全文HTML
--> --> -->2.1.电化学稳定性
在以往的认识中, 氧化物固体电解质具有宽电压窗口的特点. 以石榴石型固体电解质Li7La3Zr2O12(LLZO)为例, 大多数研究中采用循环伏安法测得LLZO的电化学窗口大于5 V. 然而, 采用热力学方法计算得到的电化学稳定窗口比循环伏安法测得的电化学窗口要窄. 如图1(a)所示, 采用第一性原理计算了多种类型的固体电解质的电化学和化学稳定性[17], 其中LLZO的电化学窗口为0.05 — 2.91 V. 从图1(b)中可以看出, LLZO在0.05 V以下被还原成Li2O、Zr3O和La2O3, Zr3O可能进一步被还原成Zr; 在2.91 V以上, LLZO被氧化成Li2O2, Li6Zr2O7和La2O3 [18]. 出现这一矛盾的原因主要是因为热力学稳定窗口是在完全理想的平衡条件下计算得到, 而循环伏安以及充放电测试过程中动力学因素占主导, 需要考虑的动力学过程包括反应界面的电荷转移、离子输运[19,20]和固相扩散等[21]. 由于固体电解质与电极材料的固固界面动力学特性差, 两相之间发生反应需要相当长的时间. 因此在常规的CV测试以及充放电测试中观察不到理想状态下的平衡反应及其产物, 这一差异在氧化物型陶瓷固体电解质中尤为凸显. 实际上, 在固态锂电池中, 只要固体电解质与电极界面满足动力学稳定的临界条件, 即可以认为两者是稳定的. 尽管目前关于固体电解质电化学稳定性热力学与动力学过程之间的竞争还需要开展详细实验进行确认, 但是研究人员在评价某一种电解质材料时应当注意区分热力学稳定窗口和动力学稳定窗口的差异.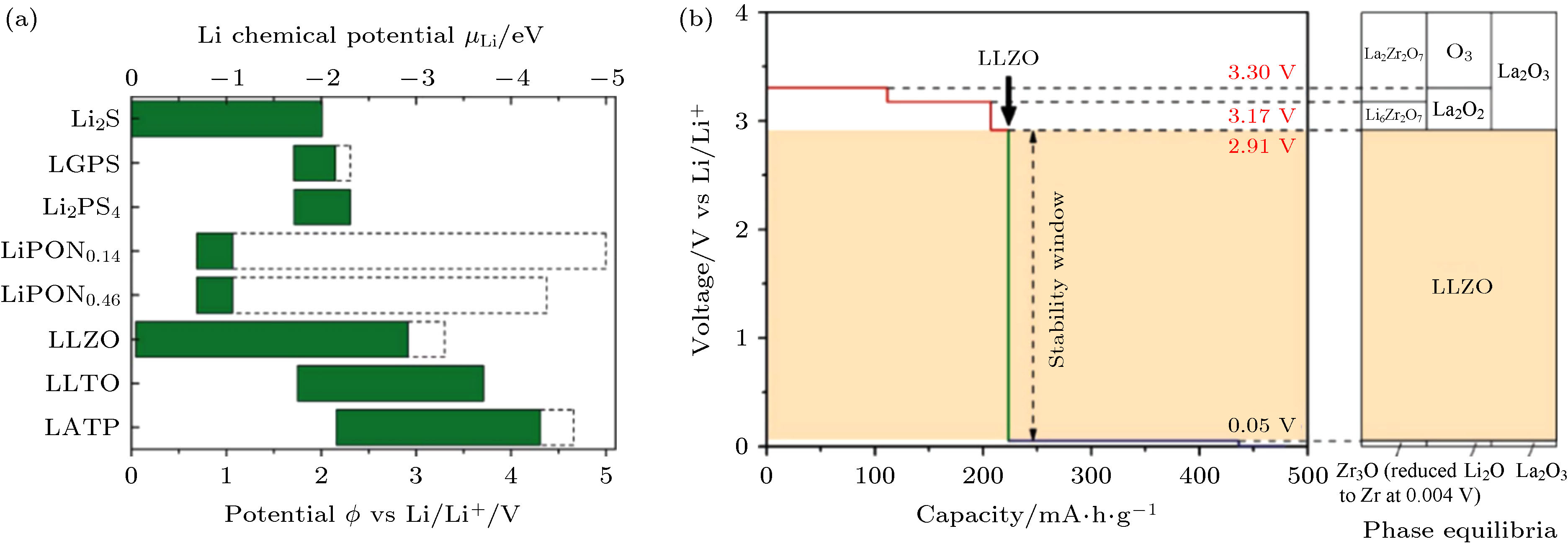 图 1 (a) 常见氧化物与硫化物固体电解质的热力学稳定窗口[17]; (b) Li7La3Zr2O12的热力学稳定窗口第一性原理计算结果[18]
图 1 (a) 常见氧化物与硫化物固体电解质的热力学稳定窗口[17]; (b) Li7La3Zr2O12的热力学稳定窗口第一性原理计算结果[18]Figure1. (a) The thermodynamics stable window of oxide and sulfide solid electrolytes[17]; (b) the first principles calculation results of the thermodynamics stable window of Li7La3Zr2O12[18].
2
2.2.离子界面输运机制
通过元素掺杂等方法, 立方相LLZO陶瓷电解质的室温离子电导率目前最高可达2.03 × 10–3 S/cm[22]. 由于脆性较大, LLZO与电极之间难以形成良好的固固接触. 以LLZO为无机填料、以聚合物为基体的复合电解质膜具有优异的柔韧性和可加工性, 是解决LLZO/电极界面接触的有效方法. 然而, LLZO复合电解质膜的室温离子电导率一般比LLZO低1—2个数量级, 离子迁移数通常小于0.5. 目前提高LLZO复合电解质膜离子电导率的常用方法是通过引入塑化剂等组分降低聚合物的结晶度, 但离子迁移数、电化学稳定性与机械强度会随之降低. 调控LLZO/聚合物界面使得复合电解质接近LLZO本征的离子电导率和离子迁移数, 首先需要深入理解锂离子在LLZO/聚合物界面的输运机制.关于LLZO-PEO复合电解质锂离子输运机制早在2016年即引起关注. Zhang等[23]采用常见的聚氧化乙烯PEO作为聚合物基体, 对比研究了添加与不添加锂盐以及不同LLZO粒径对复合电解质的离子电导率等性质的影响. 结果表明在不添加锂盐的情况下, LLZO-PEO复合电解质膜的离子电导率达到2 × 10–4 S/cm. 这一数值比纯LLZO低1个数量级, 比添加LiTFSI的LLZO-PEO复合电解质低1/3, 却远高于PEO-LiTFSI (5.2 × 10–7 S/cm), 说明LLZO-PEO复合电解质中的离子传导不同于LLZO陶瓷电解质的晶粒-晶界模式, 也不同于聚合物电解质的高分子链段运动传导模式. 图2(a)对比了微米、亚微米和纳米三种尺度的LLZO对复合电解质离子电导率的影响规律. 可以看出, 随着LLZO体积百分比的增大, 离子电导率出现峰值后逐渐降低. 究其原因, LLZO与PEO的接触界面存在空间电荷层(图2(b)), 该界面是锂离子的传导路径之一. 当LLZO添加量达到渗流阈值, LLZO颗粒之间形成连通网络(如图2(c)或2(e)), 锂离子可通过LLZO/PEO界面的空间电荷层实现快速传输; 当LLZO添加量大于(图2(d))或小于(图2(f))该阈值, 离子电导率均低于极大值[24]. 另外, 离子电导率的极大值随着LLZO颗粒尺寸的降低出现在更低的LLZO:PEO比例中(图2(a)), 这是因为LLZO颗粒尺寸的减小带来LLZO/PEO界面的增大. 然而, LLZO纳米颗粒由于表面能高、与聚合物的表面能相差较大, 颗粒之间团聚严重, 并且颗粒与聚合物分子之间的界面接触有待提高. Huang等[25]通过在LLZO颗粒表面包覆了一层可以同时与无机物和有机物发生化学键合的聚多巴胺, 改善了纳米LLZO的团聚及其在PEO基体中的分散性. 与未包覆前的LLZO相比, LLZO@PDA-PEO复合电解质膜的室温离子电导率提高约了1倍. 关于包覆层PDA对复合电解质内部界面的离子输运影响机制还有待进一步探究, 但该项研究提出的方法对有机无机复合电解质的均匀制备有指导意义.
 图 2 (a) 不同尺寸的锂镧锆钽氧(LLZTO)颗粒与聚环氧乙烯(PEO)制备复合电解质的室温离子电导率与LLZTO添加量的关系[23]; (b) LLZO-PEO复合电解质内部界面结构示意图; (c) LLZO与PEO界面的空间电荷层连接成快速离子传输通道的示意图; LLZO-PEO复合电解质内形成渗流通道的过程: LLZO添加量小于渗流阈值(d); 到达渗流阈值(e)和超过渗流阈值(f)的示意图[24]
图 2 (a) 不同尺寸的锂镧锆钽氧(LLZTO)颗粒与聚环氧乙烯(PEO)制备复合电解质的室温离子电导率与LLZTO添加量的关系[23]; (b) LLZO-PEO复合电解质内部界面结构示意图; (c) LLZO与PEO界面的空间电荷层连接成快速离子传输通道的示意图; LLZO-PEO复合电解质内形成渗流通道的过程: LLZO添加量小于渗流阈值(d); 到达渗流阈值(e)和超过渗流阈值(f)的示意图[24]Figure2. (a) The Li+ conductivity of LLZTO/PEO composite electrolytes as a function of different particle sizes and volume fractions of LLZTO[23]; (b) schematic diagram of LLZO nanoparticle in the PEO composite electrolyte; (c) the fast ionic conduction pathway along the space charge regions. Schematics illustration of the percolation behavior along the LLZTO/PEO interface: (d) Volume fraction of LLZO is less than the percolation threshold; (e) the onset and (f) the disruption of the percolation[24].
材料的表面性质与体相性质有较大差异, 并且往往成为影响材料性能的主要因素. LLZO由于表面碱性大, 易与空气中的H2O和CO2反应形成碳酸锂薄层[26, 27], 导致离子电导率下降和界面阻抗增大. 针对这一问题, 研究人员提出快速酸处理[28]、高温热处理[29]和机械抛光[30]等方法有效清除LLZO表面的碳酸锂等污染物. 近期, Huo等[26]采用酸处理法获得了表面无碳酸锂的LLZO, 并系统对比了LLZO颗粒表面有无Li2CO3对聚合物复合电解质膜的离子电导率和锂离子迁移数等方面的影响. 作者制备了不同LLZO含量的复合电解质膜, 主要的离子传输路径随着LLZO含量的提高发生如图3(d)所示的变化. 无Li2CO3的复合电解质膜的离子电导率(图3(a))和电化学稳定性(图3(b))均高于有Li2CO3的复合电解质膜. 另外, 由于无Li2CO3的LLZO与PEO之间的路易斯酸碱作用增强, 添加20 wt% LLZO的复合电解质膜的锂离子迁移数为0.5, 略高于有Li2CO3的LLZO-PEO(约0.47). 值得注意的是, 复合电解质膜的锂离子迁移数随着LLZO质量百分比的提高反而下降(图3(c)), 无Li2CO3的LLZO固含量为80 wt%的LLZO-PEO复合电解质膜离子迁移数约为0.46. 可见, 增大高锂离子迁移数的无机填料含量(LLZO的离子迁移数为1), 并不能提高复合电解质膜的锂离子迁移数. 这说明无机填料的体相性质很难成为复合电解质膜内锂离子传输路径的主要影响因素, 进一步提升复合电解质离子电导率和离子迁移数等参数需要从LLZO表面改性或聚合物分子结构设计入手.
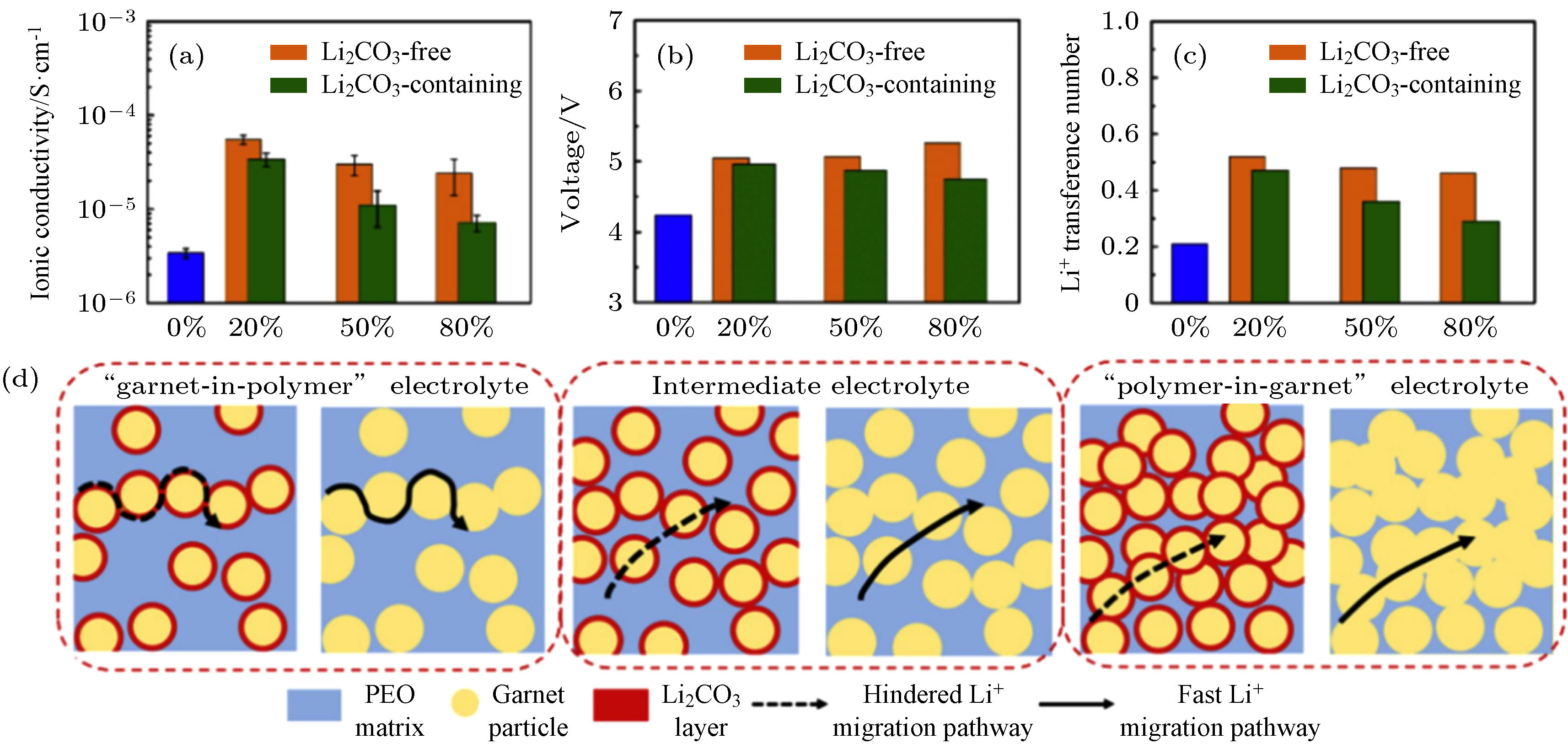 图 3 有无Li2CO3的LLZO与PEO形成复合电解质膜的 (a)离子电导率; (b)电化学窗口; (c)锂离子迁移数的对比; (d)Li2CO3对复合电解质中锂离子传输途径影响的示意图[26]
图 3 有无Li2CO3的LLZO与PEO形成复合电解质膜的 (a)离子电导率; (b)电化学窗口; (c)锂离子迁移数的对比; (d)Li2CO3对复合电解质中锂离子传输途径影响的示意图[26]Figure3. Comparation of (a) ionic conductivities; (b) electrochemical windows and (c) Li+ transference numbers of LLZO/PEO composite electrolyte with and without Li2CO3 on LLZO surfaces; (d) schematic illustration of the Li2CO3 coatings on the Li+ conduction pathway in the LLZO/PEO composite electrolytes[26].
2
2.3.力学特性
固态电解质的力学特性直接影响着固态电池的机械稳定性. LLZO陶瓷片质地坚硬, 在抗压和耐针刺方面具有显著优势. 然而基于陶瓷电解质的固态电池组装面临许多挑战: 由于脆性较大, 固体电解质陶瓷片的厚度很难被加工至100 μm以下, 并且在受到弯折或撞击时容易碎裂. 融合了LLZO刚性和聚合物柔性的复合电解质膜, 具有优异的综合力学特性, 可适应柔性电池和动力电池等多种应用场景. 崔光磊研究课题组[31]提出“刚柔并济”的设计思路, 制备出LLZO与聚碳酸丙烯酯(PPC)复合的自支撑柔性电解质膜. 在此基础上, 该课题组制备了LLZO和PVDF-PVAC复合的电解质膜. 如图4(a)所示, 刚性的LLZO与PVDF提供高机械强度, 柔性的聚乙酸乙烯酯(PVAC)提高离子电导率和电化学稳定性. 通过添加少量环丁砜(TMS)对复合电解质膜中的PVAC选择性浸润, 提高离子电导率的同时改善复合电解质膜与电极的界面接触. 基于这种复合电解质膜的钴酸锂软包电池, 在被剪断、多次撞击和150 ℃高温存储后仍可以正常工作, 说明含有刚性LLZO填料的复合电解质可以显著提高电池的机械稳定性和安全性[32].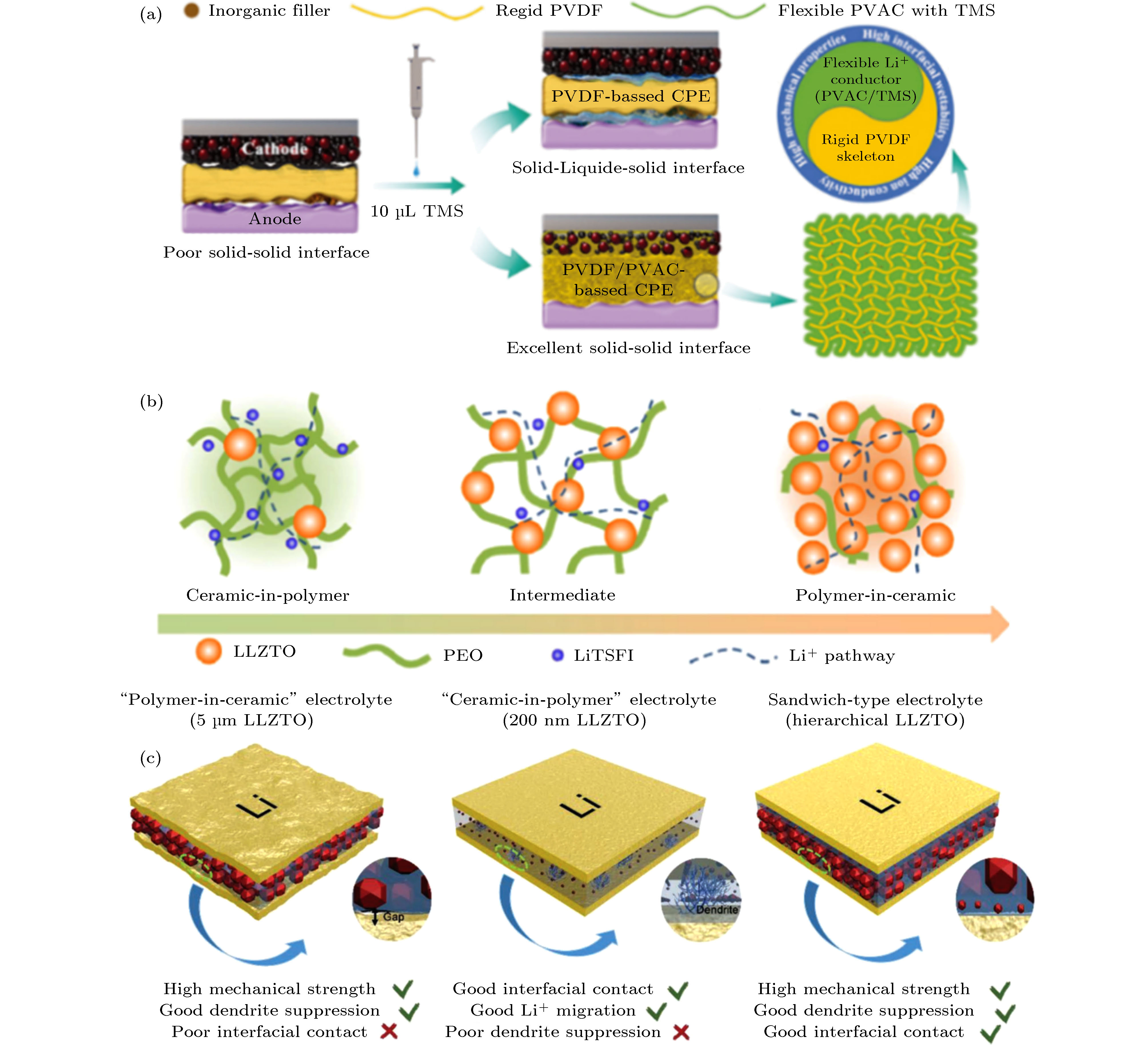 图 4 (a) “刚柔并济”的有机无机复合电解质膜的构建思路[32]; (b) “ceramic-in-polymer”(CIP)和“polymer-in-ceramic”(PIC)复合电解质膜的结构示意图[33]; (c) 以CIP为外层、以PIC为内层的“外柔内刚”分级复合电解质膜[34]
图 4 (a) “刚柔并济”的有机无机复合电解质膜的构建思路[32]; (b) “ceramic-in-polymer”(CIP)和“polymer-in-ceramic”(PIC)复合电解质膜的结构示意图[33]; (c) 以CIP为外层、以PIC为内层的“外柔内刚”分级复合电解质膜[34]Figure4. (a) Schematics illustration of the rigid-flexible organic/inorganic composite electrolyte[32]; (b) “ceramic-in-polymer” (CIP) and “polymer-in-ceramic” (PIC) composite electrolyte[33]; and (c) hierarchical sandwich-type composite electrolytes[34].
那么, 是否意味着LLZO含量越高, 复合电解质膜的力学特性越好? 早在2009年, Syzdek等[35]提出PIC型复合电解质. 传统的ceramic-in-polymer(CIP)型复合电解质以聚合物为连续相, 陶瓷填料离散地分布聚合物相中. 在PIC型复合电解质中, 陶瓷颗粒形成连续相, 起到增强复合膜刚性的作用. 近期北京科技大学范丽珍与Goodenough课题组[33]研究了LLZO陶瓷颗粒添加量对复合电解质膜力学特性的影响. 如图4(b)所示, 20 wt%添加量的CIP具有很好的柔韧性. 随着LLZO添加量的提高, PIC脆性逐渐增强, 当LLZO含量为80 wt%时复合膜容易发生脆裂. 为提高PIC电解质膜的柔韧性, 作者在PIC膜中添加了5 wt%的聚乙二醇(PEG), 获得兼具机械强度和柔韧性的自支撑电解质膜. Huo等[34]进一步研究了不同LLZO含量对复合膜抑制锂枝晶能力的影响, 设计了一种以CIP为外层、以PIC为内层的分级复合电解质膜. 如图4(c)所示, 外层CIP采用200 nm的LLZO填料, 起到提高离子电导率和改善电解质/电极界面接触的作用; 内层PIC采用5 μm的LLZO填料, 起到防止锂枝晶刺穿的作用. 基于这种分级复合膜的金属锂对称电池, 可在室温下以0.2 mA/cm2的电流密度稳定循环400 h以上, 表现出优异的循环稳定性和抑制锂枝晶生长的特性. 综上所述, 深入理解影响固态电解质膜力学特性的关键因素为设计机械性能稳定的固态锂电池提供了新的思路.
3.1.电解质/正极界面
固态正极/电解质界面决定着固态电池实际能量密度和功率密度, 是固态电池器件开发最难解决的问题之一. LLZO与正极界面的主要问题集中在如何减小界面阻抗、形成离子电子共导电的稳定界面中间相和抑制体积膨胀等方面, 深入理解这些问题背后的物理机制是构建理想的固态正极/电解质界面的前提. 本文尝试从界面稳定性、界面能级匹配和界面应力三个方面, 探讨LLZO固体电解质与正极界面中存在的关键问题及改善策略.(1)界面稳定性
固态正极发生氧化还原反应, 需要锂离子和电子同时到达表面. 由于固体电解质没有流动性, 在制备固态正极时需要在电极内构建贯通的离子电子共导通网络[36—38]. 目前以LLZO为离子导体的固态正极制备方法主要有涂布[12]、高温煅烧[39]和表面包覆[38,40]等. Du等[12,41]借助传统的电极涂布法将LLZO颗粒和锂盐LiTFSI加入正极浆料中, 在PVDF的黏合作用下, LLZO、LiTFSI与导电碳包裹正极, 形成复合正极中的离子电子导电网络. 这种方法简单高效, 避免了正极与LLZO在高温下的反应. 但是由于烘干温度低, 复合正极中可能残留少量的浆料溶剂导致电池充电存在明显的副反应. 从图5(a)可以看出该电池首圈库伦效率仅为80 %, 后续循环的库伦效率为95%. 高温煅烧(>500 ℃)可以保证LLZO与正极形成良好的物理接触, 但是会引起界面处的元素互扩散[42]. 如图5(b)所示, Goodenough课题组[13]借助飞行时间二次离子质谱(TOF-SIMS)发现LLZO/LiCoO2界面在700 ℃处理1 h后, 由于LLZO中的掺杂元素Al扩散至LiCoO2, 界面处的LLZO由立方相转变为四方相. Ren等[43]采用更加苛刻的煅烧条件(烧结时间10 h), 对比研究了LLZO与常见的商业化正极材料的界面元素互扩散能力. 实验结果表明当高于临界温度时, LLZO与正极材料均会发生反应. 其中, 层状正极材料(LiCoO2和LiNi0.33Co0.33Mn0.33O2)的反应临界温度略高(约700 ℃), 橄榄石(LiFePO4)和尖晶石(LiMn2O4)结构的正极材料约为500 ℃. 为解决这一问题, Han等[36]提出在对固态正极进行高温煅烧前引入低熔点的锂离子导体LCBO. 在烧结过程中, LCBO与LiCoO2和LLZO表面的Li2CO3反应形成离子导电中间相, 在复合正极内形成稳定的离子导电网络(图5(c)). 然而由于LCBO的电子电导率低, 该电池在室温下仅能以微安级的电流维持循环(5.75 μA/cm2). 进一步提高电池性能, 需要在界面处引入或者原位生成电子导电中间相. Kato等[37]采用脉冲激光沉积在LLZO/LiCoO2界面引入了10 nm的金属Nb中间层(图5(d)), 大幅降低界面阻抗并有效抑制了界面元素互扩散. Bi等[38]采用溶胶凝胶法在LiCoO2表面包覆一层Nb-LLZO, 并在固态复合正极中加入导电碳和微量的离子液体PP13 TFSI. 如图5(e)所示, PP13 TFSI在电池循环过程中发生分解生成LiF, Li3N和Li2S等物质, 与导电碳共同组成复合正极的离子电子混合导电网络.
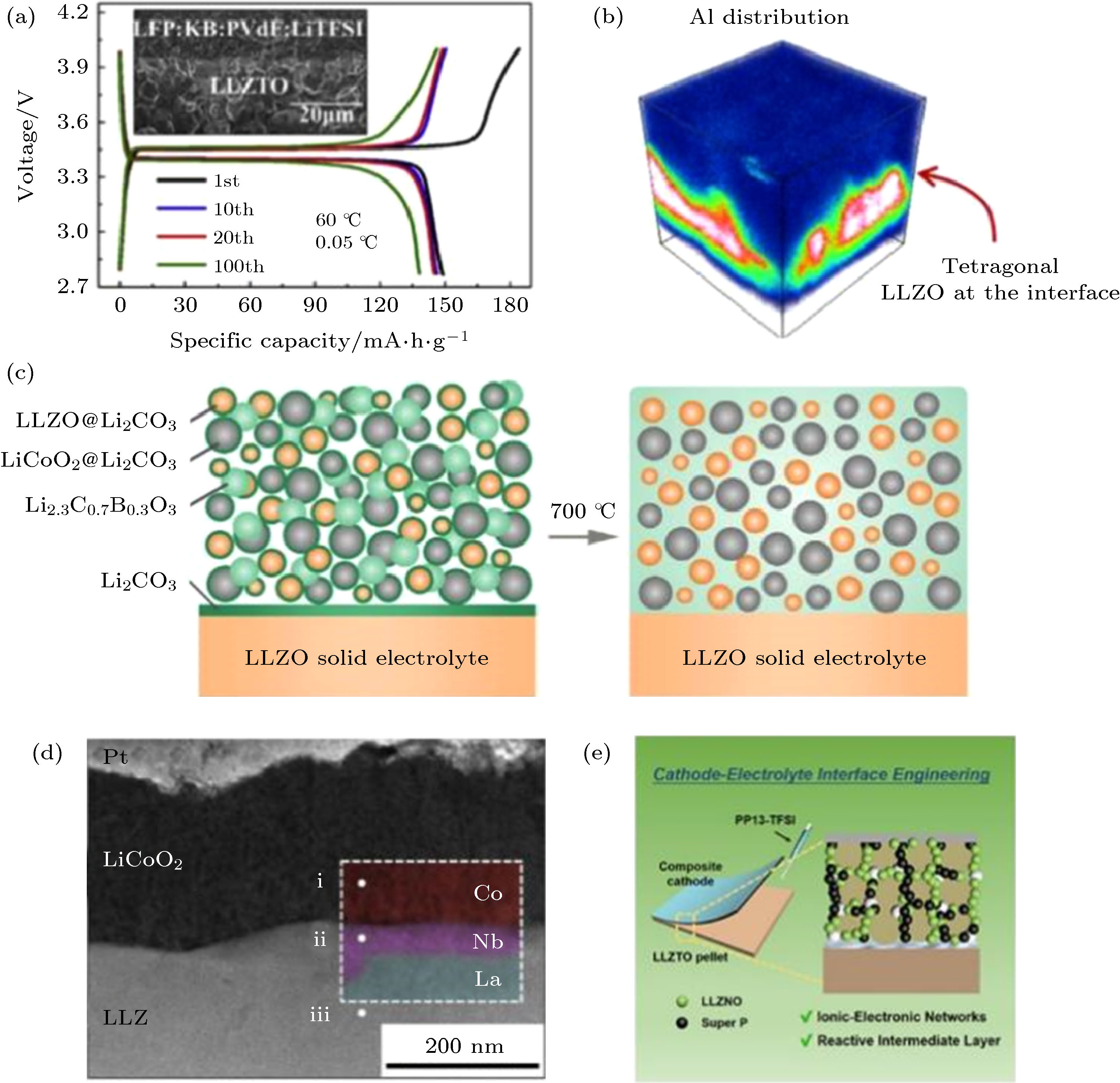 图 5 (a) 采用传统电极涂布法制备的固态复合正极及其充放电曲线[12]; (b) LLZO/LiCoO2界面发生元素互扩散形成四方相LLZO[13]; (c) 引入低熔点助烧剂Li2.3C0.7B0.3O3(LCBO)后, LLZO与LiCoO2形成离子导电的稳定界面[36]; (d) 金属Nb中间层有效抑制LLZO与LiCoO2间的元素互扩散[37]; (e) 结合表面包覆和原位电化学法在固态正极中形成离子电子共导电网络[38]
图 5 (a) 采用传统电极涂布法制备的固态复合正极及其充放电曲线[12]; (b) LLZO/LiCoO2界面发生元素互扩散形成四方相LLZO[13]; (c) 引入低熔点助烧剂Li2.3C0.7B0.3O3(LCBO)后, LLZO与LiCoO2形成离子导电的稳定界面[36]; (d) 金属Nb中间层有效抑制LLZO与LiCoO2间的元素互扩散[37]; (e) 结合表面包覆和原位电化学法在固态正极中形成离子电子共导电网络[38]Figure5. (a) Galvanostatic charge/discharge curves and cross-sectional SEM of the solid garnet batteries with the slurry-casting composite cathodes[12]; (b) three-dimensional element mapping of the LLZO /LiCoO2 interface enabled by TOF-SIMS[13]; (c) Ionic-conducting LLZO/LiCoO2 interfaces were achieved by introducing low melting-point Li2.3C0.7B0.3O3 (LCBO) [36]; (d) cross-sectional STEM image of a Nb-modified interface between LLZO and LiCoO2[37]; (e) illustration of the composite cathode configuration with garnet partially coated LiCoO2 and ionic liquid intermediate layer[38].
(2)界面能级匹配
由于固体电解质与正极材料的锂化学势存在差异, 两相界面处存在电势差, 电荷载流子为平衡界面电势会重新分布形成空间电荷层. 空间电荷层的存在引起锂离子越过界面的扩散势垒增大, 导致固体电解质与正极界面阻抗过高. Takada团队[44]采用第一性原理计算研究了硫化物电解质与氧化物正极界面的空间电荷层效应. 如图6(a)所示, 在电化学势的驱动下, Li+从硫化物电解质向LiCoO2扩散导致靠近界面的硫化物Li+浓度降低, 在充电起始阶段LiCoO2开始脱锂, LiCoO2界面的富锂区域消失, 导致空间电荷层厚度进一步增大. 在硫化物/氧化物界面引入两者化学势接近的中间层LiNbO3, 可以降低Li+在界面处的扩散势垒并抑制空间电荷层在充电态的扩展, 有效缓解硫化物电解质与氧化物正极界面的高阻问题. 之前人们一般认为氧化物电解质与氧化物正极材料因化学势相差不大, 空间电荷层效应不明显, 因此相关的研究鲜有报道. 但在实际的氧化物电解质电池中, 电解质/正极界面同样表现出较大的界面阻抗, 其中的空间电荷层贡献引起越来越多的关注. Wagermaker等[45]采用第一性原理定量研究了LLZO与LiCoO2之间的空间电荷层效应. 模拟结果预测LLZO/LiCoO2空间电荷层的厚度为纳米级, 除非界面处的Li+被完全耗尽, 否则空间电荷层效应对界面离子输运影响可以忽略(图6(b)). 这一预测结果对电极/电解质界面构筑的指导意义还有待在实验上进一步验证. 最近, 周豪慎、何平课题组与Wagermaker等[46]合作, 采用固态核磁等实验手段, 定量研究了LGAP与氧化物正极界面的空间电荷层效应, 这种研究思路值得借鉴到LLZO与正极界面的研究中. 通过调控氧化物正极中的锂化学势, 结合固态核磁技术定量研究了氧化物电解质/氧化物正极(LAGP/LixV2O5)界面的空间电荷层效应对界面处Li+交换活化能和交换电流密度的影响. 如图6(c)和6(d)所示, 锂化学势相差较大的LixV2O5和LAGP之间存在空间电荷层, 表现出强度较弱的核磁交换峰, 锂离子交换活化能为0.515 eV; 锂化学势接近的LixV2O5和LAGP之间不存在空间电荷层, 对应的二维核磁交换谱表现出强度很高的交换峰, 锂离子交换活化能为0.315 eV, 相应的电极/电解质界面阻抗显著降低. 以上研究表明, 电解质/正极界面处的化学势的差异是导致空间电荷层产生的根源, 结合材料的特点, 构建能级匹配的两相界面是改善界面离子输运特性的有效手段.
 图 6 (a)硫化物电解质/氧化物正极材料界面[44]; (b)氧化物固体电解质/氧化物正极材料界面的空间电荷层示意图[45]; (c) LiV2O5/LAGP和(d) Li2V2O5/LAGP界面的空间电荷层与对应的固态核磁共振交换谱[46]
图 6 (a)硫化物电解质/氧化物正极材料界面[44]; (b)氧化物固体电解质/氧化物正极材料界面的空间电荷层示意图[45]; (c) LiV2O5/LAGP和(d) Li2V2O5/LAGP界面的空间电荷层与对应的固态核磁共振交换谱[46]Figure6. Schematics of the space-charge layer effect at the interface between (a) sulfide electrolyte and oxide cathode[44]; (b) the interface between oxide electrolyte and oxide cathode[45]. Illustration of space-charge layer effect on Li+ transport between (c) LiV2O5/LAGP and (d) Li2V2O5/LAGP interfaces with the determination of activation energy of Li+ exchange enabled by 6Li 2-D exchange NMR spectrum[46].
(3)应力应变
基于嵌入脱出反应的正极材料在发生锂离子脱嵌时伴随着体积形变, 导致正极材料和固体电解质的固固界面产生应力并发生破裂, 表现为界面阻抗的剧增. 因此构建固态复合正极不仅要考虑界面的相稳定性与能级匹配问题, 解决界面力学失效问题同样重要. 硫化物和聚合物电解质的质地柔软, 可以通过施加外部压力缓解由于电极体积形变带来的应力问题. 氧化物陶瓷电解质由于质地坚硬, 受挤压易碎裂导致电池短路, 难以通过施加外力解决电极/电解质界面应力应变问题. 如图7(a)所示, LLZO陶瓷片与LiCoO2固态复合正极之间的界面在数圈循环后发生开裂, 阻抗随之增大[47, 48], 作者提出在LLZO陶瓷型固态电池应当使用如Li4Ti5O12的零应变电极材料[49]. 另外, 理论计算表明LLZO与LiCoO2之间由于晶格失配导致界面产生11.9%的压应变[50], 表面包覆或者在正极和LLZO之间引入界面缓冲层是潜在的解决方案. 聚合物电解质弹性模量高, 可以吸收正极体积形变产生的界面应力[29], 然而大多数聚合物的耐氧化电位较低限制了高电压正极材料在固态电池中的应用. 最近夏永姚课题组[48]提出采用具有弹性的低熔点助烧剂Li2.985B0.005OCl吸收正极与LLZO之间的应力. 如图7(b)所示, 将反钙钛矿结构的固体电解质Li2.985B0.005OCl原位生长在LiCoO2和LLZO表面, 在高于Li2.985B0.005OCl熔点、低于LiCoO2和LLZO反应温度的400 ℃进行热压烧结, 获得界面接触紧密的固态复合正极. 与Li3BO3或LiF作为助烧剂的固态复合正极相比, Li2.985B0.005OCl复合正极表现出更高的库仑效率和更好的循环稳定性.
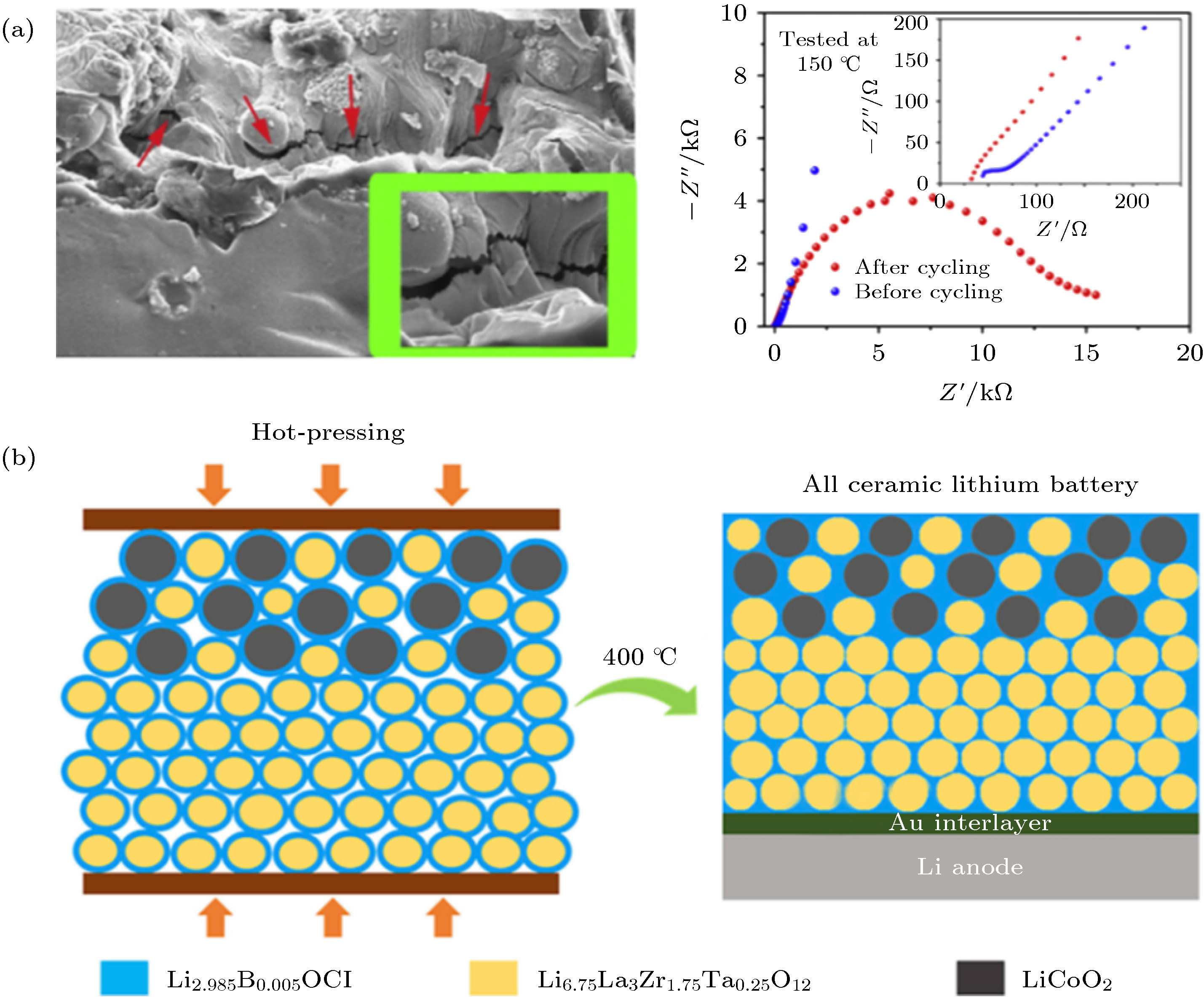 图 7 (a) LiCoO2固态复合正极与LLZO陶瓷片界面在循环后发生开裂造成界面阻抗显著增加[47]; (b) 以具有弹性的无机离子导体Li2.985B0.005OCl作为低熔点助烧剂制备的块体陶瓷型固态锂电池[48]
图 7 (a) LiCoO2固态复合正极与LLZO陶瓷片界面在循环后发生开裂造成界面阻抗显著增加[47]; (b) 以具有弹性的无机离子导体Li2.985B0.005OCl作为低熔点助烧剂制备的块体陶瓷型固态锂电池[48]Figure7. (a) Cross-sectional SEM image and impedance spectra of all-solid-state batteries after cycling which indicate the cracks of the solid-solid interface between the composite cathode and LLZO ceramics pellet[47]; (b) illustration of garnet-based all-ceramic Li battery enabled by high-1 elastic Li2.985B0.005OCl solder[48].
2
3.2.电解质/负极界面
(1)锂金属贯穿机制在基于液态电解液的锂电池中, 金属锂在反复沉积和剥离过程中出现枝晶进而刺穿隔膜造成电池短路的问题由来已久. 人们期望在固态锂电池中使用杨氏模量远高于金属锂的LLZO陶瓷电解质以克服金属锂的上述问题, 然而实验表明LLZO陶瓷片在较低的电流密度下即被锂贯穿[51]. 实际上, 金属锂在LLZO陶瓷电解质中的贯穿机制与锂枝晶在隔膜或聚合物电解质中的刺穿机制有很大不同. 从当前研究进展来看, 导致LLZO陶瓷电解质被贯穿的因素主要有自身缺陷、界面物理形貌和界面电荷转移三个方面. 如图8(a)所示, 尽管通过热压等方法可以获得致密度高于99.6%的LLZO陶瓷片, 但陶瓷电解质的晶界、裂纹和气孔很难彻底消除, 晶粒内部也存在大量的晶格缺陷. 这些宏观和微观的缺陷不可避免地导致陶瓷电解质内部锂离子传输的不均匀性, 为锂的成核生长提供了机会. 一旦电流密度超过临界值, 锂会迅速沿着缺陷连续生长直至贯穿LLZO陶瓷片[51]. 由于陶瓷电解质与锂金属的物理接触直接影响两者之间电接触, 而界面电场的分布是否均匀决定了锂在界面成核生长的均匀性. 如前所述, LLZO暴露于空气中会在表面形成碳酸锂等杂质, 这些导电性差的物质导致原本亲锂的LLZO表面与金属锂的浸润性变差[53], 造成界面电场分布不均匀. 针对这一问题研究人员提出化学腐蚀[28]、高温处理[54]和物理打磨[55, 56]等方法去除LLZO表面碳酸锂层, 显著降低LLZO/Li界面电阻和提升临界电流密度. 但是这些方法可能带来表面粗糙度和缺陷的增加, 不利于锂在界面处的均匀沉积. 引入中间层是改善界面接触和均匀化界面电场分布的有效策略. 根据导电性的差异, 一般将中间层分为电子导电、离子导电以及混合导电中间层. 以单质金属为主的电子导电中间层通过与金属锂发生合金化反应来改善界面接触, 但是合金/去合金化过程伴随着体积形变以及e-和Li+在LLZO界面的聚集, 多次循环后仍会出现锂金属贯穿. 如图8(c)所示, 离子导电中间层可以阻挡e-向LLZO迁移, 抑制锂金属在LLZO陶瓷内沉积, 但是离子导体的高阻界面限制了临界电流密度的提高. 混合导电中间层综合了电子导电和离子导电中间层的优点, 改善界面接触、均匀化界面电场分布的同时, 阻挡电子注入LLZO. 基于这一思想, 高健等[53]制备了一种外层为Ti掺杂LLZO、内层为LLZO的三层结构陶瓷电解质片, 外层的Ti掺杂LLZO在充放电过程中转变为离子电子混合导电中间相, 可以均匀化电场分布, 防止出现局部过电势. 如图8(d)所示, 得益于离子电子混合导电特性, 外层的Ti掺杂LLZO还可以“固定”Li+和e-对, 阻挡e-向内层LLZO陶瓷迁移, 有效抑制了锂金属贯穿(图8(d)). 总结以上, 减少LLZO陶瓷缺陷、改善界面接触和引入中间层等方法均可一定程度抑制锂的不均匀沉积, 但是当电流密度超过临界值仍会出现锂金属贯穿. 因此, 在实际应用某一种改善策略于LLZO固态电池时, 应当先标定该体系的临界电流密度, 将运行电流密度控制在临界值以下才能确保电池的安全性.
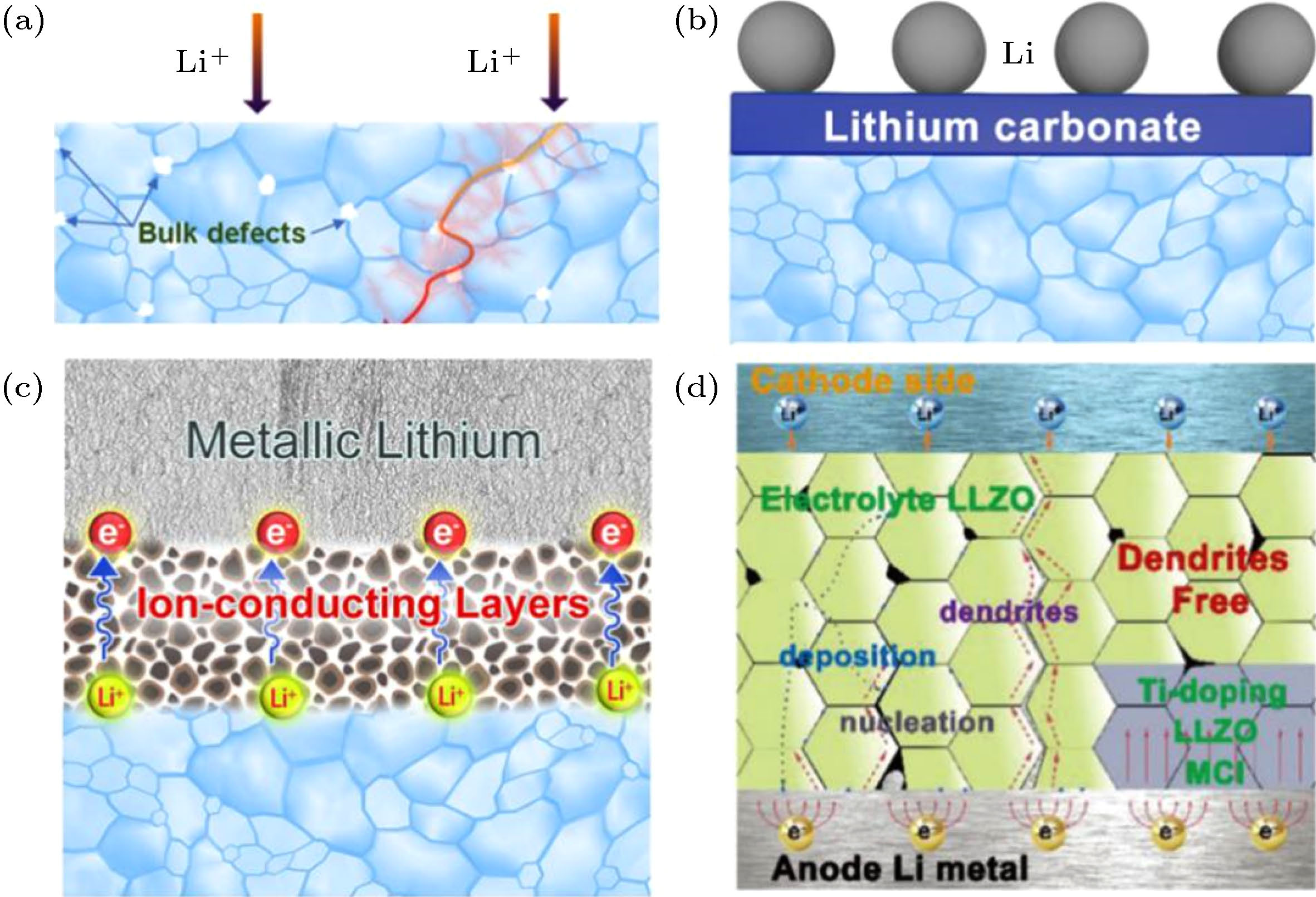 图 8 (a) 低致密度且有晶格缺陷的LLZO陶瓷电解质示意图[52]; (b) 表面碳酸锂等污染物对LLZO陶瓷亲锂性的影响; (c) 理想的纯离子导体中间层有效抑制金属锂贯穿, (d) 在LLZO陶瓷电解质近锂侧掺Ti, 在陶瓷本体部分形成混合离子电子导电界面起到均匀化电场、阻挡电子注入和抑制锂成核等作用[53]
图 8 (a) 低致密度且有晶格缺陷的LLZO陶瓷电解质示意图[52]; (b) 表面碳酸锂等污染物对LLZO陶瓷亲锂性的影响; (c) 理想的纯离子导体中间层有效抑制金属锂贯穿, (d) 在LLZO陶瓷电解质近锂侧掺Ti, 在陶瓷本体部分形成混合离子电子导电界面起到均匀化电场、阻挡电子注入和抑制锂成核等作用[53]Figure8. (a) Illustration of low-dense LLZO ceramic with lattice defects[52]; (b) the wettability of Li metal against LLZO ceramic electrolyte with Li2CO3 contamination; (c) the inhabitation effect of Li-penetration with ionically conducting intermediate layer; (d) Ti-doping electronic/ionic interphase is formed at the lithium anode side to homogenize local electrical field and hinder the e– infiltration, which is effective to inhibit the Li-penetration through LLZO ceramic pellet[53].
(2)应力应变
LLZO/负极界面出现力学失效的原因主要有两方面, 一是锂贯穿固体电解质, 二是负极的体积形变. 锂贯穿固体电解质是力学与电化学相互作用的结果: 固体电解质的力学特性会引发锂贯穿, 而锂贯穿固体电解质会产生新的力学问题. Kazyak等[57]采用原位光学显微成像技术等技术详细研究了锂贯穿LLZO陶瓷电解质的力学-电化学耦合机制. 结果显示锂在固体电解质内部的生长有多种形态, 作者将其归为四类, 分别是直线状(straight)、树枝状(branching)、散裂状(spalling)、和扩散状(diffuse). 不同的锂贯穿形态显然难以用统一的生长机制来解释, 力学-电化学耦合机制是常见的解释之一: 在相对较高的电流密度下, 锂沿着LLZO陶瓷的裂纹方向迅速沉积(图9(a)), 锂的沉积导致裂纹进一步扩展, 最终锂贯穿LLZO陶瓷造成电池短路. Yet-Ming Chiang课题组[58]在单晶LLZO中同样发现类似现象. 他们认为锂的沉积是在提供电子的金属集流体上开始的, 并且会逐渐长大以填充集流体和LLZO之间的任何空间. 由于固体电解质表面裂纹和孔洞处的粗糙度高、局部电场强度分布不均匀, 锂金属会优先沉积在这些位置. 一旦裂纹和孔洞被完全填充, LLZO/锂金属界面的应力会随着锂的沉积持续增加. 为了确定应力累积的程度, 作者提出图9(b)所示的简化分析模型. 实验数据和模拟结果表明, 锂贯穿固体电解质的过程类似格里菲斯(Griffith)裂纹扩展, 与液态电解质中的锂枝晶生长机制有很大不同. 并且, Monroe和Newman[14]提出的机械强度抑制锂枝晶生长的观点, 不适用于LLZO陶瓷电解质这种高剪切模量的脆性材料. 硅负极是未来电池能量密度继续提升的关键材料, 然而巨大的体积膨胀限制了它在固态电池中的应用. 针对硅负极在固态电池中的力学失效问题, Qi课题组[59]基于第一性原理模拟结果提出如图9(c)的分析模型. 在充电过程中, LiCoO2脱去50%的Li+时伴随2%的体积收缩, 这些Li+嵌入Si负极中伴随23%的体积膨胀; 与此同时, LiCoO2与LiPON形成界面中间相伴随8%的体积收缩, Si/LiPON界面形成中间产物伴随27%的体积收缩. 这说明, 相比于硅负极的体积变化, 固体电解质/负极界面反应对界面应力的产生有更大的影响. Huo等[60]设计了一种可以与硅负极形成稳定界面并且缓解界面应力的柔性复合固态电解质膜. 如图9(d)所示, 与刚性的LLZO陶瓷电解质相比, 弹性高的LLZO/PPC复合电解质膜可以吸收硅负极体积形变带来的应力, 保证硅负极在充放电循环过程中与复合电解质膜保持紧密接触.
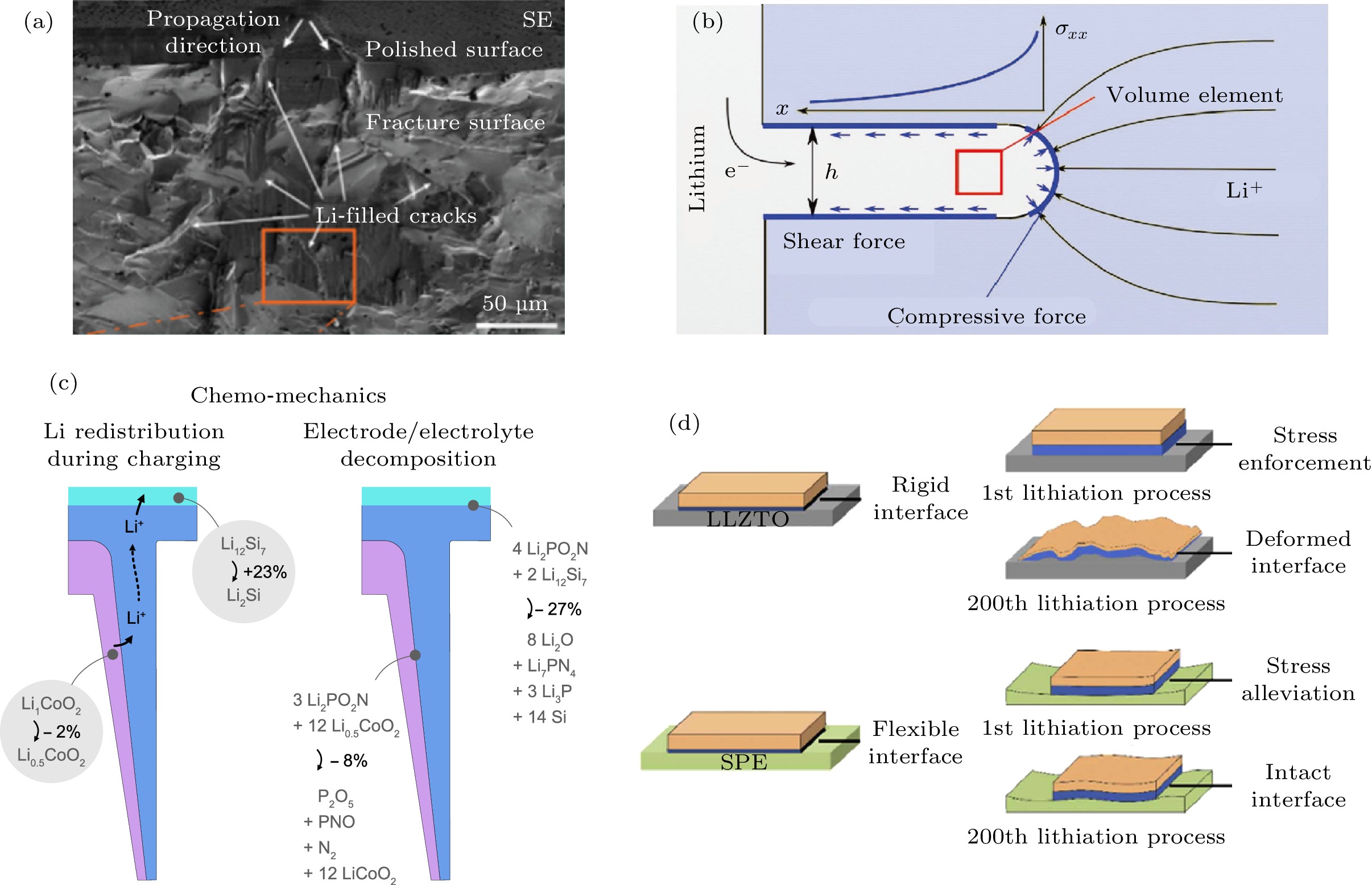 图 9 (a) 锂贯穿LLZO陶瓷电解质的微观形态分析[57]; (b) LLZO/Li负极界面应力累积程度的简化模型[58]; (c) 硅负极/固体电解质界面应力应变产生的化学-力学耦合机制[59]; (d)柔性复合固态电解质缓解硅负极产生的应力应变问题[60]
图 9 (a) 锂贯穿LLZO陶瓷电解质的微观形态分析[57]; (b) LLZO/Li负极界面应力累积程度的简化模型[58]; (c) 硅负极/固体电解质界面应力应变产生的化学-力学耦合机制[59]; (d)柔性复合固态电解质缓解硅负极产生的应力应变问题[60]Figure9. (a) Morphology of LLZO ceramic pellet after Li penetration[57]; (b) simplified model of a Li filament in a solid electrolyte which predicts the aggerating stress at Li/LLZO interface[58]; (c) chemo-mechanical coupling mechanism that cause strain/stress at the interface between Si anode and solid electrolyte[59]; (d) illustration of the flexible and rigid interfaces between Si anode and flexible composite electrolyte which alleviate the interfacial strain/stress [60].
(3)热稳定性
锂离子电池在热滥用、电滥用和机械滥用等情况下会发生热失控, 引发正负极与电解液之间的剧烈放热反应, 造成冒烟、起火甚至爆炸等安全事故. 采用热稳定性高的氧化物固体电解质替代有机电解液, 被认为是解决热失控、提高安全性的根本途径. 然而关于固态电池安全性的研究大多停留在固体电解质材料层面, 从固体电解质/金属锂界面或全电池的角度探讨固态电池实际安全性的研究较少. NASICON型(LATP, LAGP)和钙钛矿型(LLTO)电解质由于Ti4+和Ge4+易被还原, 不能直接与金属锂负极接触. Kang等[61]研究了LAGP与金属锂之间的化学反应稳定性和热稳定性. 如图10(a)所示, LAGP/Li的界面阻抗随着时间延长而持续增大, 对Li/LAGP/Li对称电池进行直流偏压测试后(4 V)陶瓷片出现碎裂, 作者推测界面处生成了由Li2O2, Li2CO3和C等组成的离子电子混合导电中间相. 将LAGP陶瓷片与金属锂加热至200 ℃以上, 界面中间相发生分解引发剧烈的放热, 最终出现燃烧等热失控现象(图10(a)). 相比之下, LLZO表现出优异的对锂稳定性[62]. Wolfenstine等[63]将LLZO(Li7La3Zr2O12)浸没于在300—350 ℃的熔融金属锂中, 未发生热失控现行. 浸没24 h以后, LLZO陶瓷片表面由纯白色转变为灰黑色, 但XRD结果显示LLZO仍保持立方相结构未生成其他产物. 对比原始LLZO陶瓷片, 电子顺磁共振谱在浸没锂后的LLZO陶瓷片表面检测到被缺陷俘获的孤对电子. 作者据此提出熔融锂中的e-和Li+会扩散至LLZO中, e-被氧缺陷俘获形成色心, 导致LLZO颜色发生变化; Li+进入晶格导致晶胞体积增加并产生应力, 造成LLZO陶瓷片出现裂纹. 近期, 李泓课题组[62]采用加速量热仪(accelerating rate calorimetry, ARC)定量研究了四种常用的氧化物固体电解质(LATP, LAGP, LLTO和LLZO)与金属锂接触后的热失控行为. 如图10(b)所示, 在升温过程中, LATP, LAGP, LLTO与金属锂均发生热失控, 只有LLZO与金属锂未出现明显放热行为. 结合第一性原理计算和X射线衍射分析, 作者提出氧化物固体电解质的热失控机制: 首先, LATP, LAGP和LLTO与金属锂接触发生界面反应; 随着温度升高, 界面反应释放热量促进固态电解质分解产生活性氧; 金属锂达到熔点后与活性氧剧烈反应产生大量热量, 加速固体电解质分解并释放更多活性氧, 最终出现热失控. 通过对比四种氧化物电解质在ARC测量过程中的最大自加热速率dT/dt (℃/min), 得出LAGP < LATP < LLTO < LLZO的热稳定性顺序. 以上研究表明, 固体电解质材料的热稳定性高并不意味着电池安全性高, 通过调控界面反应特性提高固态电池整体热稳定防止出现热失控是未来的重要研究方向.
 图 10 (a) NASICON型氧化物固体电解质LAGP陶瓷片与金属锂的热稳定性研究[61]; (b)采用加速量热仪研究LAGP, LATP, LLTO与LLZO四种氧化物固体电解质与金属锂接触时的热稳定性[62]
图 10 (a) NASICON型氧化物固体电解质LAGP陶瓷片与金属锂的热稳定性研究[61]; (b)采用加速量热仪研究LAGP, LATP, LLTO与LLZO四种氧化物固体电解质与金属锂接触时的热稳定性[62]Figure10. (a) Thermal stability between lithium metal and NASICON-type LAGP solid electrolyte[61]; (b) accelerating rate calorimeter test results of four oxide electrolytes and Li metals, and schematics of the thermal runaway reaction between oxide electrolyte and Li metal[62].
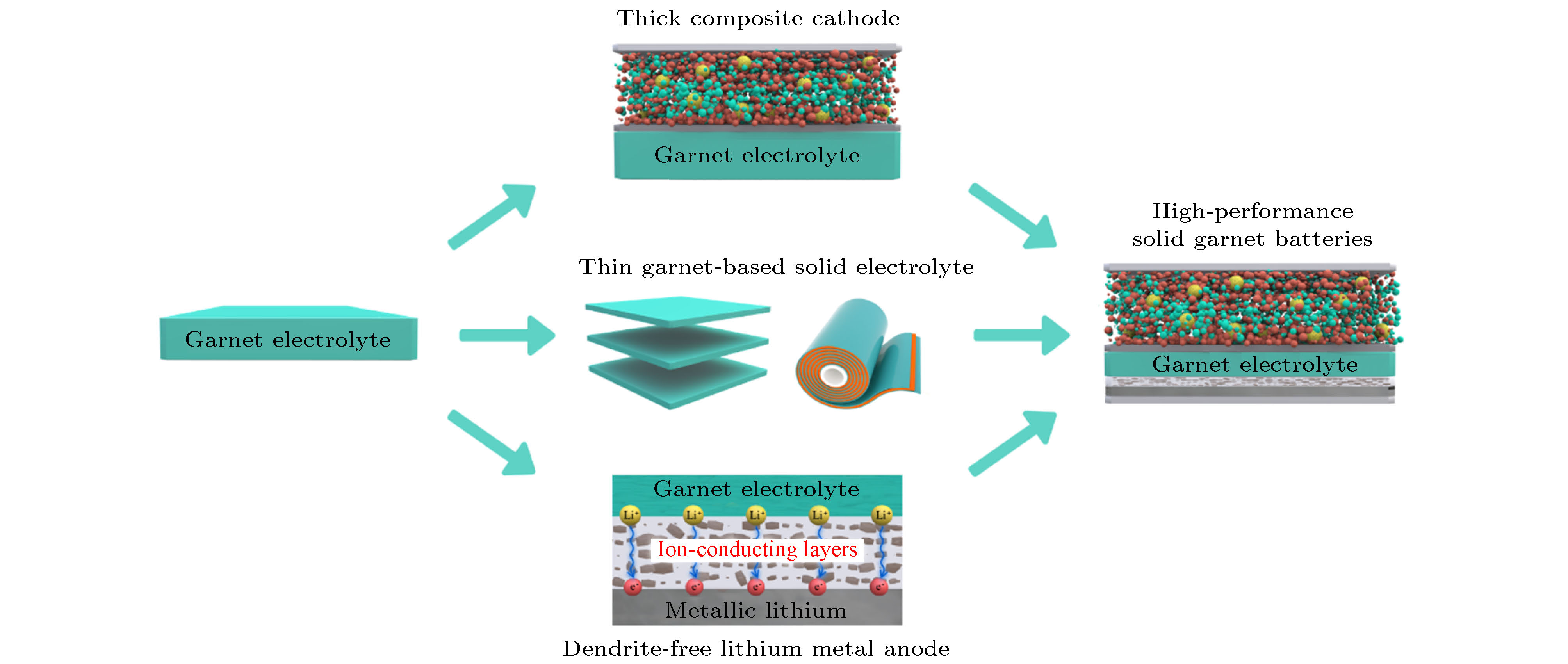 图 11 能量密度大于400 Wh/kg的石榴石型固态电池开发策略[64]??????????????????????????????????
图 11 能量密度大于400 Wh/kg的石榴石型固态电池开发策略[64]??????????????????????????????????Figure11. Development strategies of solid garnet batteries with energy density higher than 400 Wh kg-1[64].????????????????????????????
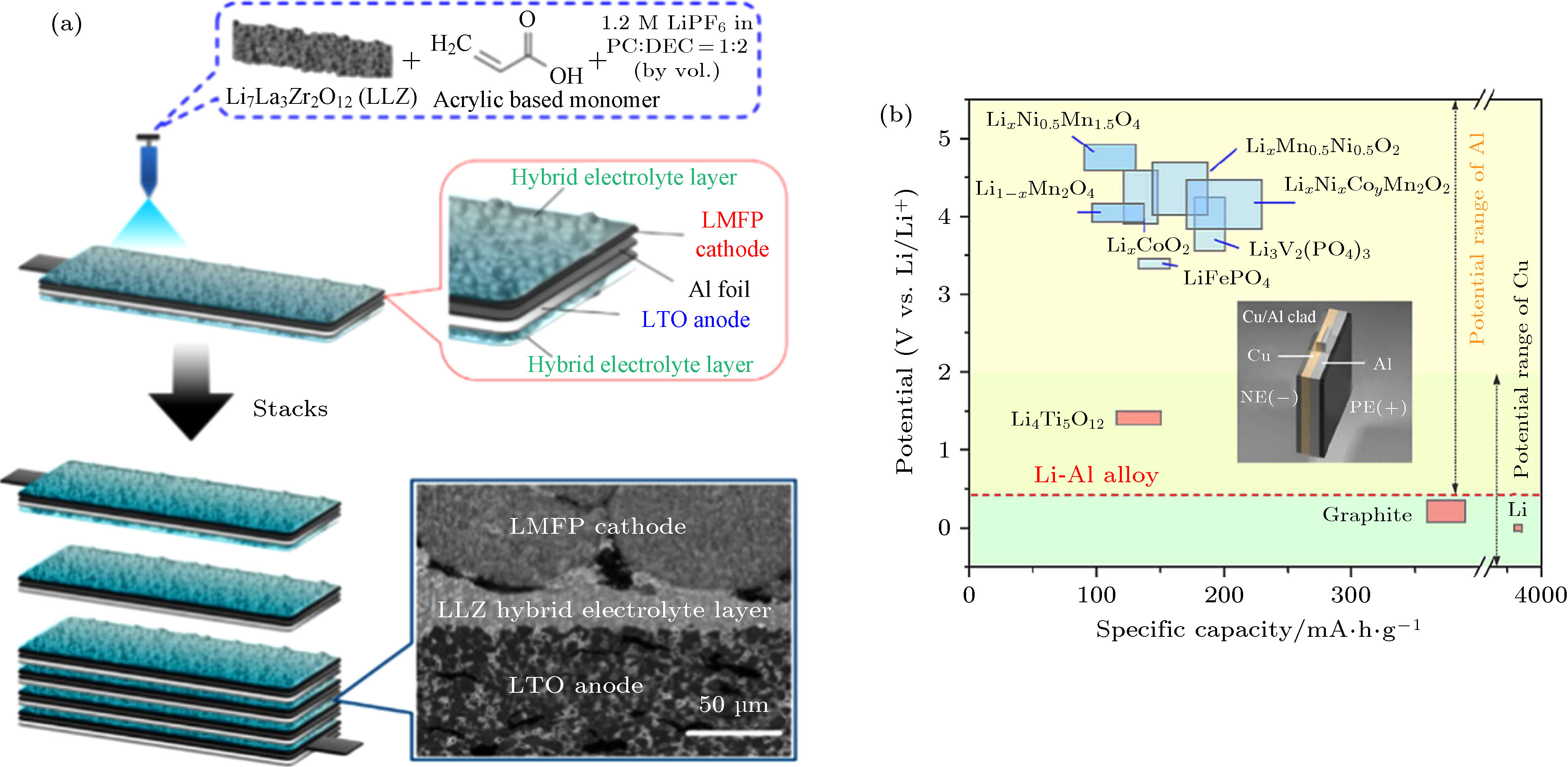 图 12 (a)由LMFP正极、LLZO基混合电解质层和LTO负极组成的双极性准固态电池的截面示意图和截面SEM图像[69]; (b)集流体Al和Cu的稳定电位范围以及Cu/Al双极性集流体的示意图[65,69]
图 12 (a)由LMFP正极、LLZO基混合电解质层和LTO负极组成的双极性准固态电池的截面示意图和截面SEM图像[69]; (b)集流体Al和Cu的稳定电位范围以及Cu/Al双极性集流体的示意图[65,69]Figure12. (a) Cross-sectional illustration and SEM images of bipolar quasi-solid-state batteries with LMFP cathode, LLZO-based hybrid electrolyte and LTO anode[69]; (b) the electrochemical stable window of Al foil and Cu foil, and the schematics of Cu/Al bipolar current collector[65,69].
双极性电池结构, 是指相邻电芯的正负极共用同一集流体, 无需使用外部导线连接直接在电池模组中形成串联. 这种设计最早出现在意大利科学家伏打(Alessandro Volta)1800年发明的伏打电堆中, 在铅酸电池和镍氢电池中也有应用, 可以减少集流体、包装以及外部电气连接等电池组件的使用, 显著提升电池组的输出电压和能量密度, 并降低电池制造成本. 此外, 双极性集流体可以缩短电子传输距离, 从而提高电池的倍率特性[65-70]. 然而由于电解液的电压窗口小于两端集流体的电压差, 电解液发生分解并产气, 导致电池的内部接触变差、输出电压下降甚至出现电池胀包等现象. 与电解液相比, 固体电解质电压窗口宽且不接触集流体, 是构建双极性结构电池的理想电解质材料. 东芝公司研究团队[69]设计了一种输出电压为12 V的双极性准固态电池. 如图12(a)所示, 超薄的LLZO混合电解质由LLZO颗粒与丙烯腈单体和碳酸酯类电解液原位聚合而成, 厚度仅为3 μm, 其中凝胶电解质质量百分比为4 wt%. 得益于超薄电解质层优异的离子传导能力和凝胶电解质良好的界面融合能力, 由LiMn0.8Fe0.2PO4(LMFP)正极和Li4Ti5O12(LTO)负极组成的全电池表现出优异的电化学性能, 工作温度范围为–40—80 ℃, 在60 ℃循环200圈的容量保持率为85%. 由于LTO的工作电压约1.5 V高于Li-Al合金电位, LTO可与LMFP共用Al箔作为集流体, LTO/LLZO/LMFP单体电芯可以轻松实现堆叠, 获得累加的电池输出电压. 如图12(b)所示, 工作电压在Cu箔和Al箔电压窗口交叉范围内的电极材料很少, 为构建更高输出电压和更高能量密度的电池, 必须开发宽电压窗口并且兼容正负极材料的双极性集流体. 复合金属(clad metal)由两种以上的金属材料复合而成, Cu/Al复合箔是一种潜在的双极性集流体. Shin等[68]首次使用Al/Cu(10 μm/10 μm)集流体, 组装出以金属锂为负极以LiNi0.6Co0.2Mn0.2O2为正极、输出电压12 V的双极性固态锂电池. 此外, 碳(碳纳米纤维/管、碳布、石墨烯纸等)和导电聚合物(例如聚吡咯和聚噻吩)等轻薄的导电材料也是颇有前景的双极性集流体材料[71].
基于双极性电池结构, Jia等[64]提出如图13(a)的固态电池能量密度计算模型, 建立了固态电解质厚度与复合固态正极载量的设计标准. 由于不同的固态电解质密度存在差异, 实现高于400 Wh/kg的能量密度需要达到的厚度不同. 在图13(a)所示6单元堆叠的双极性固态电池模型中, 作者使用厚度为100 μm的NCM811固态复合正极和厚度为50 nm的金属锂箔, 以15 μm的Cu/Al复合箔为双极性集流体. 设定复合正极的压实密度为3 g/cm3, 对应的载量为30 mg/cm2, 面容量为6.3 mAh/cm2. 如图13(b)所示, 为了满足400 Wh/kg的要求, LiPON、硫化物、LLZO陶瓷电解质与LLZO复合电解质的厚度分别为34 μm, 41 μm, 16 μm和30 μm. 显而易见, 在相同的能量密度要求下, 密度小的固体电解质可允许的厚度大. 然而, 密度大的LLZO陶瓷电解质由于硬度高脆性大, 很难加工至16 μm; 对于LLZO复合电解质膜, 30 μm是常见厚度. 可见, 基于复合电解质膜的高比能大容量固态电池技术路线具有更高的可行性. 需要注意的是, 固体电解质的厚度设计不仅要考虑对能量密度的影响, 还应结合离子电导率和面电阻对固体电解质厚度的要求. Zhao等[72]指出尽管LiPON室温离子电导率只有2 × 10–6 S/cm, 却能够应用于薄膜型固态锂电池并实现上万次循环. 考虑到固体电解质层的面电阻对电池容量和倍率的影响, 只有将LiPON做到厚度小于1 μm、面电阻低于50 Ω·cm2, 电池才能在室温下发挥正常的电化学性能, 这一准则同样适用于其他类型的固体电解质材料. ??????????????????????????
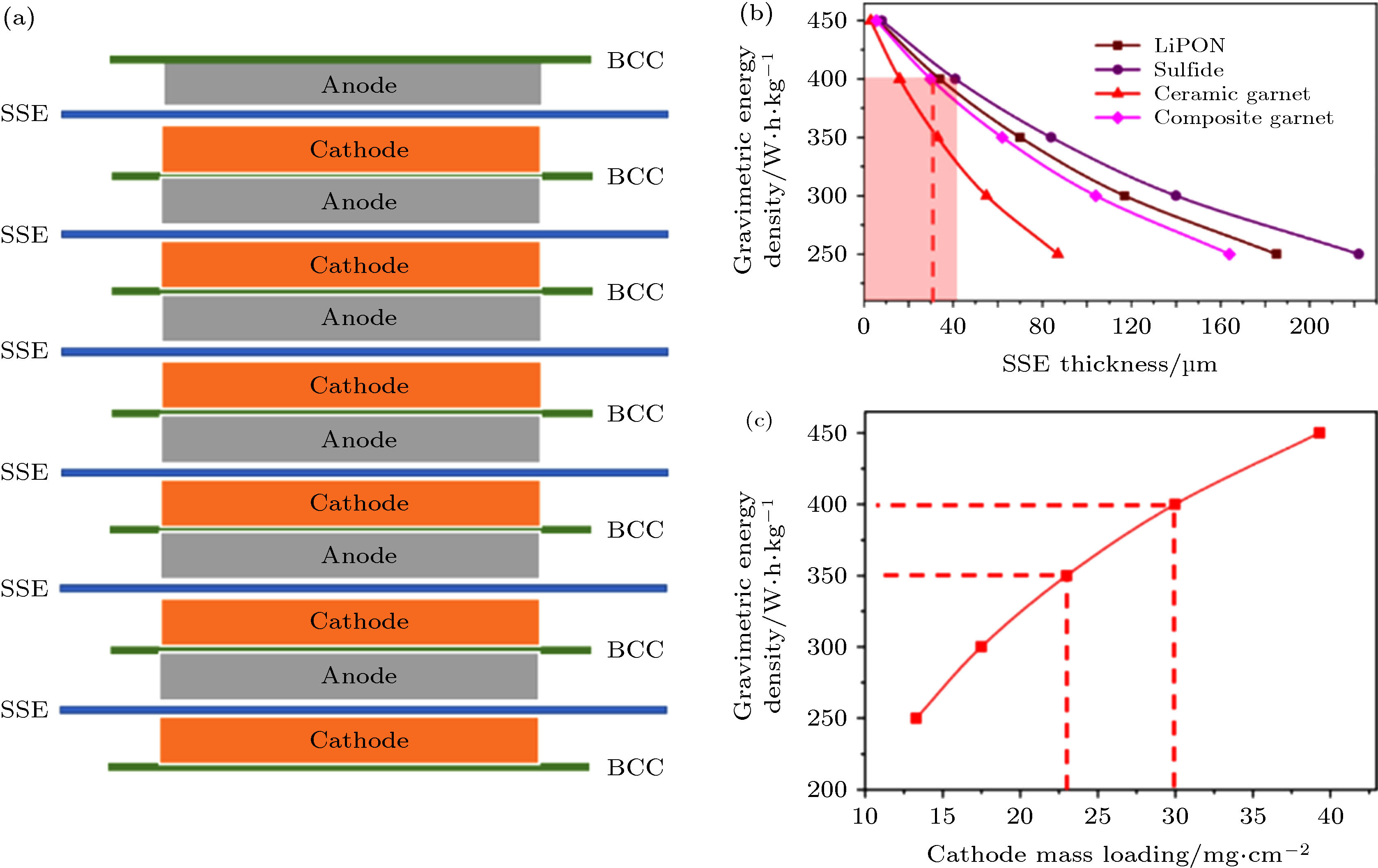 图 13 (a)固体电解质(solid state electrolyte, SSE)与固态正负极以及双极性集流体(bipolar current collector, BCC)组成双极性电池结构的固态电池示意图; (b)基于双极性电池结构, 四种固体电解质(LiPON、硫化物、LLZO陶瓷电解质与LLZO复合电解质)厚度与质量能量密度的关系, (c)固态复合正极的载量与质量能量密度的关系[64]??????????????????????????????????????
图 13 (a)固体电解质(solid state electrolyte, SSE)与固态正负极以及双极性集流体(bipolar current collector, BCC)组成双极性电池结构的固态电池示意图; (b)基于双极性电池结构, 四种固体电解质(LiPON、硫化物、LLZO陶瓷电解质与LLZO复合电解质)厚度与质量能量密度的关系, (c)固态复合正极的载量与质量能量密度的关系[64]??????????????????????????????????????Figure13. (a) Schematics of bipolar solid-state batteries composed of solid-state electrolyte (SSE), cathode, anode and bipolar current collector (BCC). (b) Based on bipolar battery architecture, the gravimetric energy density as a function of thickness of four solid electrolytes (LiPON, sulfide, LLZO ceramic electrolyte and LLZO composite electrolyte), and (c) the mass loading of composite cathode[64].????????????????????????????
在大多数固态电池的报道中, 固态正极的载量通常小于3 mg/cm2, 不能充分体现固态电池在能量密度方面的优势. 为了提高能量密度, 增加复合正极的载量势在必行. 基于双极性电池模型, 采用30 μm厚的LLZO复合电解质、NCM811正极、金属锂负极和Cu/Al双极性集流体, 计算得到固态复合正极载量与质量能量密度的函数关系(图13(c)). 由图可见, 实验室常用的低载量正极(< 3 mg/cm2)远低于高能量密度固态电池的要求. 当固态复合正极的载量达到30 mg/cm2以上时, 质量能量密度密度才能突破400 Wh/kg, 按照3 g/cm3的压实密度换算出正极厚度约为100 μm. 然而, 提高正极厚度不仅增大了离子/电子的扩散距离和传输不均匀性、导致电池倍率特性和循环稳定性变差, 还会带来金属锂负极体积变化增大和锂枝晶生长加剧等问题[73,74]. 考虑到固体电解质/正极的固固界面阻抗高, 固态电池的高载量复合正极构筑比液态锂离子电池面临更多挑战. 为获得高载量的固态复合正极, 研究人员提出三维复合结构和低温共烧等方法, 分别将固态复合正极载量提高至10.5 mg/cm2[75]和217.9 mg/cm2[76], 低温共烧法制备的固态复合正极的厚度最高可达1300 μm. 尽管这些方法能否实际应用于固态电池工程化还有待验证, 但是固态锂电池仍然是突破当前能量密度困境的有效途径.
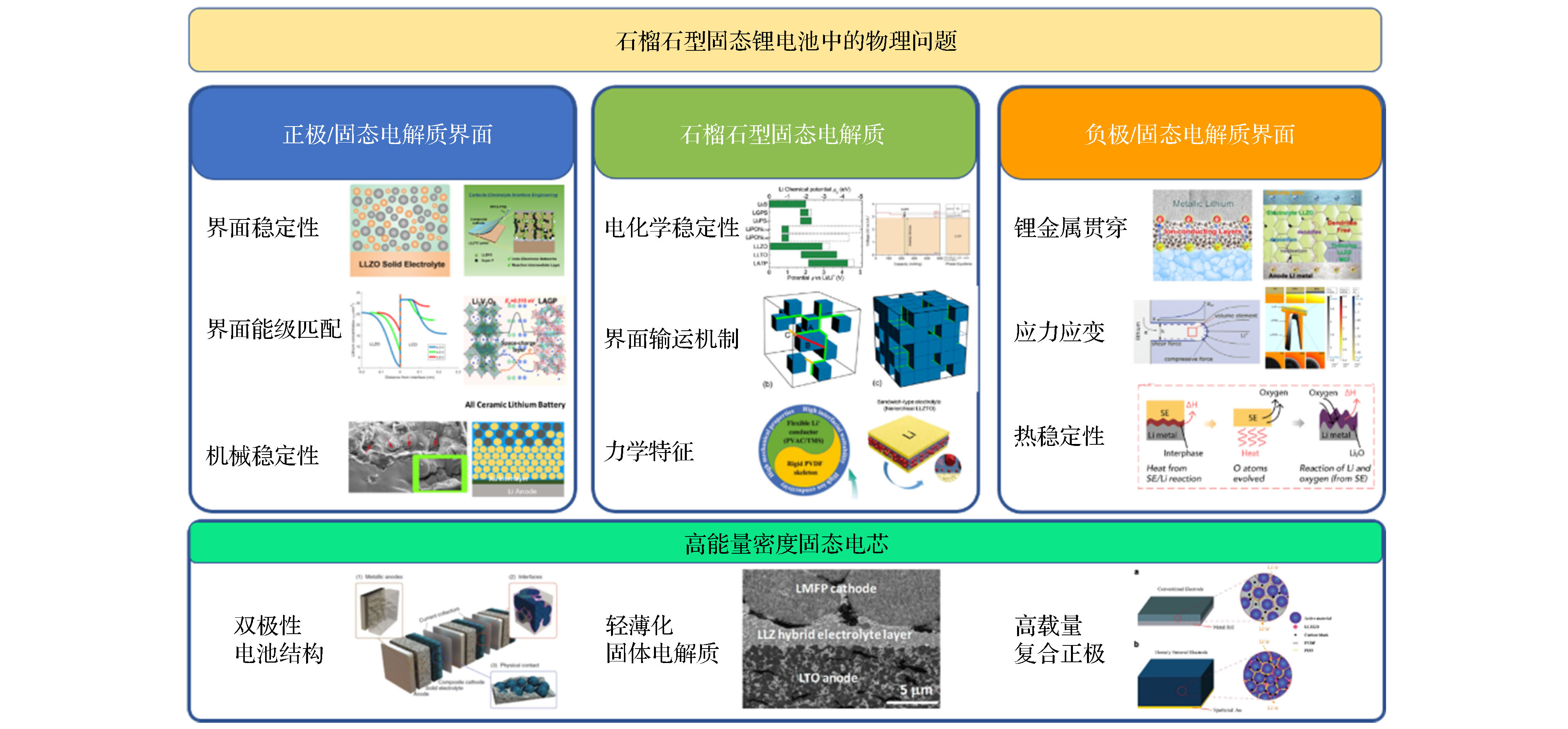 图 14 石榴石型固态锂电池中的关键物理问题(内嵌图出自参考文献)
图 14 石榴石型固态锂电池中的关键物理问题(内嵌图出自参考文献)Figure14. Key physical issues involved in solid garnet batteries (the insets excerpted from references).
1)开发新材料 结合高通量计算与实验方法, 探索新组成和新结构的快离子导体材料. 正如在锂离子电池中通过正极材料表面改性有效提升电池性能, 固态电池也应该探索合适的电极材料和表面修饰材料, 比如零应变正/负极材料、离子电子混合导电型界面中间层等.
2)材料和界面表征 对于固体电解质材料的研究, 目前大多停留在X射线衍射等晶体学方法, 对于固态电池中存在的大量“包埋”界面的表征大多停留在交流阻抗谱和恒流充放电等电化学方法. 理解和提高固体材料及固固界面的离子传导能力需要发展更多直观的表征方法, 比如借助同步辐射X射线吸收谱分析固体电解质材料的局域电子结构, 利用X射线断层成像无损地分析固固界面的动态形貌演变[77,78], 以及球差/冷冻电镜等原子尺度的表征技术. 针对固态电池失效分析, 发展与电化学结合的现场和原位技术(原位中子衍射、微分电化学质谱和加速量热仪等), 监测电池工作过程中的应力应变、产气、产热情况, 进而指导固体电解质/电极界面的构建.
3)材料加工和器件组装 进一步优化陶瓷电解质片和复合电解质膜的可加工性: 在稳定立方相的基础上融入薄膜化技术, 开发兼具刚性和韧性的陶瓷电解质片; 借鉴卷对卷的隔膜制备方法, 发展高机械强度、高离子电导率和宽电压窗口的复合电解质膜. 双极性电池结构设计、高载量复合正极的制备和固固界面融合技术, 是高比能固态电池组装和稳定运行的关键, 而电池的可靠性和一致性还依赖于工业制造流程的高度自动化. 另外, 制定固体电解质材料和固态电池相关的国家技术标准, 会促进固态电池的实用化研发, 应是接下来重点考虑的问题.
