杂位置示意图

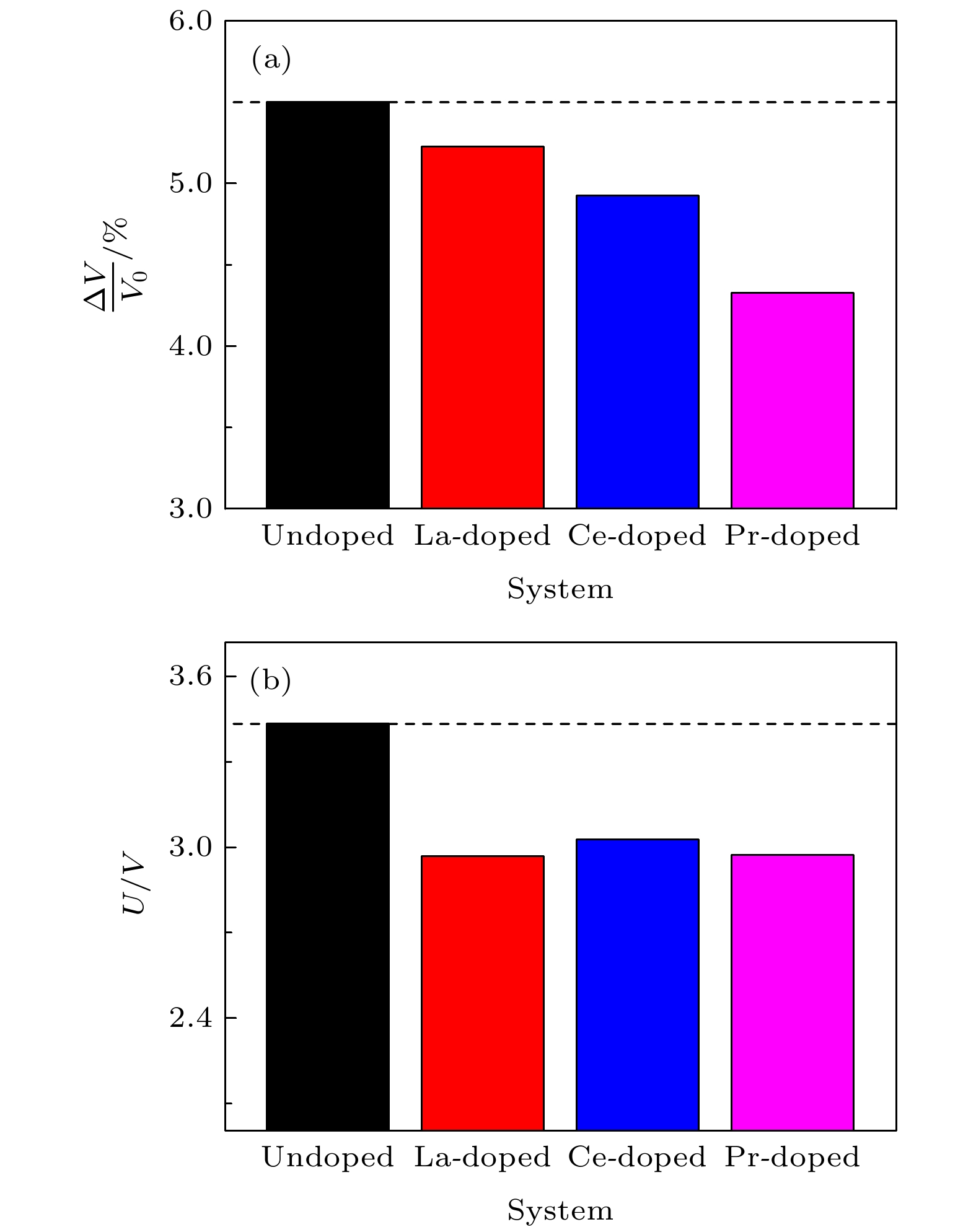 图 2 LiFePO4 (a)完全脱锂后的体积变化率与(b)平均脱锂电位
图 2 LiFePO4 (a)完全脱锂后的体积变化率与(b)平均脱锂电位







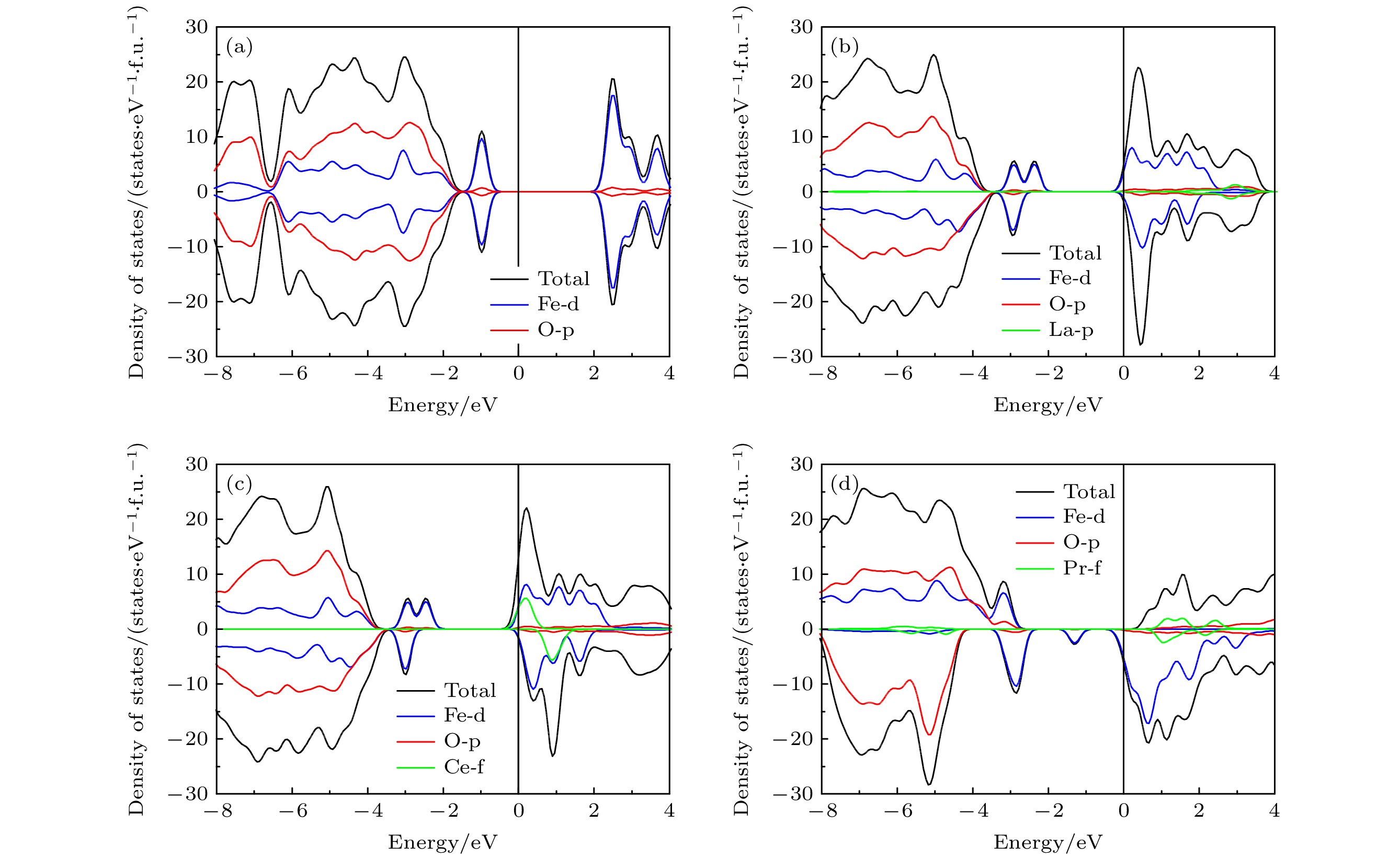 图 3 LiFePO4的电子态密度图 (a)未掺杂; (b) La掺杂; (c) Ce掺杂; (d) Pr掺杂
图 3 LiFePO4的电子态密度图 (a)未掺杂; (b) La掺杂; (c) Ce掺杂; (d) Pr掺杂 图 4 稀土元素(La, Ce)掺杂后的LiFePO4中不同的Li离子迁移路径
图 4 稀土元素(La, Ce)掺杂后的LiFePO4中不同的Li离子迁移路径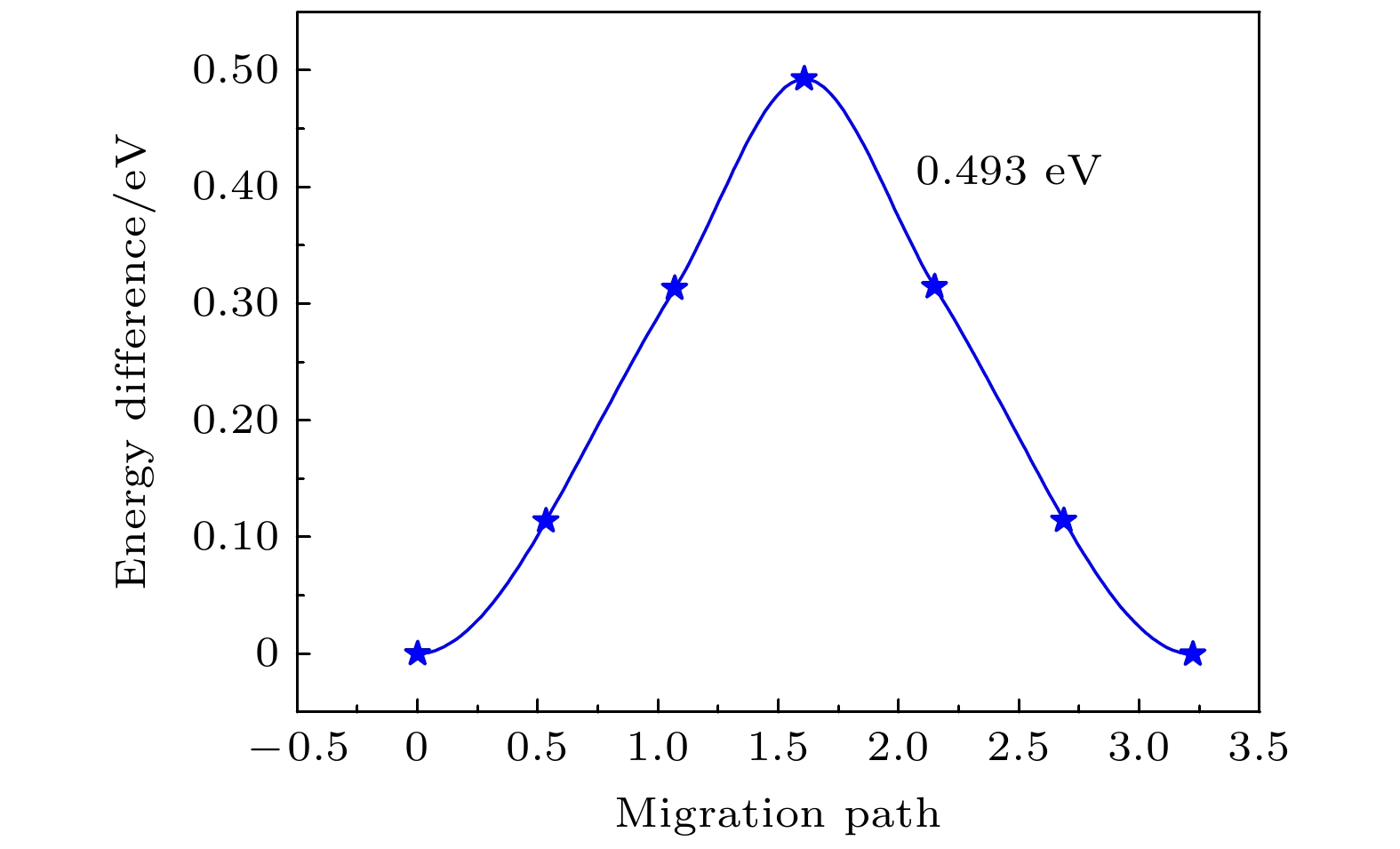 图 5 未稀土掺杂的LiFePO4中Li离子迁移的能量分布
图 5 未稀土掺杂的LiFePO4中Li离子迁移的能量分布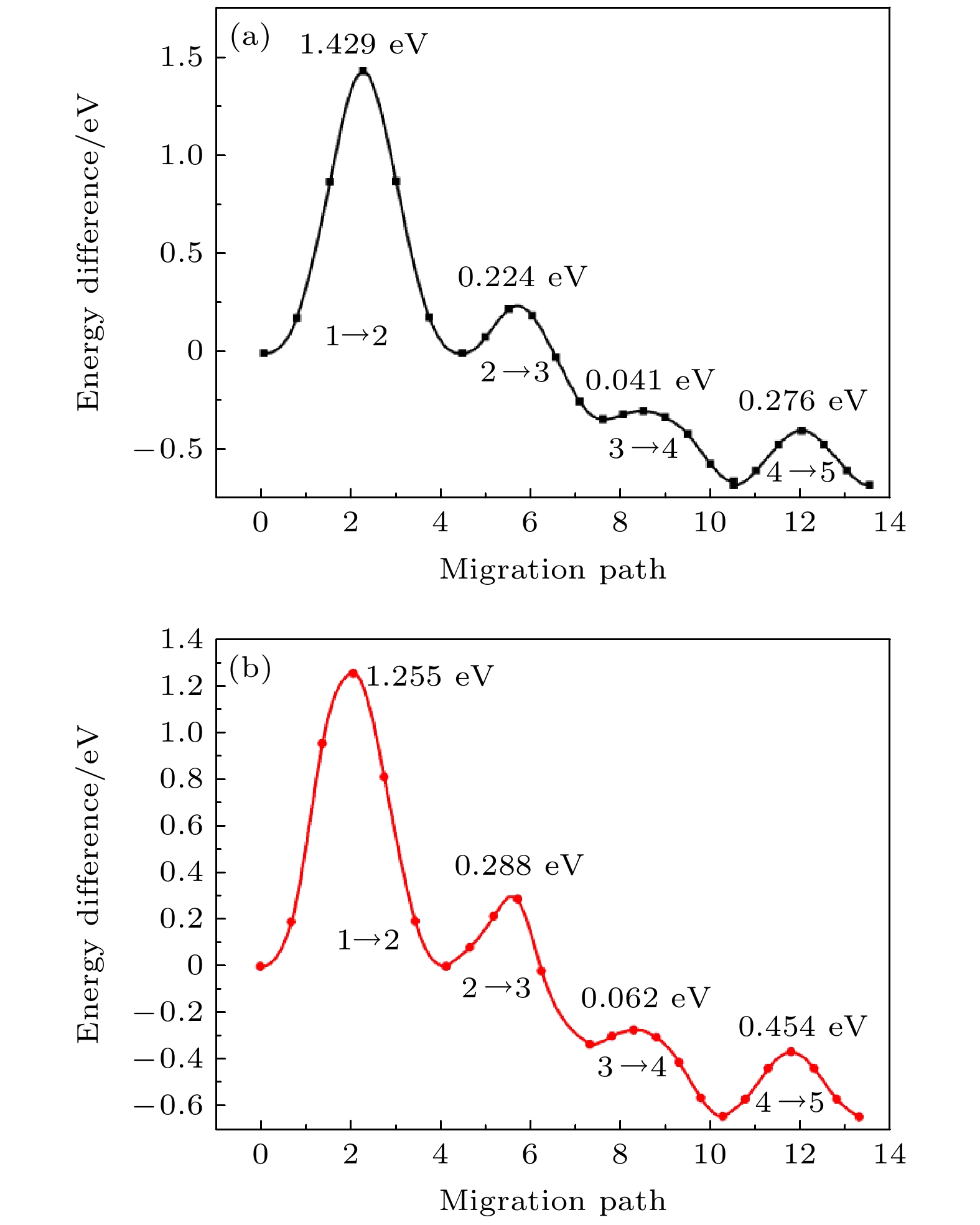 图 6 Li离子在La和Ce掺杂LiFePO4中的迁移路径和势垒 (a) La掺杂; (b) Ce掺杂
图 6 Li离子在La和Ce掺杂LiFePO4中的迁移路径和势垒 (a) La掺杂; (b) Ce掺杂