全文HTML
--> --> -->因为有利于系统能量的降低确保稳定结构形成, 一般而言, 原子半径大的、表面能小的金属原子倾向于占据纳米粒子的表面. 在理论上, 当前主要采用静态的理论方法研究, 如蒙特卡罗[9,10]和全局优化方法[11]. 在实验上, 通过透射电子显微镜观察到了三层的核壳结构Au-Pd体系[7], Henglein[12]采用氢还原和辐射分解方法分别合成了Au核/Pt壳和Pt核/Au壳的纳米团簇. 通过原子沉积生长理论模拟获得了Mg核/Al壳、Fe核/Al壳和Pd核/Ag壳纳米团簇[2,13,14].
Ni是过渡金属基催化剂中应用最广泛的元素, 与其他金属形成双金属体系的能力最强, 因此研究人员对双金属镍催化剂进行了大量的研究[15]. 尽管在纳米合金的生长方面已经进行很多的理论数值研究[5,16-18], 但是关于基底结构对生长影响的研究却非常有限. 与Al原子相比, 金属Ni原子具有更小的原子半径和更大的表面能. 原子半径小的占据有利于释放团簇内部的应力, 从而增强纳米粒子的稳定性[19]. 从偏析的理论角度, Al原子应该倾向于纳米粒子的表面, 形成Ni-核/Al-壳结构. 因此本文选择NiAl双金属体系作为研究对象, 探究不同的Ni基底构型对Al原子在Ni团簇表面的生长影响. 得到NiAl的形成热为一个负值, 表明NiAl应该倾向于形成混合的合金结构, 因此探究不同的基底构型对生长的影响是非常有必要的. 在纳米尺寸范围内, 对于FCC晶体含有原子数为923, 正二十面体(Icosahedra, ICO923)、截角十面体(Decahedra, DEC923)和截角八面体(Cuboctahedra, CUB923)这三种几何结构是能够稳定存在的结构. 由于这三种结构具有相同的原子数, 但是却拥有截然不同的几何结构, 并且通过本文所采用的EAM势[20], 采用共轭梯度法对三种基底结构进行能量最小化, 计算得出系统的总能量也较为接近(ICO923, –4.14 eV/atom; DEC923, –4.12 eV/atom; CUB923, –4.12 eV/atom), 因此适合用来进行比较研究. 首先通过对单个Al吸附原子在ICO923, DEC923和CUB923结构的Ni基底表面的扩散行为进行对比研究, 随后从原子的角度分析了不同基底结构对生长的影响. 研究结果能够为实验上控制和合成特定结构的纳米合金提供一定的理论依据.
为了去研究Al吸附原子在不同Ni基底构型上的扩散行为, 单个金属Al吸附原子被放在结构分别为ICO923, DEC923和CUB923的Ni基底表面. 然后在正则系综下(canonical ensemble, NVT), 对整个体系逐渐升温并追踪Al吸附原子的运动轨迹, 随后使用肘弹性带方法(nudged elastic band, NEB), 计算出对应扩散路径的能量势垒. NEB方法是求解给定初始状态和最终状态之间过渡的最小能量路径的有效方法. 在NEB方法中, 在初态和末态之间线性地插入中间图像链, 每一幅图像对应着原子从初始位置到最终位置特定的几何形状, 这些图像通过弹性弹簧相互连接, 同时进行优化, 其中初始位置和最终位置保持不变[4,25]. 在当前的模拟中, 分子动力学(MD)模拟采用LAMMPS程序[26]中的NEB方法计算最小能量路径上的扩散势垒, 在NEB中使用了二十个中间图像. 在扩散过程中, 能量最高的图像对应系统的鞍点Esad, 系统的扩散势垒Ea可以被定义为Ea = Esad – Emin, 这里的Emin表示系统初态或者末态的能量.
对于单个Al原子在Ni基底(CUB923, DEC923和ICO923结构)上的生长注入模拟是在Lammps软件中[26]进行的. 在生长过程中原子的注入方式是Al原子随机沉积到Ni基底的表面, 其中注入原子的速率为100 ps/atom, 所选取的系综是NVT系综. 系统的运动方程遵循牛顿定律, 原子运动的数值积分算法采用的是具有时间反演对称的Verlet算法, 时间步长?t = 1 fs, 这与原子振动的时间尺度是相同的, CUB, DEC和ICO结构的壳层数与原子数之间的关系如下[27]:
 图 1 含有923个原子的CUB (a), DEC (b)和ICO (c)结构示意图, 三种结构是以壳层组成的非晶结构, 其中DEC和CUB由{111}和{100}面组成, 而ICO仅由{111}面组成
图 1 含有923个原子的CUB (a), DEC (b)和ICO (c)结构示意图, 三种结构是以壳层组成的非晶结构, 其中DEC和CUB由{111}和{100}面组成, 而ICO仅由{111}面组成Figure1. CUB (a), DEC (b) and ICO (c) microstructure with 923 atoms. The three clusters are a non-crystalline structure organized in shells. The DEC and CUB consist of {111} and {100} facets, and the ICO has only {111} facets.
 图 2 对于ICO, DEC和CUB结构, 总原子数和表面原子数所占的比例与壳层数的函数关系
图 2 对于ICO, DEC和CUB结构, 总原子数和表面原子数所占的比例与壳层数的函数关系Figure2. For the ICO, DEC and CUB structures, the total atomic number and percent of the surface atoms as a function of the shell number.
3.1.表面能和形成热
表面能(Esurf)定义为一个特定的晶体平面单位面积上的表面多余自由能, 是表面物理的基本物理量之一, 它决定了晶体的平衡形态, 对晶体的形成、粗化和生长起着重要的作用, 其计算表达式如下[28,29]:
| 元素 | 晶体结构 | Esurf/mJ·m–2 | ||
| 晶面 | 本工作 | FP[25] | ||
| Ni | FCC | (111) | 1875 | 2011 |
| (100) | 1964 | 2426 | ||
| (110) | 2148 | 2368 | ||
| Al | FCC | (111) | 933 | 939 |
| (100) | 994 | 1081 | ||
| (110) | 1063 | 1090 | ||
表1金属Ni和金属Al的表面能(Esurf). 列出了对应的第一性原理(FP)[28]数据
Table1.The surface energy (Esurf) for the two different material of Ni and Al. For comparison, the first principle (FP) calculations[28] are listed.
表2中列出了NiAl体系形成B2结构和L12结构的金属化合物的形成热, 与对应的实验数据和第一性原理计算结果有很好的一致性, 说明我们使用的势函数能够应用于不同浓度比例的NiAl合金, 其形成热

| 化合物 | 结构 | ?H/eV·atom–1 | |||
| 本工作 | FP | EMP | EXP | ||
| NiAl | B2 | –0.61 | –0.67 | –0.67 | |
| NiAl3 | L12 | –0.26 | |||
| Ni3Al | L12 | –0.45 | –0.44 | –0.46, -0.48 | –0.49, -0.43 |
表2NiAl不同化合物的形成热(?H). 为了对比列出了对应的第一性原理[32](FP)和其他理论方法[33] (EMP)以及对应实验结果[34-36] (EXP)
Table2.The calculated heat of formation (?H), structural of NiAl in varying crystal structures. For comparison, the first-principle (FP)[32] and other calculations with empirical methods (EMP)[33] and available experimental values (EXP)[34-36] are also listed.




2
3.2.单个Al原子在Ni团簇的表面扩散
金属原子的扩散机制主要分为两类, 一类是跳跃机制, 即吸附原子从一个平衡位置跨过能量势垒进入另外一个新的平衡位置进行扩散; 另一类则是交换机制, 当吸附原子到达台阶边缘, 它会推出台阶边缘原子然后占据其留下的空位, 随后边界原子成为新的吸附原子在邻面上进行扩散[37].为研究单个金属Al原子在Ni基底(即CUB923, DEC923和ICO923)表面的扩散行为, 将单个Al原子添加到Ni基底的{111}面的中心作为扩散的起始点, 如图3所示, 我们给出了Al原子在不同Ni基底表面的扩散路径.
 图 3 对于Al 吸附原子在CUB923, DEC923和ICO923结构的Ni基底表面上, 通过跳跃和交换机制, 吸附原子从{111}面向{100}面或向{111}面扩散的路径 (a) Hopping CUB{111} → {100}; (b) Exchange CUB{111} → {100}; (c) Exchange CUB{111} → {111}; (d) Exchange DEC{111} → {100}; (e) Hopping DEC{111} → {100}; (f) Exchange DEC{111} → {111}; (g) Hoping DEC{111} → {111}; (h) Exchange ICO{111} → {111}; (i) Hoping ICO{111} → {111}. 橙色的球表示铝原子, 灰色的球表示镍原子
图 3 对于Al 吸附原子在CUB923, DEC923和ICO923结构的Ni基底表面上, 通过跳跃和交换机制, 吸附原子从{111}面向{100}面或向{111}面扩散的路径 (a) Hopping CUB{111} → {100}; (b) Exchange CUB{111} → {100}; (c) Exchange CUB{111} → {111}; (d) Exchange DEC{111} → {100}; (e) Hopping DEC{111} → {100}; (f) Exchange DEC{111} → {111}; (g) Hoping DEC{111} → {111}; (h) Exchange ICO{111} → {111}; (i) Hoping ICO{111} → {111}. 橙色的球表示铝原子, 灰色的球表示镍原子Figure3. For the Al adatoms on the surfaces of the Ni CUB923, DEC923, and ICO923, the diffusion path of the adatom from the {111} facet to the {100} facet or to the {111} facet via the hopping and exchange mechanisms: (a) Hopping CUB{111} → {100}; (b) Exchange CUB{111} → {100}; (c) Exchange CUB{111} → {111}; (d) Exchange DEC{111} → {100}; (e) Hopping DEC{111} → {100}; (f) Exchange DEC{111} → {111}; (g) Hoping DEC{111} → {111}; (h) Exchange ICO{111} → {111}; (i) Hoping ICO{111} → {111}. The orange balls show the Al adatoms and the gray balls show the Ni atoms.
3
3.2.1.Al原子在Ni CUB923表面的扩散行为
在CUB923的表面, Al吸附原子能够通过交换机制或者跳跃机制从{111}面扩散到{100}面, 然而对于Al吸附原子从{111}面扩散到相邻的{111}面上仅能通过交换机制, 这三种类型的扩散路径如图3(a)—图3(c)所示.Al吸附原子在Ni CUB923基底两个相邻表面之间有三种可能的扩散路径, 对应扩散路径上的能量势垒如图4(a)和图4(b)所示, 图中的横坐标已经进行了归一化处理, 其中0.0和1.0分别对应于吸附原子扩散中的初始位置和最终位置, 扩散路径上系统能量的最低点被定义为0. 对比于在平面上的扩散, 其中一个额外的势垒在台阶的边缘存在, 即Ehrlich-Schwoebel(ES)势垒[34]. 对于吸附原子从{111}面扩散到{100}面通过跳跃和交换机制对应的ES势垒分别为0.79和0.38 eV. 对比可知, 跳跃机制的ES势垒[38]远高于交换机制, 因此吸附原子从{111}面扩散到{100}面应该优先发生交换, 当吸附原子从{111}面通过交换机制扩散到相邻的{111}面时存在一个巨大的ES势垒[38] (1.26 eV), 表明吸附原子从{111}面扩散到相邻的{111}面只可能在高温下发生.
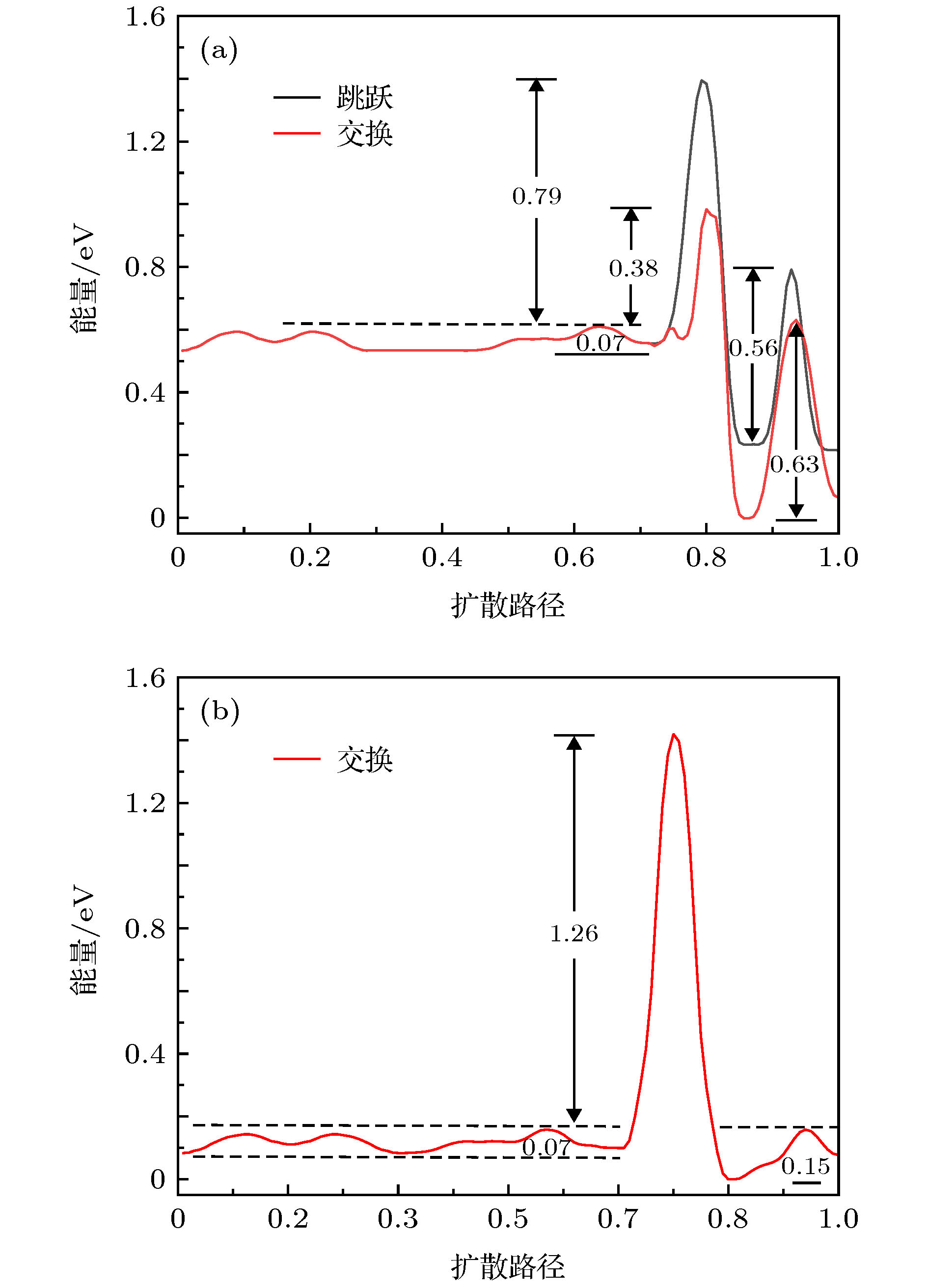 图 4 Al吸附原子在Ni CUB923基底表面扩散对应的能量势垒曲线 (a) {111} → {100}; (b) {111} → {111}. 扩散路径如图3(a)—图3(c)所示
图 4 Al吸附原子在Ni CUB923基底表面扩散对应的能量势垒曲线 (a) {111} → {100}; (b) {111} → {111}. 扩散路径如图3(a)—图3(c)所示Figure4. For the Al adatom on the surface of the Ni CUB923, the system energies as a function of reaction coordinate corresponding to the diffusion path shown in Fig.3(a)-Fig.3(c): (a) {111} → {100}; (b) {111} → {111}.
3
3.2.2.Al原子在Ni DEC923表面的扩散行为
如图3(d)—图3(g)所示, Al吸附原子在Ni DEC923基底两个相邻表面之间有四种可能的扩散路径, 其对应系统的能量作为扩散路径的函数已经在图5中给出. 如图5所示, 吸附原子在台阶边缘从{111}面扩散到{100}面, 对于交换和跳跃机制, 其ES势垒分别为0.52 和0.78 eV, 结果与在CUB923表面扩散行为类似, 即跳跃的能量势垒高于交换势垒, 应当优先发生交换. 然而, 对于吸附原子在{111}面与相邻{111}面之间扩散, 交换的势垒(0.87 eV)高于跳跃的势垒(0.54 eV). 这主要与不同面之间的几何结构有关, 在扩散过程中台阶越平滑则跳跃势垒越小[4]. 对于DEC结构, 两个相邻{111}面之间的二面角大于{111}与{100}面之间的二面角, 通过几何结构之间的关系能够解释上述所观察到的现象. 图 5 Al吸附原子在Ni DEC923基底表面扩散对应的能量势垒曲线 (a) {111} → {100}; (b) {111} → {111}. 扩散路径如图3(d)—图3(g)所示
图 5 Al吸附原子在Ni DEC923基底表面扩散对应的能量势垒曲线 (a) {111} → {100}; (b) {111} → {111}. 扩散路径如图3(d)—图3(g)所示Figure5. For the Al adatom on the surface of the NiDEC923, the system energies as a function of reaction coordinate corresponding to the diffusion path shown in Fig.3(d)-Fig.3(g): (a) {111} → {100}; (b) {111} → {111}.
3
3.2.3.Al原子在Ni ICO923表面的扩散行为
由于ICO结构仅由二十个{111}面组成, 因此吸附原子只会在相邻的两个{111}面之间扩散, 如图3(h)和图3(i)所示. 对应系统的能量作为扩散路径的函数, 如图6所示. 对于跳跃机制, 通过计算得到的ES势垒为0.52 eV, 然而对于交换机制, 其ES势垒为0.73 eV. 如上所述, 对于ICO结构, 由于相邻的{111}面之间的台阶相对比较平滑, 因此吸附原子在基底表面扩散通过跳跃机制更加有利. 图 6 Al吸附原子在Ni ICO923基底表面扩散对应的能量势垒曲线, 扩散路径如图3(h)和图3(i)所示
图 6 Al吸附原子在Ni ICO923基底表面扩散对应的能量势垒曲线, 扩散路径如图3(h)和图3(i)所示Figure6. For the Al adatom on the surface of the Ni ICO923, the system energies as a function of reaction coordinate corresponding to the diffusion path shown in Fig. 3(h) and Fig.3(i).
3
3.2.4.扩散行为差异的原因
如上所述, Al吸附原子通过交换或跳跃两种机制分别在三种不同Ni基底上表面扩散, 最低的ES势垒分别为0.38 eV (Exchange CUB{111}→{100}), 0.52 eV (Exchange DEC{111}→{100})和0.52 eV (Hoping ICO{111}→{111}). 由此可知, 对于CUB和DEC两种基底, Al吸附原子的最低ES势垒所对应的机制为交换机制, 这将不利于Ni核Al壳的形成. 通过对比三种结构的面间扩散势垒可知, 当Al吸附原子从{111}面扩散到{100}面时, 对应的交换势垒小于跳跃势垒, 当在两个相邻的{111}面间扩散时, 其对应的交换势垒大于跳跃势垒. 这种行为的差异除了上述提到的几何结构原因, 还有另外一个重要的原因, 即在台阶边缘处的原子配位数大小. 其中对于{111}面和{100}面之间, 台阶边缘上的原子配位数为7, 而对于两个相邻的{111}面之间, 台阶边缘上的原子配位数为8. 扩散势垒的大小通常由打破吸附原子与基底原子间的成键所需要耗费的能量决定[39]. 当吸附原子在两个相邻的{111}面之间进行交换扩散, 则需要打破更多的键, 因此需要更多的能量.吸附原子在{100}面上的扩散势垒远大于在{111}面上的扩散势垒. 最主要的原因是, 对于FCC金属, {111}面相比于{100}面排布得更紧凑, 因此{111}面更加平滑, 对应的跳跃势垒更低. 通过计算吸附原子在不同的基底表面扩散的能量势垒的差异, 可以预测相比于CUB和DEC基底, 在ICO结构基底的表面沉积Al原子将获得最好的核壳结构.
2
3.3.Al吸附原子在Ni基衬底的聚集生长
33.3.1.基底结构对生长的影响
系统地研究了金属Al原子在不同Ni基底表面的扩散机制, 进一步分析在不同Ni基底的团簇生长情况是非常必要的. 由于在台阶边缘处有一个较大的ES势垒[38], 为进一步研究不同Ni基底Al原子团簇的生长情况, 我们选取的生长的模拟温度为室温, 即300 K. 根据Baletto等[8]的描述, 利用纳米颗粒表面原子数(



如图7所示, 对于三种不同的基底结构生长,







 图 7 T = 300 K, 在不同的Ni基底表面生长,
图 7 T = 300 K, 在不同的Ni基底表面生长, 

Figure7. On the different substrates of Ni core, the


为了更加清楚地看到这一生长过程, Al原子在ICO结构的Ni基底表面生长序列如图8所示. 从图8(a)—图8(e), 对应沉积Al原子的数量分别为100, 200, 300, 400和500. 从图可知, 沉积的Al原子倾向于占据台阶边缘和顶角位置, 随着沉积的Al原子数(Ndep)增加, 然后开始聚集成岛. 这一现象的主要原因是Al原子在扩散过程中相遇, 随后相互成键, 以此降低系统的能量. 随着注入的Al原子数超过400, 最终得到一个ICO结构的Ni-核/Al-壳纳米团簇, 如图8(d)所示.
 图 8 T = 300 k, Al原子在Ni ICO923基底上的生长序列 (a) Ndep = 100; (b) Ndep = 200; (c) Ndep = 300; (d) Ndep = 400; (e) Ndep = 500. 橙色和灰色的球分别表示Al原子和Ni原子
图 8 T = 300 k, Al原子在Ni ICO923基底上的生长序列 (a) Ndep = 100; (b) Ndep = 200; (c) Ndep = 300; (d) Ndep = 400; (e) Ndep = 500. 橙色和灰色的球分别表示Al原子和Ni原子Figure8. Growth sequence of Al atoms growth on the ICO923 of Fe at T = 300 k: (a) Ndep = 100; (b) Ndep = 200; (c) Ndep = 300; (d) Ndep = 400; (e) Ndep = 500. The orange and gray balls show the Al atoms and the Ni atoms, respectively.
3
3.3.2.温度对生长的影响
为进一步探究温度对生长的影响, 选取NiICO923作为生长的基底, 在300, 500, 700和900 K下, 通过沉积Al原子在Ni基底的表面, 如图9所示.





 图 9 在T = 300, 500, 700和900 K, Al原子在Ni ICO923基底表面生长,
图 9 在T = 300, 500, 700和900 K, Al原子在Ni ICO923基底表面生长, 

Figure9. At T = 300, 500, 700 and 900 K, for the growth of Al atoms on the ICO923 of Ni, the


如图9所示, 生长温度为300, 500, 700和900 K时, 对应的缺陷数Ndef分别是30, 60, 110和158. 说明当生长的温度逐渐升高, Al吸附原子拥有更高的能量, 更加容易进入Ni核里面与金属Ni原子成键, 而不是与自身成键. 与之前计算NiAl的形成热为负值一致.
通过计算NiAl原子的异质配位数NAl-Ni的大小来表征NiAl合金化的程度[40], 如图10所示. 随着温度的升高, NAl-Ni逐渐增加, 表明合金化程度越强, NiAl键的数量越来越多, 插图对应于不同生长温度下NiAl纳米粒子的最终构型. 随着温度的升高, NiAl纳米粒子的表面逐渐开始合金化. 然而, 值得注意的是, NiAl纳米粒子的核内部仍然是纯Ni原子, 如图10中对应的截面图.
 图 10 异质配位数(NAl-Ni)随温度的变化, 插图对应于每个生长温度下最终构型. 橙色的球表示Al原子, 灰色的球表示Ni原子
图 10 异质配位数(NAl-Ni)随温度的变化, 插图对应于每个生长温度下最终构型. 橙色的球表示Al原子, 灰色的球表示Ni原子Figure10. The variation of hetero-coordination number (NAl-Ni) with temperature, and the inset correspond to the final configuration at each growth temperature. The orange and gray balls show the Al atoms and the Ni atoms, respectively.
随着Al原子数量的增加, 沉积的Al原子开始聚集. 对于Ni团簇上的Al原子, 在较低温度下在Ni基底表面沉积Al原子, 可以获得良好的Ni核/Al壳结构. 相对于CUB和DEC基底, ICO基底对应的核壳团簇缺陷数较小. 随着生长温度的升高, NiAl双金属表面逐渐合金化.
