1.Nanoscale Physics and Devices Laboratory, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China 2.School of Physical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China 3.Department of Physics, Renmin University of China, Beijing 100049, China
Fund Project:Project supported by the National Natural Science Foundation of China (Grant No. 61674045) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB30000000)
Received Date:15 December 2020
Accepted Date:10 February 2021
Available Online:26 June 2021
Published Online:05 July 2021
Abstract:In this review paper, we introduce representative research work on single atomic/molecular manipulations by atomic force microscopy (AFM), which possesses extraordinary ability to resolve atomic and chemical bonds, and charge density distributions of samples. We first introduce the working principle of AFM, then focus on recent advances in atom manipulation at room temperature, force characterization in the process of atom/molecule manipulation, and charge manipulation on insulating substrates. This review covers the following four aspects: 1) the imaging principle of AFM and the atomic characterization of typical molecules such as pentacene and C60; 2) the mechanical manipulation and atomic recognition capability of AFM at room temperature; 3) the characterization of forces in the process of surface isomerization and adsorption configuration changes of the molecules; 4) the manipulation of charge states and the characterization of single and multiple molecules on insulating substrates. The capability of manipulation by AFM in these fields widens the range in atomic/molecular manipulation, which can provide new and well-established schemes for the analysis and precise control of the manipulation process, and can further contribute to the construction of nanoscale devices, such as “molecular switches” and storage components. Keywords:atomic force microscopy/ atomic/molecular manipulations/ manipulation mechanism/ manipulation of charge state/ atom identification
AFM通过探测针尖-样品间的相互作用力来表征样品表面形貌和局域功函数[47,48]等信息. 相比于STM, AFM不依赖于隧穿电流, 因此可以探测绝缘基底上的样品. 常规AFM探针由硅悬臂和悬臂一侧的针尖组成. 悬臂的特征参数包括弹性系数k, 本征频率$ {f}_{0} $, 以及品质因子Q等. AFM按工作时针尖与样品是否接触, 大致可以分为接触式AFM(contact AFM)及非接触式AFM(non-Contact AFM, NC-AFM)两种模式. 其中接触式AFM的探针尖端与样品接触, 利用悬臂的机械形变量作为反馈信号, 直接测量表面轮廓, 该模式下针尖与样品相互作用力很强, 经常会由针尖造成样品表面的不可逆损伤, 因此主要应用在表征“硬”材料领域[49]. 本文主要介绍基于NC-AFM的工作. NC-AFM的悬臂在机械激励下以本征频率振动, 根据反馈信号的不同, 可以分为振幅调制和频率调制两种工作模式. 在振幅调制模式下, 保持悬臂振动频率为本征频率, 针尖和样品的相互作用引起振幅变化, 以振幅为反馈信号表征样品表面形貌. 在频率调制模式下, 保持悬臂振幅恒定, AFM针尖接近样品时, 针尖与样品间的相互作用力梯度变化会引起针尖悬臂的振动频率偏离其本征振动频率, 频率偏移$ \Delta f $经由带通滤波器相位偏移后获得, 并反馈给激励信号, 新的振荡频率$ f={f}_{0}+\Delta f $由锁相环(phase-locked loop, PLL)获得, 其中$ \Delta f $可以作为成像信号[53]. $ \Delta f $和针尖-样品间距离z的依赖关系如图1(b)中$ \Delta f\left(z\right) $曲线所示. 根据Sader-Jarvis公式[54,55], 利用积分$ \Delta f\left(z\right) $曲线的方法, 可以进一步得到$ F\left(z\right) $(图1(c))和$ E\left(z\right) $关系. 图 1 频率调制AFM成像原理图及频率偏移-距离曲线$ \Delta f\left(z\right) $和对应的力-距离曲线$ F\left(z\right) $[50,51] (a) 一氧化碳修饰的qplus型NC-AFM示意图, 悬臂以振幅A振动, 悬臂振动频率偏移其本征频率$ {f}_{0} $的值$ \Delta f $反映了针尖-样品相互作用力梯度[50]; (b) $ \Delta f\left(z\right) $曲线; (c) 隧穿电流$ {I}_{t} $(红色)、短程力(绿色)、长程力(深蓝色)和合力(黑色)随距离z变化的示意图[51]; (d) qPlus型力传感器的光学显微镜照片; (e) qPlus型力传感器的模型示意图[52] Figure1. Functional principle of frequency modulation AFM and $ \Delta f\left(z\right) $ as well as the corresponding force curve $ F\left(z\right) $[50,51]: (a) Schematic of a qPlus-based NC-AFM with a CO-tip; the cantilever oscillates at an amplitude of A and the tip-sample force-induced frequency shift of the cantilever from its natural resonance frequency $ {f}_{0} $ is $ \Delta f $[50]; (b) frequency shifts in FM-AFM; (c) plot of tunneling current $ {I}_{t} $ (red), short range force (green), long range force (dark blue) and total force (black) as a function of distance z between center of front atom and plane defined by the centers of surface atom layer[51]; (d) optical microscope photograph of qPlus sensor; (e) schematic diagram of qPlus sensor model[52].
通过使用合适的原子/分子使针尖尖端功能化, 可以提高NC-AFM的空间分辨率至亚原子级[62-67]. 功能化针尖通过针尖活性、针尖尺寸、针尖弛豫和针尖偶极等方面影响AFM图像的衬度和分辨率[68]. 在本文介绍的实验中, 通常使用一氧化碳分子(carbon monoxide, CO)修饰的AFM针尖[8,62,69-75]. CO修饰的AFM针尖具有化学惰性, 以短程泡利排斥力为主要成像机制, 可以实现对表面原子/分子的无损扫描, 对多种分子结构和化学反应进行详细表征[76]. 使用CO修饰的AFM可以对有机分子进行化学键的成像, 通过定性地比较不同化学键的$ \Delta f $大小和表观键长[77,78]来区分键级, 并进一步解析不同电荷状态的分子的结构和电子性质. 更强的$ \Delta f $对比度意味着更大的泡利斥力, 即该化学键处具有更高的电荷密度, 其键长更短. 在更小的针尖-样品距离下, CO的倾斜会导致AFM图像畸变, 使得表观键长变大[77]. 此外, 环境的局域背景势差异将导致CO受背景力影响而倾斜, 同样会影响表观键长. 针尖施加的偏压V变化时, 电场强度变化, 也会使CO倾斜. 所以仅当分子所处的局域环境相似且偏压相同时, 键长的比较才有意义[13]. 图2(a)所示是使用CO修饰的针尖在恒高模式下得到的并五苯分子的频率偏移NC-AFM图像, 呈现出清晰的碳-碳(C—C)键[62,79]. 图2(d)—图2(g)分别是六苯并蔻(hexabenzocoronene, HBC)结构模型在z = 3.7 ?和z = 3.5 ?时的AFM图像及理论计算得到的分子平面上z = 2.5 ?时电子密度分布. 具有不同键级的键i和键j (图2(d))表现出不同的亮度与键长(图2(e)和图2(f)), 这意味着通过使用CO修饰的功能化针尖的NC-AFM可以区分多环芳香烃和富勒烯中C—C键的不同键级[77]. 图 2 并五苯分子[62,79]及六苯并蔻[77]的化学键分辨NC-AFM图 (a)—(c) Cu(111)表面上并五苯分子的恒高NC-AFM图像、表征示意图及三维力谱; (d) 六苯并蔻结构模型; (e)针尖-样品距离为z = 3.7 ?时在NaCl(2 ML)/Cu(111)上的HBC分子的恒高AFM图像, 扫描振幅A = 0.35 ?; (f) 针尖-样品距离为z = 3.5 ?时在NaCl(2 ML)/Cu(111)上的HBC分子的恒高AFM图像; (g)分子平面上z = 2.5 ?时电子密度分布计算图[77] Figure2. Pentacene imaged with CO-tip AFM[62,79] and Hexabenzocoronene model[77]: (a)–(c) constant height NC-AFM image, characterization schematic diagram and three-dimensional force spectrum of pentaphenyl molecules on Cu(111); (d) hexabenzocoronene model; (e), (f) constant height AFM measurements (A = 0.35 ?) on HBC on Cu(111) at z = 3.7 ? and 3.5 ?; (g) calculated electron density at a distance of 2.5 ? above the molecular plane[77].
32.1.2.AFM探测电荷分布 -->
2.1.2.AFM探测电荷分布
AFM另一个很重要的应用是探测样品表面局域功函数分布的信息, 即开尔文力探针显微镜(Kelvin probe force microscopy, KPFM). 如图3(a)—图3(c), 当针尖与样品非常接近时, 由于两者的费米能级不同, 会产生电子的转移直至两边费米能级相等. 此时针尖与样品存在接触电势差V * = (Φ1–Φ2)/e, 当针尖处于样品某点处时, 通过对针尖-样品施加补偿电压(V *)并同时测量悬臂的频率偏移$ \Delta f $, 得到该点的$ \Delta f\left(V\right) $曲线. 该$ \Delta f\left(V\right) $曲线是一条抛物线[53,80], 当$ V=V ^*$, 即补偿电压等于接触电势差时, $ \Delta f $的绝对值最小. 因此, 接触电势差V *可以直接由$ \Delta f\left(V\right) $抛物线确定. 应用KPFM功能可以表征带不同电荷的吸附原子[47], 使用CO修饰的针尖提高分辨率后还可以探测单分子内的电荷分布[48,53]. 图 3 KPFM原理图及分子局域电荷分布[47,53] (a)—(c) 两个未接触的金属拥有共同的真空能级${E_{{\rm{VAC}}}}$, 当两个金属接触时, 其费米能级对齐; 在二者间施加$ V=V ^*$时, 两个接触金属的接触电势差被补偿[53]. (d) 双势垒隧穿结的简单静电学模型: 针尖和样品间简单静电学等效电路模型, C0代表针尖-基底电容, C1代表针尖-吸附原子电容, C2代表吸附原子-基底电容[47,53]. (e) Au原子充电前后$ \Delta f\left(V\right) $曲线图($ d$ = 5.8 ?, A = 0.6 ?). 黑色曲线代表数据拟合的抛物线, 黑色箭头指出LCPD的值V *[47] Figure3. Schematic illustration of the Kelvin principle and local charge distribution[53]: (a)–(c) Two different metals which are not connected to each other share the same vacuum level ${E_{{\rm{VAC}}}}$; when the two materials are connected, their Fermi levels align, accompanied by an electron flow to the material with the greater work function; the contact potential difference can be compensated by applying a dc voltage $ V=V ^*$[53]. (d) Simple electrostatic model for the double-barrier tunnel junction; schematic illustration of the tip and sample system and equivalent circuit of the electrostatic model: ${C_0}$, ${C_1}$ and ${C_2}$ denote the tip–substrate capacitance, the tip– adatom capacitance and the adatom–substrate capacitance, respectively[47,53]. (e) $ \Delta f\left(V\right) $ spectra measured above a Au atom before and after charge switching (d = 5.8 ?, A = 0.6 ?)[47]. Solid black lines show parabolic fits to the measured data and the resulting LCPD values V * are indicated by arrows[47].
NC-AFM具有电子/空穴注入能力, 可以用于诱发表面化学反应; 相较于STM表征仅能反映样品费米能级附近的电子态密度信息, NC-AFM表征可以反映样品的实际形貌. 应用NC-AFM, 能够同时获得分子轨道成像及化学键成像, 得到化学反应前驱体、中间产物及最终产物详细的结构特征, 如C-C键的形成与断裂及其键级信息等, 从而可以更加深入地了解反应路径和反应机理. 2016年, Schuler等[6]利用针尖操纵第一次在表面上实现了可逆的Bergman环化反应. Schuler等在实验上以NaCl(2ML)/Cu(111)绝缘基底上的9, 10 -二溴蒽(9, 10-dibromoanthracene, DBA)分子为例, 将针尖置于分子上方并施加合适的电压后, DBA分子将脱去两个溴原子形成双自由基分子. 继续对该双自由基分子施加1.7 V的电压, 将发生Bergman环化反应, 形成一个六元环与十元环融合在一起的环化二炔烃结构. 促进该Bergman环化反应发生的阈值电压值与环化二炔烃的最低未占分子轨道(lowest unoccupied molecular orbital, LUMO)轨道能量非常接近, 这表明该反应是由电子注入激发的, 在这个反应体系中, 通过针尖施加高于阈值电压的电压脉冲, 环化二炔烃与双自由基分子可以互相转化. 2018年, Pavlicek等利用针尖操纵诱导1, 1 -二溴烯烃形成聚炔烃[5], 该反应类似于通常在溶液中进行的Fritsch–Buttenberg–Wiechell重排反应(FBW)[84]. Pavlicek等通过在NaCl(2ML)/Cu(111)表面利用针尖对分子进行电子注入, 操纵1, 1 -二溴烯烃依次脱溴(bromine, Br), 导致C-C的骨架重排, 形成了聚炔烃. R2C = C?—X这类化合物的结构既可以是直线型的, 也可以是弯曲的, 这将取决于R和X的种类, Pavlicek等发现C = C?—Br自由基是非直线型的, 这是实验上第一次直接“看到”乙烯自由基. 利用针尖操纵的方法, 还可以制备出一些其他方法无法得到的化学结构, 如由纯碳构成的C18分子环[4]. Kaiser等[4]将环形碳结构的反应前驱体, 环碳氧化物C24O6(图4(a))置于绝缘基底NaCl上, 通过针尖操纵的方法: 将针尖置于分子附近, 对样品施加+3 V的电压并维持数秒, 使其逐步脱去两个、四个、六个CO酮羰基基团(图4(f), 图4(k), 图4(p)), 最终形成目标产物环形碳C18(图4(p))[4]. 进一步地, 通过对键级的分析, 可以确定环形碳结构分子C18是具有九重对称性的单键-三键交替连接的结构, 而不是十八重对称的连续二键连接结构. 上述结果充分表明AFM可以操纵合成含多个C原子的分子并解析其复杂的化学结构. 图 4 针尖诱导的表面化学反应: 前驱体脱羰反应形成环碳C18[4]. 第一列为前驱体及中间产物的结构示意图. 第二列和第三列分别对应使用CO修饰AFM在$ \Delta z $较小和$ \Delta z $较大时AFM表征图像, z的零点设置为STM模式下I = 0.5 pA, V = 0.2 V. (l)(m)(q)(r)中下方明亮的点对应于CO分子. 第四列和第五列对应体相DFT计算的分子构型. 第二行(f)—(j)、第三行(k)—(o)对应最常见的反应中间产物. 第四行(p)—(t)对应环碳C18. “模拟.远”、“模拟.近”对应同一行的“AFM.远”、“AFM.近”. 所有图像的标尺与图(b)中标尺保持一致 Figure4. Precursor and products generated by tip-induced decarbonylation[4]. Structures are shown in column 1. AFM images (columns 2 and 3) were recorded with a CO-functionalized tip at different tip offsets $ \Delta z $, with respect to an STM set point of I = 0.5 pA, V = 0.2 V above the NaCl surface. (a)–(e) Precursor; ((f)–(j) and (k)–(o)) the most frequently observed intermediates; the bright features in the lower part of (l), (m), (q), and (r) correspond to individual CO molecules; columns 4 and 5 show simulated AFM images based on gas-phase DFT-calculated geometries; (p)–(t) cyclo carbon. The difference in probe height between “sim. far” and “sim. close” corresponds to the respective difference between “AFM far” and “AFM close”. The scale bar in (b) applies to all experimental and simulated AFM images.
2005年, Sugimoto[12]第一次在室温下实现了使用AFM横向操纵原子, 获得“人造原子图案”. 这项工作被称之为“原子镶嵌”, 即在样品表面特定位置嵌入几个原子, 在室温下该结构依然能够在较长一段时间内保持稳定存在. Sugimoto[12]使用小覆盖度锡Sn吸附的锗Ge(111)-c(2 × 8)表面作为针尖横向操纵原子的研究对象. 因为Ge(111)-c(2 × 8)表面重构的Ge原子与基底Ge原子的成键相较于硅Si(111)-(7 × 7)体系[107]来说较弱, 所以选用锗基底. Sn/Ge(111)-c(2×8)体系与之前研究过的铅Pb/Ge(111)-c(2 × 8)[108,109]有很多的相似之处, 不同的是在室温下Pb吸附原子可以在Ge(111)-c(2 × 8)表面自由扩散; 而Sn在Ge(111)-c(2 × 8)表面的扩散势垒比Pb原子更高, 室温下它的最高扩散速度仅为5.7 × 10–7 /s/atom, 比Pb/Ge(111)-c(2 × 8)体系小了几个数量级[12]. 将微量Sn沉积到Ge(111)-c(2 × 8)表面并退火(退火温度 < 400 K), 在其AFM形貌图中, Sn原子比周围的Ge原子更亮, 即AFM针尖与Sn原子有更强的吸引相互作用. 室温下Sn和Ge吸附原子之间的交换率很低, 在针尖与原子之间的吸引力作用下, 通过扫描可以诱导Sn原子定向扩散到邻近吸附位置[12]. Sn和Ge吸附原子的协同交换作用也与针尖的扫描方向有关. 将Sn吸附原子中心与最临近的Ge原子中心连线作为快速扫描的方向, 针尖从Sn吸附原子位置开始, 向Ge原子方向扫描, 逐步减小针尖-表面间距离, 会增大针尖-表面吸引相互作用直至大于操纵原子所需要的力, 使Sn原子沿快扫方向替代Ge原子. 通过反转扫描的方向并重复上述操作, Ge原子可以反向替代Sn原子, 也就是说该横向原子操纵原子协同交换作用是可逆的[12]. 扫描方向为两个吸附原子的连线时, 未观察到Sn原子与线外的相邻Ge原子的交换作用, 这表明由半导体针尖尖端产生的势阱对在交换过程中能量势垒减少的贡献是非常局域的. 另外, “操纵原子阈值”大小对针尖尖端构型敏感, 对于相同的悬臂和相同的振幅, 即使两次操纵的针尖尖端构型仅发生轻微变化, 实现吸附原子交换时力的阈值大小也不相同. 上述实验结论均与理论预测[110]的在半导体上横向操纵原子条件一致[12]. 该实验首次表明, 利用半导体针尖尖端和表面之间的吸引相互作用力, NC-AFM技术能够横向操纵表面上的单个原子, 并在适当的条件下, 在室温下逐个操纵原子创建人造原子结构. 除横向操纵外, AFM还可对原子进行垂直方向的操纵, 即从表面移除一个原子, 或将一个原子沉积在原子空位中. 使用AFM扫描样品时, 针尖与样品表面的机械接触可促使针尖“捡起”一个或多个原子, 导致针尖尖端被样品表面原子“污染”, 这样的针尖可以被理解成一根原子级的“蘸水笔”, 可以在不同的样品表面“写”出特定的纳米结构[111]. Sugimoto等[16]使用在Si(111)–($\sqrt {\rm{3}} \!\times\! \sqrt {\rm{3}}$)R30°基底上的Sn单层作为针尖纵向操纵原子的研究对象. 如图5(a), Si替代Sn单层中的几个原子后, Si缺陷展现出较暗的衬度, Sugimoto等假设此时针尖尖端的Si原子也被基底上的一个Sn原子取代. 图5(b)展现了可逆的过程. 这种在尖端和表面之间强结合原子的垂直交换与之前报道的使用STM电压脉冲诱导的原子互换[85]有着不同的机制, 同样的, 也与之前报道的利用AFM针尖与样品的相互吸引来进行原子横向操纵的机制[8,12,112]不同. 这种纵向操纵的机制可以理解为: 当针尖和表面极近时, 形成了针尖-表面混合结构, 同时发生针尖尖端原子与表面原子交换. 有些AFM针尖可以交替沉积Si和Sn, 有的针尖只能沉积两种中的一种. 在上述实验中, 有29%的针尖可以操纵原子垂直交替[16]. 图 5 AFM在垂直基底方向纵向操纵针尖-基底原子交换[16] (a) 针尖接近(黑色)和远离(红色)基底上的Si原子(右侧白色圆框标识)时的频率偏移$ \Delta f\left(z\right) $曲线, 在这一过程中来自针尖的Sn原子取代了原来在基底上的Si原子; (b) 针尖接近(黑色)和远离(红色)(a)中沉积的Sn原子(右侧黑色圆框标识)时的频率偏移$ \Delta f\left(z\right) $曲线; (c) 在混合半导体表面使用上述操纵方法在低原子浓度处沉积或移除原子, 实现“写”原子标记; (d) 操纵过程中针尖与基底的结构模型. 垂直交换原子的操纵方法包含了针尖和表面间多原子的复杂相互作用. 图中硅Si, 锡Sn, 氢H原子分别用黄、蓝、白球表示, 上半部分代表针尖尖端模型, 下半部分代表表面原子分布模型 Figure5. Atom exchange by vertical manipulation of AFM[16]: (a) Frequency shift $ \Delta f\left(z\right) $ signal upon approach (black) and retraction (red) of the tip over the Si atom marked with a white circle in the left inset image, in this process, the Sn atom from the tip replace the Si atom in the substrate; (b) frequency shift $ \Delta f\left(z\right) $ signal upon approach (black) and retraction (red) of the tip above the Sn atom deposited in (a), pointed out by a black circle (left inset); (c) series of topographic images showing the creation and remove of atomic patterns displaying the symbol of silicon, implementing “write” atomic markers; (d) structural model of tip and substrate during manipulation. These vertical-interchange manipulations involve complex multi-atom contacts between tip and surface. Tin and hydrogen atoms are represented by yellow, blue and white spheres, respectively, and the tip apex and surface models correspond to the atomic arrangements in the upper and lower halves respectively.
短程相互作用对化学元素种类非常敏感[40], 因此可以通过测量[125-127]AFM针尖与样品间的短程力, 实现对绝缘体、半导体和金属表面的原子级分辨率的化学元素识别[51,128-131]. 通过进一步的标准化方法可排除不同探针针尖对实验结果的影响[40]. 运用这一技术, 可以实现对组成元素相近、优先吸附位一致的材料(如合金等)的实空间原子级分辨[51,128,129]. Sugimoto等[40]使用这一方法实现了Si, Sn, Pb三种元素的识别. 在AFM形貌图中, Si(111)表面单层的Sn和Pb衬度有差异, 将AFM针尖分别置于Sn和Si原子上方获得力学谱线图, 其中每组曲线都是通过将针尖与基底轻微接触改变针尖尖端的组成和构型后, 表征表面Sn和Si原子获得. Sugimoto等发现在多组探测中, 同一针尖探测到的Si和Sn的最大吸引力值比率保持固定. 如图6(b)所示, 使用测量值与Si的力谱曲线中$ \left|{F}_{{\rm{S}}{\rm{i}}\left({\rm{S}}{\rm{e}}{\rm{t}}\right)}\right| $的比值将测量结果标准化, 平均相对吸引力比率约为0.77 ± 0.02. 实验证明这种方法同样适用于Pb与铟In. 由于探测两个原子时采用同一针尖, 所以使用两个原子的最大吸引力比率可以将针尖的影响抵消. 理论计算与实验结果相符, 同时模拟结果表明$ F\left(z\right) $曲线最大吸引力值仅取决于短程化学相互作用, 由表面原子元素类别、针尖尖端构成及针尖-基底相对取向决定[40]. 图 6 用AFM实现Si, Sn, Pb原子的元素分辨[40] (a) 针尖分别与Si(111)表面生长的Sn和Pb单原子层的短程相互作用力探测; (b)使用Si曲线的最小短程力的绝对值($ \left|{F}_{{\rm{S}}{\rm{i}}\left({\rm{S}}{\rm{e}}{\rm{t}}\right)}\right| $)将(a)中力曲线标准化 Figure6. Resolution of Si, Sn, Pb by AFM[40]: (a) Images of a single-atomic layer of Sn and Pb grown, respectively, over a Si(111) substrate; probing short-range chemical interaction forces; (b) the same force curves as in (a), but the curves in each set are now normalized to the absolute value of the minimum short range force of the Si curve(|FSi(set)|).
Ternes等[8]使用AFM来测量探针尖端施加在单个吸附原子或分子上的纵向力和横向力, 发现移动一个原子所需的力强烈地依赖于吸附物本身以及基底表面. 其中, 在金属基底上移动金属原子时, 横向力起到了主要作用. 在大多数的AFM操作中, 针尖尖端的原子和被吸附原子或分子之间形成了局部化学键, 这使得吸附物能够随着针尖运动, 从表面结合位点“跳”到另一个结合位点. Ternes等[8]证明了不同的轨道杂化情况使得在不同基底上移动钴Co原子所需的力显著不同, 如在铂Pt(111)表面, 针尖拉动Co原子需要210 pN的横向力, 而在Cu(111)基底上移动Co原子时, 横向力大小仅为17 pN. 此外, 在上述两种基底上移动Co原子时, 所需的力是球对称的; 而移动CO分子时, 力明显与球对称的情况不同, 组成也更加复杂[8]. 图7(b)展示了针尖以恒定的高度在Pt(111)表面的Co原子的正上方移动, 逐渐减小针尖高度并反复扫描, 直至Co原子被移动, 扫描方向对应于基底上最易于移动的方向: 即两个相邻的三重对称性中空位点之间的连线[8,54]. 由力梯度${k_z}$ ≈ $(2{k_{\rm{0}}} / {f_{\rm{0}}}) \times \Delta f $积分得到的垂直力${F_z}$(图7(c))可以分解为背景力${F_{\rm{B}}}$和由吸附物引起的力$F_z^*$, 其中${F_{\rm{B}}}$[128]主要来源于长程范德瓦尔斯力, 随针尖-样品距离减小而增大, 并且不受针尖所处的横向位置影响. $F_z^*$是短程力, 随针尖降低而更快增加(图7(f)), 当针尖在引起吸附物移动的z值阈值附近并再靠近15 pm时, $F_z^*$数值增加一倍. 当针尖位置在不发生力耗散的范围内时, 通过对$F_z$沿z方向积分得到势能U, 逐点做谱可得到势能U的分布, 而对U在x方向求微分可得到横向力${F_x}$[8]. 图 7 AFM测量在不同基底上操纵不同原子位移的作用力[8] (a) 单个吸附物的模拟AFM和STM测量. 振幅A = 30 pm的金属针尖探测金属基底上的单个Co原子或CO分子. 插图显示了针尖在z方向随时间的运动, 针尖距离基底最近时为$z'$, 最远时为$z' + 2 A$. (b) Pt(111)表面原子(灰)及吸附Co原子(红)示意图. 不断降低针尖-样品距离并在最易吸附的方向(x方向)进行连续的扫描, 直至Co原子跳跃到临近吸附位点. (c) 针尖尖端和Co原子之间的力F *可以被分解为横向力${F_x}$和垂直力$F_z^*$. 垂直力${F_z}$为$F_z^*$和背景力${F_B}$的和. (d) 测量弹性系数kz (圆和灰色线)的值, 测得的量是针尖在$z'$与$z' + 2 A$内振动时的时间平均量. (e)—(g) 分别对应由(d)中弹性系数kz得到的针尖-样品相互作用能U, 垂直力${F'_z}$, 横向力${F_x}$. 线扫描结果被针尖高度z标记. 其中(f)中的红色箭头标识出Co原子跳跃到相邻结合位点. 图(f)和图(g)中彩色线符合s波模型 Figure7. Different atoms manipulation on different substrates and tip-substrate interaction by AFM[8]. (a) Simultaneous AFM and STM measurements of individual adsorbates; an atomically sharp metal tip is oscillating in z with an amplitude A = 30 pm over a flat metal surface on which an individual Co atom or CO molecule is adsorbed. The inset graph shows the tip motion z(t) between its closest distance (z') and farthest distance (z' + 2A) from the sample. The ball models of the surfaces are scaled to match the dimensions of the images in the following panels. (b), (c) Measuring the force to move Co on Pt(111): (b) Schematic top view of the Pt(111) surface atoms (gray) and the adsorbed Co atom (red). In the following panels, constant-height line scans in the direction of easiest adsorbate motion (x direction) were taken at successively reduced tip sample separations until the Co atom hopped to the adjacent adsorption site [red circle in (b)]. (c) The force F * between tip apex and the Co atom can be divided into the lateral force ${F_x}$ and the vertical force $F_z^*$. The total vertical force ${F_z}$ is the sum of $F_z^*$ and the background force ${F_{\rm{B}}}$. (d) Stiffness kz (circles and gray lines). Note that these values are time-averaged over the cantilever oscillation between z = z' and z = z' + 2A. (e)–(g) Tip-sample interaction energy U, vertical force ${F'_z}$, and lateral force ${F_x}$ extracted from the stiffness kz data in (d). Selected line scans are labeled with the tip height z; the red arrows in (f) indicate the hop of the Co atom to the neighboring binding site. Colored lines in (f), and (g) are fits with the s-wave model.
利用扫描探针显微镜可以在单分子水平操纵其表面异构化, 同时记录操纵过程中的$ \Delta f\left(z\right) $曲线, 为研究针尖与分子间的相互作用、理解针尖操纵分子表面异构的力学机制提供重要的信息. Qi等[10]报道了使用qplus型NC-AFM对Ag(100)基底上的N, N-二甲氨基–2, 6-双蒽基-苯(DMADAB)分子施加机械力作用, 诱导其发生表面分子异构的研究[10]. DMADAB分子具有一个被平面外二甲氨基功能化的中心苯环及两个临位蒽基基团. 在分子升华沉积到基底的过程中, 蒽基基团和苯环中间的C—C键可以旋转, 形成三种表面异构体: “直线”型DMADAB-1、“L”字型DMADAB-2、“V”字型DMADAB-3. 从图8(a)和图8(b) DMADAB分子的STM图像中可以看出, 存在平面外二甲氨基的分子中心比两侧亮. 图 8 AFM针尖操纵DMADAB分子三种表面异构[10] (a), (b) Ag(100)表面DMADAB分子的两种异构体的STM图像(标尺为1 nm). (c) 分子动力学模拟的异构化过程内针尖分子相互作用的四个关键过程. (d) 针尖操纵DMADAB分子异构过程中的$ \Delta f\left(z\right) $曲线. 插图展示了DMADAB分子在操纵前(左图)后(右图)的STM图像. 标尺为1 nm, z = 0点设置为二甲氨基结上$ V=- 300$ mV, I = 10 pA. 阶段Ⅰ和Ⅱ对应于针尖接近分子的过程, z由0到–700 pm, 阶段Ⅲ对应于针尖在–700 pm处停留3 s, 阶段Ⅳ和Ⅴ对应针尖缩回的过程, 即从–700 pm到0. (e) 由阶段Ⅰ中的$ \Delta f\left(z\right) $计算得到短程力曲线, 红星标注z的位置为–476 pm, 位于阶段Ⅰ和Ⅱ的交界处, 此时$ \Delta f\left(z\right) $突然下降. 力曲线由Sader–Jarvis方法计算得出[54,55] Figure8. Three surface isomers of DMADAB molecules and surface reversible isomerization by AFM tip[10]: (a), (b) STM topographic images DMADAB-1 and DMADAB-2 on the Ag(100) substrate, respectively; scale bars: 1 nm. (c) Four typical states in tip manipulation on DMADAB taken from the SMD simulations on a successful isomerization. (d) The $ \Delta f\left(z\right) $ curve recorded during a successful manipulation on a DMADAB molecule. Insets are STM images of the DMADAB molecule before (left) and after (right) manipulation. Scale bars are 1 nm; z = 0 pm is defined as a tunneling junction height of –300 mV, 10 pA on top of the dimethylamino group. Regions I and II correspond to the tip approaching to the molecule from 0 to –700 pm. Region III is where the tip stays at –700 pm for 3 s; Rregions IV and V correspond to the tip retracting from–700 to 0 pm. (e) The short-range force curve calculated from the $ \Delta f\left(z\right) $ curve in region I; the red star marks the z position at –476 pm, where $ \Delta f\left(z\right) $ suddenly drops at the boundary of regions I and II; the force curve is calculated via the Sader-Jarvis method[54,55].
Shiotari等[11]发现可通过针尖与分子间的力矩来操纵一氧化氮(NO)分子在Cu(110)基底上的吸附构型, 使其在沿着$ \left[001\right] $方向“平躺”、直立, 沿$ \left[00\bar 1\right] $方向“平躺”三种状态之间可控切换[11]. Shiotari等使用qplus型NC-AFM在液氦温度零偏压下记录$ \Delta f\left(z\right) $曲线, 通过Sader–Jarvis[54,55]公式对$ \Delta f\left(z\right) $积分并扣除长程力背景后, 得到垂直力曲线$ F\left(z\right) $和势能$ U\left(z\right) $[11]. Shiotari等[11]发现NO修饰的针尖可以操纵Cu(110)基底上直立吸附的NO分子, 使其“平躺”, 并通过改变针尖在NO分子上方横向的相对位置(图9(a)—图9(c)), 控制NO“平躺”的方向: $ \left[001\right] $或$ \left[00\bar 1\right] $(图9(b), 图9(c)中黄色箭头所示). 实验过程中施加的偏压为0 V, 说明NO表面吸附构型变化(关-开)与电子注入无关. 使用Cu针尖进行上述操纵, 并不能改变NO表面构型, 而通常会在Cu靠近分子时, 由于吸引形成NO针尖, 这证实了针尖与NO分子间的排斥力在改变直立吸附的NO构型变化中至关重要[11]. 图 9 Cu(110)表面NO分子三种吸附构型及在针尖诱导下的相互转化[11]. (a)—(c) 针尖诱导的直立NO到平卧NO的构型转换过程 (a) Cu(110)表面使用tNO针尖(tilting NO针尖)表征两个直立NO分子的STM图像; (b) 针尖在图(a)中红点位置处接近表面后, 与(a)同样区域的STM图像; (c) 针尖在图(b)中红点位置继续接近表面后, 与(a) (b)同样区域的STM图像. (d), (f) 分别对应(a)$ \to $(b), (b)$ \to $(c)过程中的$ \Delta f\left(z\right) $曲线. (e), (g) 分别对应(a)$ \to $(b), (b)$ \to $(c)过程中的$ U\left(z\right) $曲线, 针尖接近表面和缩回过程分别用红色和蓝色线表示. (h) Cu(110)上直立NO分子附近处tNO针尖探测的$ U(x, z) $分布图. 插图记录了目标分子的STM图像, 蓝绿色线代表x方向, 黑色箭头标明作用于针尖的力$ F(x, z) $分布, 绿色区域标明NO由直立到平卧构型变化的区域. (i)构型变化前原子结构侧视图 Figure9. Three adsorption configurations of NO molecules on Cu(110) and $ \Delta f\left(z\right) $, $ U\left(z\right) $ curves of the tip-induced conversion of NO molecules[11]. (a)?(c) Tip-induced conversion of upright NO into flat-lying NO: (a) STM images of two upright NO molecules on Cu(110) using a tNO tip; (b) STM images of the same area following approach of the tip to the surface over the red point in (a); (c) STM images of the same area after the tip approached the surface over the red point in (b). (d)[(f)] $ \Delta f\left(z\right) $ and (e)[(g)] $ U\left(z\right) $ for (a)$ \to $(b)[(b)$ \to $(c)], the tip approach and retraction are indicated in red and blue, respectively. (h) $ U(x, z) $ map recorded with a tNO tip near an upright NO on Cu(110). Inset shows an STM image of the target. The cyan line represents the x region of the map; black arrows represent force vectors $ F(x, z) $ acting on the tip; green line indicates the region where the NO configuration changes from upright to flat lying. (i) Depicting the side-view atomic structure immediately before conversion.
分子的电荷状态影响着分子的物理化学性质, 如构型、反应活性和芳香性等, 这对表面合成、催化、光转换和分子电子学应用具有重要意义[13]. 在较厚的绝缘层薄膜上, 分子可以有多种稳定的电荷状态[15]. Fatayer等[13]通过CO功能化的qplus型NC-AFM控制在多层NaCl上的有机分子的电荷状态, 并解析其在中性、阳离子、阴离子和二价阴离子状态下构型、吸附几何及键级关系. 这项工作为研究单个分子在不同电荷态下的化学结构变化开辟了道路. Fatayer等[13]选取偶氮苯作为研究对象, 采用NaCl(> 20 ML)/Cu(111)作为绝缘基底. 偶氮苯的恒高AFM图像(图10(b))表明, 两个苯环相对于NaCl表面来说都略微倾斜出平面. 此外, 吸附在NaCl上的偶氮苯两侧的苯环是近似平行的, AFM图像中, 相同侧的苯环边更明亮[13]. 图 10 偶氮苯分子两种电荷状态下$ \Delta f\left(V\right) $曲线和AFM图像[13](a) 偶氮苯分子上的$ \Delta f\left(V\right) $曲线. 扫描电压V由1 V到3 V. 插图显示偶氮苯的化学结构. (b) V = 0.5 V时的A0恒高AFM图像. (c) V = 2.5 V时的A–1恒高AFM图像. 针尖-样品间距离相对于(b)降低了0.3 ?. (d), (e) 分别对应A0和A–1的模拟恒高AFM图像. 标尺为5 ?. (f), (h) 分别对应A0和A–1的原子模型俯视图. (g), (i)分别对应A0和A–1的化学结构 Figure10.$ \Delta f\left(V\right) $ curve and AFM images of azobenzene molecules at two charge states[13]: (a) $ \Delta f\left(V\right) $ spectrum recorded on top of an azobenzene molecule. Voltage was ramped from 1 to 3 V. The inset shows the chemical structure of azobenzene. (b) Constant-height AFM image of A0 at V = 0.5 V. (c) Constant height AFM image of A–1 at V = 2.5 V, tip-sample distance reduced by 0.3 ? with respect to (b). (d), (e) Simulated AFM images of on-surface A0 and A–1, respectively. All scale bars correspond to 5 ?. (f), (h) Top view of the atomic models of A0 and A–1, respectively. (g), (i) Chemical structures of A0 and A–1, respectively, with wedged bonds representing out-of-plane conformations.
用A表示偶氮苯, 当利用AFM针尖对中性的A0施加 > 2 V的偏压时(图10(a)), 得到阴离子状态的A–1. A–1两侧的苯环仍然倾斜出平面, 但倾斜的方向相反. 在AFM图像中, 表现为两个苯环的相对边更明亮(图10(c)), 这意味着偶氮苯的构型发生了变化. 交替改变偶氮苯的带电荷状态并持续对其进行AFM表征, 可以发现这种电荷构象转换具有可逆性. AFM测量结果与由第一性原理计算得到的A0(图10(f))和A–1 (图10(h))各自的AFM模拟[142]结构(图10(d)和图10(e))一致. 两种氧化态的A都是反式构象的, 但它们的几何结构有微小的差异. A0是平面的, 整个分子平面相对于基底平面倾斜17°. 而A–1是非平面的, 其苯环相对于基底平面在相反方向倾斜了约4°. 从A0转换到A–1时偶氮基团(N = N)被还原, 这改变了π共轭体系, 并引起了平面畸变, 可以利用这一点来解释平面构象到非平面构象的转换. 偶氮苯的例子表明, 电荷状态变化会引起分子构型的变化, 导致分子的部分倾斜[13]. Fatayer等[13]还研究了卟啉类化合物在不同氧化态下的芳香性和偶联途径. 卟啉类化合物的母化合物是卟啉(F), 一种完全共轭无取代基的平面大环(图11(a)). 中性F是芳香性的, 由轮烯模型[143]可知其包含18π电子(4n + 2)的芳香性共轭偶联(图11(a)所示共振结构中红色键), 绕过了吡咯内的NH基团和最外层的CH = CH基团. 经过两次还原后, 大环共轭转变为反芳香式的, 如图11(b)红色标注, 包含了整个吡咯的外围. 大环π共轭的变化和杂环π回路影响了整个分子的芳香性. $ \Delta f\left(V\right) $谱显示了三种不同的氧化态, 即中性F, 阴离子态F–1, 二价阴离子态F–2. AFM图像如图11(c), 图11(e)和图11(g))所示, 相应的拉普拉斯滤波图像如图11(d), 图11(f)和图11(h)所示[13]. 图 11 卟啉分子三种电荷状态下的AFM图像、$ \Delta f\left(V\right) $曲线及键的变化[13] (a) 中性卟啉F0的化学结构; (b) 负离子卟啉F–2的化学结构, 红色通路标记的是每种电荷状态下卟啉分子的环形共轭通路; (c), (d) F0的恒高AFM图像及Laplace滤波后恒高AFM图像; (e), (f) F–1的恒高AFM图像及Laplace滤波后恒高AFM图像, 针尖样品的距离比图(c) 和图(d)中大0.5 ?; (g), (h) F–2的恒高AFM图像及Laplace滤波后恒高AFM图像, 针尖样品的距离比图(c) 和图(d)中大0.4 ?; (i) 卟啉F的$ \Delta f\left(V\right) $谱, 颜色灰度不同代表不同的电荷状态; (j) 卟啉中随电荷状态变化, 键长发生变化的键a, c, l1, l2 Figure11.$ \Delta f\left(V\right) $ curve and AFM images of porphine molecules at three charge states and the change of the bonds[13]: Chemical structure of (a) neutral (F0) and (b) dianionic (F–2) porphine, the red path shows the expected annulene-type conjugation pathway for each charge state. constant-height and corresponding Laplace-filtered AFM images of (c) and (d) F0, (e) and (f) F–1, and (g) and (h) F–2. The constant-height AFM images in (e) and (g) are taken at tip-sample distances larger by 0.5 ? and 0.4 ?, respectively, than the AFM image in (c). (i) $ \Delta f\left(V\right) $ spectrum of F; colored regions indicate the charge states. (j) Highlighted bonds in F. The bonds a, c, ${l_1}$, ${l_{\rm{2}}}$ in porphyrins change with the charge state.
实验上使用两个金属电极“夹住”功能化分子[91,146-148]来测量其电导[149,150], 并形成电子元器件[151]. 金属-分子-金属结对微观细节非常敏感, 因此在原子尺度上精确调控结的构型至关重要. Steurer等[15]利用qplus型NC-AFM探测单电子在NaCl绝缘基底上弱耦合的并五苯分子间的转移, 证实了AFM针尖不仅可以控制分子的电荷状态, 还可以探测分子-分子间的电荷转移[15]. 并五苯分子吸附在> 20 ML厚的NaCl上, 电子在分子与Cu(111)导电基底间的隧穿被抑制, 仅能在针尖-分子间或者分子-分子间转移. 而当针尖与分子间距足够大时, 电子在针尖-分子间的运动也被抑制, 因此在NaCl绝缘基底上的分子的电荷状态将保持. 将针尖置于分子上方并且持续调节样品偏压(ramping the sample bias), 通过电子/空穴注入占据分子最低未占据轨道(LUMO)或最高占据轨道(HOMO), 可以调控并五苯分子在带正电荷、保持中性和带负电荷三种状态中转换, 如图12(a)所示[15]. 图 12 并五苯分子的AFM电荷状态调控[15] (a) 电荷转换循环示意图: 中性$ \to $带负电$ \to $中性$ \to $带正电$ \to $中性, 虚线抛物线显示局部接触电位差(三角形)的变化; (b) AFM操纵最高分子占据轨道(HOMO轨道)分离一个电子的$ \Delta f\left(V\right) $谱; (c) HOMO 轨道添加一个电子的$ \Delta f\left(V\right) $谱; (d) AFM操纵最低分子未占据轨道 (LUMO 轨道) 添加一个电子的$ \Delta f\left(V\right) $谱;(e)LUMO 轨道分离一个电子的$ \Delta f\left(V\right) $谱, 每种情况的偏压变化方向如图中箭头所示; (f) 分子间的横向电荷转移, 插图显示两个临近并五苯分子的恒高AFM图像, 实验零点的参考值$ \Delta f$ = 0.5 Hz, 偏压V = 0 V, $ \Delta f\left(V\right) $曲线为在此零点基础上远离表面6 ?处, 顶部水平轴显示时间依赖性. $ \Delta f\left(V\right) $曲线各部分已由四种不同电荷状态的四条抛物线(虚线)拟合 Figure12. Charge state regulation of pentacene by AFM[15]: (a) Schematic depiction of a closed charge-switching cycle (neutral-negative-neutral-positive-neutral), the dashed parabolas visualize the change of the local contact potential difference (triangles); (b)–(e) experimental manipulation spectra for detaching/attaching a single electron from the highest occupied molecular orbital (HOMO) (b) and to the lowest unoccupied molecular orbital (LUMO) level (d) and the reverse processes (c), (e); the direction of the applied bias ramp is indicated by arrows in each case; (f) lateral charge transfer between individual molecules; the overview AFM image shown in the inset was taken in constant-height mode at a distance determined by a $ \Delta f $ set point of 0.5 Hz at a sample bias of 0 V. The $ \Delta f\left(V\right) $ curve was taken at a distance 6 ? further out from this set point. The time dependence is indicated on the top horizontal axis. The individual segments of the $ \Delta f\left(V\right) $ curve have been fitted by four parabolas (dashed lines) corresponding to the four different charge configurations.









 图 1 频率调制AFM成像原理图及频率偏移-距离曲线
图 1 频率调制AFM成像原理图及频率偏移-距离曲线















 图 2 并五苯分子[62,79]及六苯并蔻[77]的化学键分辨NC-AFM图 (a)—(c) Cu(111)表面上并五苯分子的恒高NC-AFM图像、表征示意图及三维力谱; (d) 六苯并蔻结构模型; (e)针尖-样品距离为z = 3.7 ?时在NaCl(2 ML)/Cu(111)上的HBC分子的恒高AFM图像, 扫描振幅A = 0.35 ?; (f) 针尖-样品距离为z = 3.5 ?时在NaCl(2 ML)/Cu(111)上的HBC分子的恒高AFM图像; (g)分子平面上z = 2.5 ?时电子密度分布计算图[77]
图 2 并五苯分子[62,79]及六苯并蔻[77]的化学键分辨NC-AFM图 (a)—(c) Cu(111)表面上并五苯分子的恒高NC-AFM图像、表征示意图及三维力谱; (d) 六苯并蔻结构模型; (e)针尖-样品距离为z = 3.7 ?时在NaCl(2 ML)/Cu(111)上的HBC分子的恒高AFM图像, 扫描振幅A = 0.35 ?; (f) 针尖-样品距离为z = 3.5 ?时在NaCl(2 ML)/Cu(111)上的HBC分子的恒高AFM图像; (g)分子平面上z = 2.5 ?时电子密度分布计算图[77]





 图 3 KPFM原理图及分子局域电荷分布[47,53] (a)—(c) 两个未接触的金属拥有共同的真空能级
图 3 KPFM原理图及分子局域电荷分布[47,53] (a)—(c) 两个未接触的金属拥有共同的真空能级















 图 4 针尖诱导的表面化学反应: 前驱体脱羰反应形成环碳C18[4]. 第一列为前驱体及中间产物的结构示意图. 第二列和第三列分别对应使用CO修饰AFM在
图 4 针尖诱导的表面化学反应: 前驱体脱羰反应形成环碳C18[4]. 第一列为前驱体及中间产物的结构示意图. 第二列和第三列分别对应使用CO修饰AFM在



 图 5 AFM在垂直基底方向纵向操纵针尖-基底原子交换[16] (a) 针尖接近(黑色)和远离(红色)基底上的Si原子(右侧白色圆框标识)时的频率偏移
图 5 AFM在垂直基底方向纵向操纵针尖-基底原子交换[16] (a) 针尖接近(黑色)和远离(红色)基底上的Si原子(右侧白色圆框标识)时的频率偏移











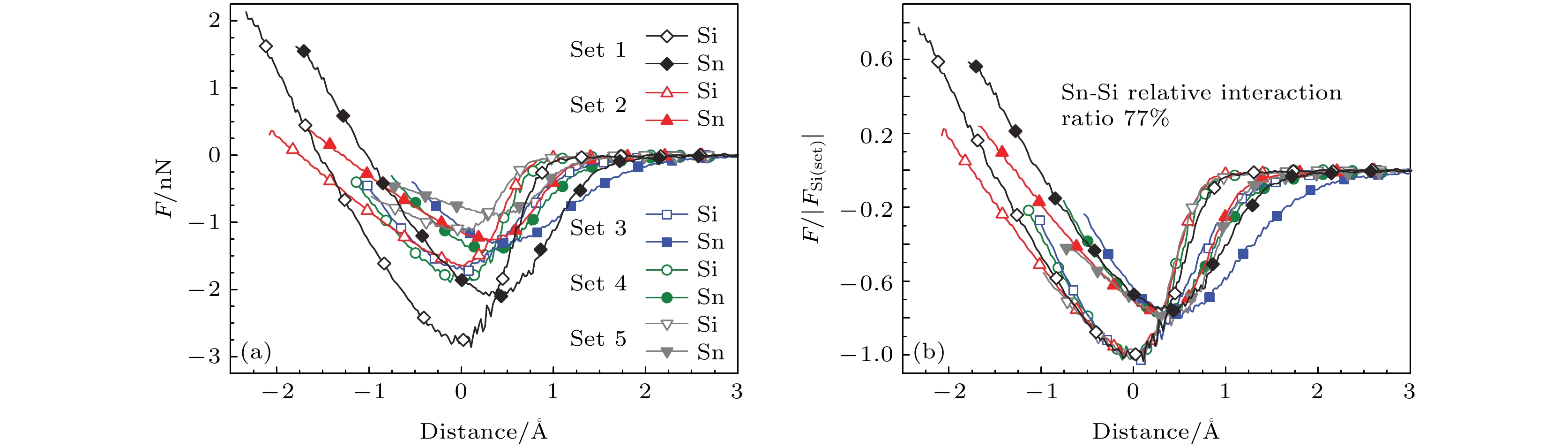 图 6 用AFM实现Si, Sn, Pb原子的元素分辨[40] (a) 针尖分别与Si(111)表面生长的Sn和Pb单原子层的短程相互作用力探测; (b)使用Si曲线的最小短程力的绝对值(
图 6 用AFM实现Si, Sn, Pb原子的元素分辨[40] (a) 针尖分别与Si(111)表面生长的Sn和Pb单原子层的短程相互作用力探测; (b)使用Si曲线的最小短程力的绝对值(
































 图 7 AFM测量在不同基底上操纵不同原子位移的作用力[8] (a) 单个吸附物的模拟AFM和STM测量. 振幅A = 30 pm的金属针尖探测金属基底上的单个Co原子或CO分子. 插图显示了针尖在z方向随时间的运动, 针尖距离基底最近时为
图 7 AFM测量在不同基底上操纵不同原子位移的作用力[8] (a) 单个吸附物的模拟AFM和STM测量. 振幅A = 30 pm的金属针尖探测金属基底上的单个Co原子或CO分子. 插图显示了针尖在z方向随时间的运动, 针尖距离基底最近时为
























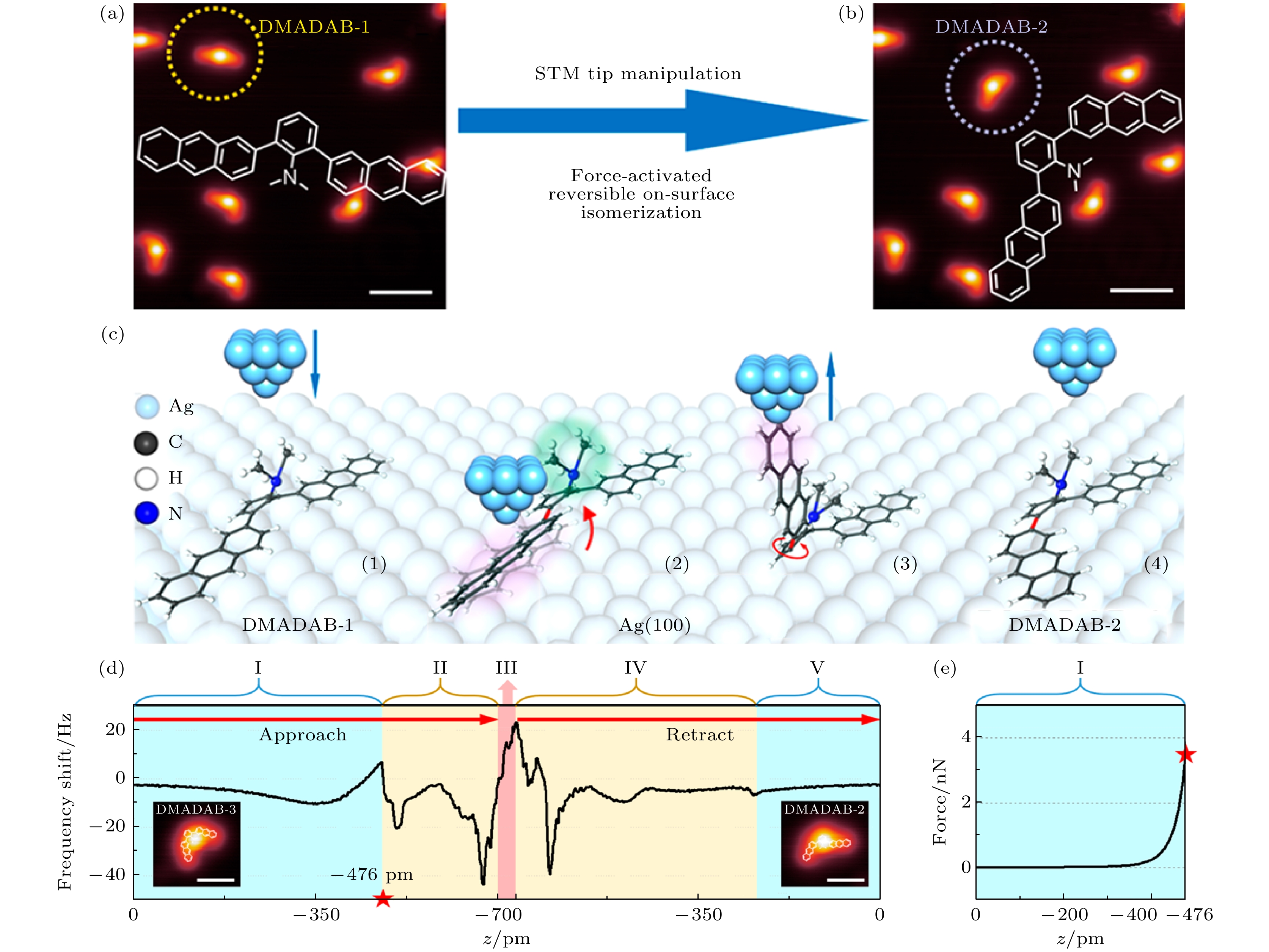 图 8 AFM针尖操纵DMADAB分子三种表面异构[10] (a), (b) Ag(100)表面DMADAB分子的两种异构体的STM图像(标尺为1 nm). (c) 分子动力学模拟的异构化过程内针尖分子相互作用的四个关键过程. (d) 针尖操纵DMADAB分子异构过程中的
图 8 AFM针尖操纵DMADAB分子三种表面异构[10] (a), (b) Ag(100)表面DMADAB分子的两种异构体的STM图像(标尺为1 nm). (c) 分子动力学模拟的异构化过程内针尖分子相互作用的四个关键过程. (d) 针尖操纵DMADAB分子异构过程中的






















 图 9 Cu(110)表面NO分子三种吸附构型及在针尖诱导下的相互转化[11]. (a)—(c) 针尖诱导的直立NO到平卧NO的构型转换过程 (a) Cu(110)表面使用tNO针尖(tilting NO针尖)表征两个直立NO分子的STM图像; (b) 针尖在图(a)中红点位置处接近表面后, 与(a)同样区域的STM图像; (c) 针尖在图(b)中红点位置继续接近表面后, 与(a) (b)同样区域的STM图像. (d), (f) 分别对应(a)
图 9 Cu(110)表面NO分子三种吸附构型及在针尖诱导下的相互转化[11]. (a)—(c) 针尖诱导的直立NO到平卧NO的构型转换过程 (a) Cu(110)表面使用tNO针尖(tilting NO针尖)表征两个直立NO分子的STM图像; (b) 针尖在图(a)中红点位置处接近表面后, 与(a)同样区域的STM图像; (c) 针尖在图(b)中红点位置继续接近表面后, 与(a) (b)同样区域的STM图像. (d), (f) 分别对应(a)



























 图 10 偶氮苯分子两种电荷状态下
图 10 偶氮苯分子两种电荷状态下




 图 11 卟啉分子三种电荷状态下的AFM图像、
图 11 卟啉分子三种电荷状态下的AFM图像、











 图 12 并五苯分子的AFM电荷状态调控[15] (a) 电荷转换循环示意图: 中性
图 12 并五苯分子的AFM电荷状态调控[15] (a) 电荷转换循环示意图: 中性





























