摘要: 采用平衡态分子模拟的方法, 从微观角度对温度
$T^* = 0.85$ —5、密度
$ \rho^* $ = 0.85—1、势参数
ε = 0.97—1和
σ = 0.8—1.3范围内22组液固共存态及液态单原子Lennard-Jones (L-J)体的黏弹性弛豫时间进行了研究, 计算了单原子L-J体的静态黏弹性(黏度
η * 、无限大频率的剪切模量
$ G_∞^* $ )及动态黏弹性(储能模量
$ G^{\prime *} $ 、损耗模量
$ G^{\prime \prime *} $ )等特性参量, 并在此基础上分析了黏弹性特征弛豫时间、Maxwell弛豫时间及原子连通弛豫时间. 此外, 本文根据系统内原子的排布情况, 应用Kramers逃逸速率理论描述原子的扩散、汇聚过程, 提出并建立了一种单原子L-J体黏弹性弛豫时间的预测方法. 结果表明: 在单原子L-J体系统中, 低温情况下, Maxwell弛豫时间与黏弹性特征弛豫时间差异明显; 原子连通弛豫时间与黏弹性特征弛豫时间结果接近, 但原子连通弛豫时间的计算过程需耗费大量时间和计算资源; 预测方法得到的弛豫时间与黏弹性特征弛豫时间的结果更为接近. 本文提出的单原子L-J体黏弹性弛豫时间的预测方法具有一定的准确性和可靠性, 可为材料黏弹性弛豫时间的研究提供一种新的思路.
关键词: Lennard-Jones体 /
分子模拟 /
黏弹性 /
弛豫时间 /
预测方法 English Abstract Viscoelastic relaxation time of the monoatomic Lennard-Jones system Wang Yang Zhao Ling-Ling Key Laboratory of Energy Thermal Conversion and Control of Ministry of Education, School of Energy and Environment, Southeast University, Nanjing 210096, China Received Date: 19 January 2020Accepted Date: 03 April 2020Published Online: 20 June 2020Abstract: Viscoelastic relaxation time is an important concept to characterize the viscoelastic response of materials, which is directly related to the interactions among the microscopic atoms of materials. Few studies have focused on the methods of characterizing viscoelastic relaxation time. To investigate how to represent viscoelastic relaxation time effectively, the viscoelastic relaxation times of the monoatomic Lennard-Jones system on 22 conditions in a range of $ T^{ *} $ = 0.85–5, ρ * = 0.85–1, ε = 0.97–1, and σ = 0.8–1.3 are discussed from a microscopic perspective by the equilibrium molecular dynamics methods. Static viscoelasticity (viscosity η * , high-frequency shear modulus $ G_{\infty}^* $ ) is calculated by the Green-Kubo formula, and the Fourier transform is applied to the calculation of dynamic viscoelasticity (storage modulus $ G'^* $ and loss modulus $ G''^* $ ). On this basis, the viscoelastic characteristic relaxation time ($ \tau _{{\rm{MD}}}^*$ ), Maxwell relaxation time ($ \tau _{{\rm{Maxwell}}}^*$ ) and the lifetime of the state of local atomic connectivity ($ \tau _{{\rm{LC}}}^*$ ) are calculated. The viscoelastic characteristic relaxation time $ \tau _{{\rm{MD}}}^*$ , defined when the two responses crossover, is the key measure of the period of such a stimulus when the storage modulus (elasticity) equals the loss modulus (viscosity). Maxwell relaxation time $ \tau _{{\rm{Maxwell}}}^* = {\eta ^*}/G_\infty ^*$ , where η * is the static viscosity under infinitely low stimulus frequency (i.e., zero shear rate), $ G_{\infty}^* $ is the instantaneous shear modulus under infinitely high stimulus frequency, and $ \tau _{{\rm{LC}}}^*$ is the time it takes for an atom to lose or gain one nearest neighbor. The result is observed that $ \tau _{{\rm{LC}}}^*$ is closer to $ \tau _{{\rm{MD}}}^*$ than $ \tau _{{\rm{Maxwell}}}^*$ . But the calculation of $ \tau _{{\rm{LC}}}^*$ needs to take into count the trajectories of all atoms in a certain time range, which takes a lot of time and computing resources. Finally, in order to characterize viscoelastic relaxation time more easily, Kramers’ rate theory is used to describe the dissociation and association of atoms, according to the radial distribution functions. And a method of predicting the viscoelasticity of the monoatomic Lennard-Jones system is proposed and established. The comparison of all the viscoelastic relaxation times obtained above shows that $ \tau _{{\rm{Maxwell}}}^*$ is quite different from $ \tau _{{\rm{MD}}}^*$ at low temperature in the monoatomic Lennard-Jones system. Compared with $ \tau _{{\rm{Maxwell}}}^*$ , $ \tau _{{\rm{LC}}}^*$ is close to $ \tau _{{\rm{MD}}}^*$ . But the calculation of $ \tau _{{\rm{LC}}}^*$ requires a lot of time and computing resources. Most importantly, the relaxation time calculated by our proposed method is closer to $ \tau _{{\rm{MD}}}^*$ . The method of predicting the viscoelastic relaxation time of the monoatomic Lennard-Jones system is accurate and reliable, which provides a new idea for studying the viscoelastic relaxation time of materials.Keywords: Lennard-Jones system /molecular simulation /viscoelasticity /relaxation time /prediction method 全文HTML --> --> --> 1.引 言 黏弹性是物质的重要性质. 一般而言, 任何材料都具有弹性和黏性, 但会因温度和作用力速率不同, 或主要表现为弹性(低温或作用时间快), 或主要表现为黏性(高温或作用时间慢)[1 ] . 黏弹性的本质是由于材料内分子运动具有弛豫特性, 当材料受外力作用时, 其分子响应与外力达不到平衡, 从而产生了黏弹性[2 ] . 因此, 黏弹性弛豫时间是表征材料黏弹性响应的重要概念, 且与物质微观原子间相互作用直接相关. 如何更好地对黏弹性弛豫时间进行表征和获取, 对物质黏弹特性的研究具有重要的意义.$ {\tau }_{\rm{MD}}^{*} $ 、Maxwell弛豫时间$ {\tau }_{\rm{Maxwell}}^{*} $ 、原子连通弛豫时间$ {\tau }_{\rm{LC}}^{*} $ . 黏弹性特征弛豫时间$ {\tau }_{\rm{MD}}^{*} $ 是材料储能模量与损耗模量的交点频率对应的时间, 其表征的是材料表现出显著黏弹性的时间尺度[3 ] . Sunthar[4 ] 指出在振荡实验中, 低频状态下(或长时间响应)汇聚物的储能模量(弹性)总大于损耗模量(黏性), 而在高频状态下(或短时间响应), 汇聚物主要展现出黏性特征, 储能模量与损耗模量的交点频率对应的时间即为黏弹性特征弛豫时间$ {\tau }_{\rm{MD}}^{*} $ . Agrawal等[5 ] 采用分子模拟的方法, 在类似的黏弹动态模量分析的基础上, 运用傅里叶变换得到了高分子物质聚脲的储能及损耗模量的数据, 其模拟结果与加载应力实验结果一致, 证明了应用分子模拟的方法可以开展黏弹性弛豫时间的研究. Maxwell弛豫时间$ {\tau }_{\rm{Maxwell}}^{*} $ 是目前最常见的用来计算物质黏弹性弛豫时间的方法, 已有研究****应用分子模拟的方法开展了Maxwell弛豫时间的相关研究. Hartkamp等[6 ] 运用Green-Kubo公式[7 ,8 ] 计算了简单原子流体的黏度、无穷大频率的剪切模量、Maxwell弛豫时间等黏弹性物理量, 并得到了与动力学理论预测类似的结果. Guillaud等[9 ] 采用TIP4P/2005f力场模拟了大范围温度下超冷水的弛豫行为, 结果显示当温度低于285 K时, Maxwell弛豫时间无法表征超冷水的弛豫行为. 近年来, 为分析黏弹性的产生机理, 有些研究****基于对分子扩散、汇聚过程的研究来描述黏弹性弛豫时间. Iwashita和Egami[10 ] 首次提出了原子连通弛豫时间$ {\tau }_{\rm{LC}}^{*} $ 的概念去描述粒子得到或者失去自身周围最近的粒子过程, 并将各类液态金属向玻璃态转化过程中的Maxwell弛豫时间和原子连通弛豫时间进行比较[11 ] , 结果显示二者在高温下比较接近, 但在低温下原子连通弛豫时间更能反映物质黏弹性的响应. Ashwin和Sen[12 ] 在分析Yukawa液体内Maxwell弛豫时间和原子连通弛豫时间的过程中指出原子连通弛豫时间是系统内剪切应力松弛的微观起源.[13 ] 的限制而无法应用于更广泛的物质[14 ] . 原子连通弛豫时间能够从微观角度解释剪切应力松弛产生的原因, 但其计算过程需要耗费大量计算资源统计原子的运动轨迹. 因此, 进一步开展计算物质黏弹性弛豫时间方法的研究具有重要的意义.${T}^{*}\!=\!0.85—5$ 、密度$ {\rho }^{*}=0.85—1 $ 、势参数$\varepsilon \!=\! 0.97—1$ 和$ \sigma =0.8—1.3 $ 范围内液固共存态及液态的单原子Lennard-Jones (L-J) 体的黏弹性进行了研究, 计算了黏弹性特性参量, 分析了黏弹性特征弛豫时间、Maxwell弛豫时间和原子连通弛豫时间. 此外, 在分析系统原子排布的基础上, 应用Kramers逃逸速率理论描述原子扩散、汇聚的过程, 提出并建立了一种单原子L-J体黏弹性预测方法, 可为材料黏弹性弛豫时间的研究提供新的思路.2.研究对象及方法 22.1.对象及方法 2.1.对象及方法 为方便对计算结果进行更好的分析, 本文中的物理量均采用“约化单位”[15 ] , 用上标“*”表示, 如${T}^{*}=Tk/\varepsilon ,\; {\rho }^{*}=\rho {\sigma }^{3}$ , $ {t}^{*}=t\sqrt{\varepsilon /m}/\sigma $ , $ {r}^{*}=r/\sigma $ , $ {p}^{*}=p{\sigma }^{3}/\varepsilon $ , $ {\eta }^{*}=\eta {\sigma }^{3}/\sqrt{m\varepsilon } $ . 其中ε 和σ 为L-J势参数, 分别表示势能为0时的原子距离、势能阱深度; m 为原子质量; T 为真实单位下的温度, 单位为K; ρ 表示密度, 单位为mol/L; p 为压强, 单位为MPa; $ \eta $ 为黏度, 单位为μPa·s.[16 ] , 计算域尺寸(模拟盒子)为16 × 16 × 16单位体积, 并按照相应的密度设置盒子内的粒子数量, 如图1 所示.图 1 L-J体计算系统Figure1. L-J fluid simulation system.[17 ] 为rij 为原子i 与j 之间的距离, εij 为势能阱的深度, σij 为两体互相作用的势能为零时的距离. 其中[18 ] .[19 ] 条件, 时间步长设为0.003. 模拟总时间步数为108 步, 系统经过5 × 107 个时间步长后达到平衡, 本文选取平衡后系统的数据进行分析, 并统计了系统内所有粒子在最后500个时间步长内的运动轨迹. 本文应用LAMMPS[20 ] 软件进行建模和具体计算.2.2.数据处理方法 -->2.2.数据处理方法 本文采用Green-Kubo公式[7 ,8 ,21 ] 计算黏度:V 表示体积; k B 为玻尔兹曼常数; T 为热力学温度; t 表示时间; ταβ 表示剪切应力张量的非对角元素, 其具体表达式为$ {r_{ij, \alpha }}$ 表示i 和j 粒子在α 方向的位移分量, $ {F_{ij, \beta }}$ 表示i 粒子作用在j 粒子上在β 方向作用力的分量, N 为粒子数, m 是粒子质量.G (t )可通过应力自相关函数公式进行计算[22 ] :6 )式转换为复数型频域剪切模量函数G (ω ):[6 ] 定义为7 )式中复数模量的实部为储能模量((9 )式)[23 ] , 其反映材料形变时的回弹能力[24 ] , 即弹性:7 )式中的虚部实质为损耗模量((10 )式)[25 ] , 反映材料形变时内耗程度, 即黏性:[26 ] 计算得到系统的扩散率D 为t 表示时间; r 表示原子的位移; 〈·〉表示组内所有原子的平均.2.3.模型验证 -->2.3.模型验证 为保证模拟的准确性, 本文主要从物质的黏度、系统原子的排布规律两方面对所建立的模型和计算方法进行验证.$ {T}^{*}= 0.6—1.5$ , $ {\rho }^{*}= 0.9$ , $ \varepsilon = 1$ , $ \sigma = 1$ 条件下的单原子L-J体系统. 不同温度时单原子L-J体的黏度模拟值与Gallie?o等[27 ] 的文献值对比示于图2 . 由图2 可知, 随着温度的升高, 单原子L-J体的黏度不断下降; 本文所计算的不同温度下单原子L-J体的黏度与文献值一致, 相对误差在5%之内, 本文的模拟方法具有一定的可靠性和准确性.图 2 不同温度下L-J体的黏度验证Figure2. Verification of viscosity of L-J fluid at different temperatures.[28 ] 经验公式的对比结果示于图3 . 由图3 可以看出, 本文中的模拟结果与相同温度密度条件下的文献数据[28 ] 十分符合, 且反映出径向分布函数曲线随温度变化的共同趋势: 在密度为1的情况下, 温度为0.9时(图3(a) ), 径向分布函数曲线拥有多个波峰波谷, 数值波动明显, 在$ r^* = 1.0 $ , 2.0, 2.8, 3.7, 4.8依次出现峰值逐渐降低的波峰, 原子排布不规则, 属于黏弹性显著的固液共存态; 当温度升高至4时(图3(b) ), 径向分布函数曲线发生明显变化, 曲线平滑, 数值波动减小, 波峰波谷数量显著减少, L-J体处于液态.图 3 L-J体的径向分布函数验证 (a) ${\rho }^{*}=1,\; {T}^{*}=0.9$ ; (b) ${\rho }^{*}=1, \;{T}^{*}=4.0$ Figure3. Verification of the radial distribution function of L-J fluid: (a) ${\rho }^{*}=1, \;{T}^{*}=0.9$ ; (b) ${\rho }^{*}=1, \;{T}^{*}=4.0$ .$ {\rho }^{*}= 1$ , $ \varepsilon = 1$ , $ \sigma = 1$ , $ {T}^{*}= 0.85—5$ 条件下单原子L-J体系统, 其径向分布函数曲线计算结果示于图4 . 由图4 可以明显看出, 随着温度的升高, 单原子L-J体的径向分布函数曲线逐渐平滑, 数值波动明显减小, 波峰波谷数量显著减少, 单原子L-J体逐渐由液固共存态转变为液态. 上述观察到的现象也与Heyes等[24 ] 对L-J体相态变化的温度密度范围划分结果一致, 故进一步验证了本文所选方法能较为精确地反映单原子L-J体内原子分布及排布情况, 能有效计算本文所需的相关参数内容.图 4 ${\rho }^{*}=1, \;\varepsilon =1,\; \sigma =1$ 时不同温度下L-J体的径向分布函数Figure4. Radial distribution function of L-J fluid at ${\rho }^{*}=1, \varepsilon =1,\; \sigma =1$ with different temperatures.3.模拟结果与讨论 23.1.各类弛豫时间讨论 3.1.各类弛豫时间讨论 为开展单原子L-J体黏弹性时间的研究, 计算了黏弹性特征弛豫时间、Maxwell弛豫时间以及原子连通弛豫时间, 并对各类黏弹性弛豫时间进行了分析.3.1.1.黏弹性特征弛豫时间计算 -->3.1.1.黏弹性特征弛豫时间计算 通过应用(9 )和(10 )式计算了$ {T}^{*}= 0.85—5$ , ${\rho }^{*}\!=\! 0.85—1$ , $ \varepsilon\! =\! 0.97—1$ , $ \sigma\! =\! 0.8—1.3$ 条件下共22组单原子L-J体的储能模量和损耗模量随频率的变化. 图5 为${T}^{*}\!=\! 0.9—1.5$ , ${\rho }^{*} \!=\! 1$ , $ \varepsilon = 1$ , $\sigma = 1$ 条件下储能模量$ G^{\prime *} $ 和损耗模量$ G^{\prime \prime *} $ 的计算结果. 由图5(a) 可以看出, 当频率较小(0 < ω * < 10)时, 储能模量小于损耗模量, 单原子L-J体主要表现为黏性特征, 储能模量和损耗模量均随频率的上升而增大, 单原子L-J体的弹性和黏性均增强; 当频率处于中间频率(10 < ω * < 20)时, 储能模量和损耗模量接近, 单原子L-J体主要表现出黏弹性特征, 储能模量随着频率的上升逐渐接近并超过损耗模量; 当频率较大时(ω * > 20), 储能模量大于损耗模量, 单原子L-J体主要表现为弹性特征, 储能模量随着频率的上升而不断增大并趋于稳定, 单原子L-J体的弹性增强, 损耗模量随着频率的上升而不断减小并趋于稳定, 单原子L-J体的黏性减弱. 图5(b) 所示为$ {\tau }_{\rm{MD}}^{*} $ 的计算方法[4 ] , 物质的黏弹性特征弛豫时间对应于储能及损耗模量相等时的频率$ {\omega }_{{\rm{c}}{\rm{r}}{\rm{o}}{\rm{s}}{\rm{s}}}^{*} $ , 即:图 5 储能模量和损耗模量曲线 (a)不同温度条件写的储能模量和损耗模量曲线; (b)粘弹性特征弛豫时间$ {\tau }_{\rm{MD}}^{*} $ Figure5. Storage modulus and loss modulus: (a) Storage modulus and loss modulus at different temperatures; (b) viscoelastic characteristic relaxation time $ {\tau }_{\rm{MD}}^{*} $ .12 )式, 在储能模量和损耗模量计算结果的基础上得到单原子L-J体的黏弹性特征弛豫时间$ {\tau }_{\rm{MD}}^{*} $ , 结果示于图6 . 由图6 可以看出: 当$ {\rho }^{*}, \varepsilon ,\;\sigma $ 保持不变时(图6(a) ), 随着$ {T}^{*} $ 的上升, $ {\tau }_{\rm{MD}}^{*} $ 不断减小, 但减小的速度不断下降; 当$ {T}^{*} $ , $ \varepsilon ,\; \sigma $ 保持不变时(图6(b) ), 随着$ {\rho }^{*} $ 的增大, $ {\tau }_{\rm{MD}}^{*} $ 不断增大; 当$ {T}^{*} $ , $ {\rho }^{*} $ 保持不变时(图6(c) 和图6(d) ), 势参数$ \sigma $ 或$ \varepsilon $ 的增大, 均会导致$ {\tau }_{\rm{MD}}^{*} $ 的增大.图 6 不同温度、密度以及L-J势参数下$ {\tau }_{\rm{Maxwell}}^{*} $ , $ {\tau }_{\rm{LC}}^{*} $ , $ {\tau }_{\rm{MD}}^{*} $ , $ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^{*} $ (a)不同温度条件; (b)不同密度条件; (c)不同势能为0时的原子距离条件; (d)不同势能阱深度条件Figure6. $ {\tau }_{\rm{Maxwell}}^{*} $ , $ {\tau }_{\rm{LC}}^{*} $ , $ {\tau }_{\rm{MD}}^{*} $ , $ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^{*} $ under different T , $ \rho $ , ε and σ : (a) Different temperatures; (b) different densities; (c) different distances between atoms when potential is 0; (d) different potential well depths.3.1.2.Maxwell弛豫时间计算 -->3.1.2.Maxwell弛豫时间计算 通过应用(4 )式和(6 )式计算了$ {T}^{*}= 0.85—5$ , $ {\rho }^{*}= 0.85—1$ , $ \varepsilon = 0.97—1$ , $ \sigma = 0.8—1.3$ 条件下单原子L-J体的黏性$ {\eta }^{*} $ 和无穷大频率剪切模量$ {G}_{\infty }^{*} $ , 并计算各条件下的Maxwell弛豫时间. Maxwell弛豫时间的计算公式[6 ] 为$ {\tau }_{\rm{Maxwell}}^{*} $ 与黏弹性特征弛豫时间$ {\tau }_{\rm{MD}}^{*} $ 进行比较, 结果示于图6 . 由图6 可以看出, 在不同的温度、密度和势参数条件下, $ {\tau }_{\rm{Maxwell}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 的变化趋势一致; 当${\rho }^{*},\; \varepsilon ,\; \sigma$ 保持不变时(图6(a) ), 随着$ {T}^{*} $ 的下降, $ {\tau }_{\rm{Maxwell}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 的差异逐渐增大, 这说明应用Maxwell弛豫时间表征黏弹性特征弛豫时间存在一定的局限性; 当$ {T}^{*} $ , $ \varepsilon , \;\sigma $ 保持不变时(图6(b) ), 随着$ {\rho }^{*} $ 的增大, $ {\tau }_{\rm{Maxwell}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 均逐渐增大, 但$ {\tau }_{\rm{Maxwell}}^{*} $ 增大的速率更快; 当$ {T}^{*} $ , ${\rho }^{*},\; \varepsilon$ 保持不变时(图6(c) ), 随着$ \sigma $ 的增大, $ {\tau }_{\rm{Maxwell}}^{*} $ 不断增大, 但与$ {\tau }_{\rm{MD}}^{*} $ 的差异越来越明显; 当$ {T}^{*} $ , ${\rho }^{*},\; \sigma$ 保持不变时(图6(d) ), 随着$ \varepsilon $ 的增大, $ {\tau }_{\rm{Maxwell}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 的差异逐渐增大.$ {T}^{*} < 1.5 $ )时, $ {\tau }_{\rm{Maxwell}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 差异明显, 应用Maxwell弛豫时间表征单原子L-J体的黏弹性特征弛豫时间存在一定的局限性.3.1.3.原子连通弛豫时间计算 -->3.1.3.原子连通弛豫时间计算 原子连通数N c 是指在原子近邻(径向分布函数的第一处谷值位置)范围内的原子数, 原子连通弛豫时间$ {\tau }_{\rm{LC}} $ [10 ] 是指系统内所有原子中发生一次扩散或者汇聚的时间. 若$ {t}_{0} $ 时刻原子连通数为$ {N}_{{\rm{c}}}\left({t}_{0}\right) $ , $ {t}_{0}+{\tau }_{\rm{LC}} $ 时刻总原子连通数为$ {N}_{{\rm{c}}}({t}_{0}+{\tau }_{\rm{LC}}) $ , 则$ {N}_{{\rm{c}}}({t}_{0}+{\tau }_{\rm{LC}}) $ = $ {N}_{{\rm{c}}}\left({t}_{0}\right)-1 $ . 本文基于计算所得的L-J体原子运动轨迹, 通过MATLAB程序统计不同时刻原子间的距离, 从而分析得出原子的扩散或汇聚的时间.图7 所示为$\rho^* = 1,\; T^* = 1,\; \varepsilon = 1$ , σ = 1物性条件下L-J体原子扩散或汇聚时间分布图, 其他条件下的单原子L-J体原子扩散或汇聚时间分布与该条件下的情况类似. 通过观察图7 可以发现, L-J体的原子扩散或汇聚时间分布符合指数函数分布的密度函数. 因此, 本文应用指数函数分布的密度函数即(14 )式去拟合L-J体的原子扩散或汇聚时间分布. 其中, (14 )式中的特征时间$ {\tau }_{\rm{LC}}^{*} $ 即为原子连通弛豫时间:图 7 $\rho^* = 1,\; T^* = 1$ , ε = 1, σ = 1条件下L-J体原子扩散或汇聚时间分布Figure7. Distribution of the dissociation or association time of L-J fluid at $\rho^* = 1,\; T^* = 1$ , ε = 1, σ = 114 )式得到了${T}^{*}= 0.85—5$ , ${\rho }^{*}\!= 0.85—1$ , $ \varepsilon = 0.97—1$ , $ \sigma = 0.8—1.3$ 条件下单原子L-J体的原子连通弛豫时间$ {\tau }_{\rm{LC}}^{*} $ , 并与黏弹性特征弛豫时间$ {\tau }_{\rm{MD}}^{*} $ 和Maxwell弛豫时间$ {\tau }_{\rm{Maxwell}}^{*} $ 进行比较, 结果示于图6 . 由图6 可以看出, 在不同的物性参数和势参数条件下, $ {\tau }_{\rm{LC}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 具有相同的变化趋势, 与$ {\tau }_{\rm{Maxwell}}^{*} $ 相比, $ {\tau }_{\rm{LC}}^{*} $ 对$ {\tau }_{\rm{MD}}^{*} $ 的预测更加准确; 当${\rho }^{*},\; \varepsilon ,\; \sigma$ 保持不变时(图6(a) ), 随着$ {T}^{*} $ 的下降, ${\tau }_{\rm{LC}}^{*},\; {\tau }_{\rm{Maxwell}}^{*}$ 与$ {\tau }_{\rm{MD}}^{*} $ 的差异均逐渐增大, 但$ {\tau }_{\rm{LC}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 的结果更加接近; 当$ {T}^{*} $ , $\varepsilon ,\; \sigma$ 保持不变时(图6(b) ), 随着$ {\rho }^{*} $ 的增大, ${\tau }_{\rm{LC}}^{*},\; {\tau }_{\rm{Maxwell}}^{*}$ , $ {\tau }_{\rm{MD}}^{*} $ 均呈现上升趋势, 但$ {\tau }_{\rm{Maxwell}}^{*} $ 增大的速率最快, 而$ {\tau }_{\rm{LC}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 的差异在减小; 当$ {T}^{*} $ , ${\rho }^{*},\; \varepsilon$ 保持不变时(图6(c) ), 随着$ \sigma $ 的增大, $ {\tau }_{\rm{Maxwell}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 的差异不断增大, 而$ {\tau }_{\rm{LC}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 的差异存在减小的趋势; 当$ {T}^{*} $ , ${\rho }^{*},\; \sigma$ 保持不变时(图6(d) ), 随着$ \varepsilon $ 的增大, $ {\tau }_{\rm{LC}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 的结果逐渐接近.3.2.预测方法及弛豫时间计算 -->3.2.预测方法及弛豫时间计算 在上述计算过程中, 发现原子连通弛豫时间对黏弹性弛豫时间的表征更加准确, 但连通性弛豫时间的计算工作量大、耗时长. 本文在分析每个原子周围原子排布的基础上, 应用Kramers逃逸速率理论[29 ] 来描述原子的扩散和汇聚过程, 建立了一种新的表征单原子L-J体黏弹性弛豫时间的方法, 避免了统计所有原子的运动轨迹时需要消耗大量时间和计算资源.图8 所示为Kramers逃逸速率理论示意图, 其中g (r )表示的是原子的径向分布函数曲线, U (r )表示的是原子的能量变化曲线. 当粒子间距离由近处远离, 粒子间能量(U (r )曲线)由第一个谷值点a 处上升至能量壁垒b 点时, 发生扩散反应, 此时的能量壁垒为$ E_b^{\rm ass} $ ; 而当粒子间距离由远处靠近, 对应能量(U (r )曲线)由第二个谷值点c 处上升至能量壁垒b 点时, 发生汇聚反应, 其能量壁垒为$ E_b^{\rm ass} $ . 该过程中的能量U 可通过径向分布函数即$ g\left(r\right) $ 曲线获得:图 8 Kramers逃逸速率理论示意图Figure8. Schematic diagram of Kramers’ rate theory.$ E_b^{\rm diss} $ 和$ E_b^{\rm ass} $ 可分别表示为k diss 和k ass 则可通过能量壁垒E b 及能量曲线波峰(壁垒)、波谷曲率相关参数ωa , ωb , ωc 和迁移率γ 等描述:N c 为原子连通数, 可通过本文计算得到的径向分布函数得到; ω 表示为γ 表示为D 为系统扩散系数, m 为单个原子的质量.N 表示系统内原子的数量.k ass 为A原子靠近A—A分子的扩散、汇聚过程(图8 )的速率常数; k diss 为A原子远离A—A分子的扩散分散过程(图8 )的速率常数. 图9 所示为不同温度、密度、势参数条件下, A原子和A—A分子的数量随时间的变化.图 9 A原子和A—A分子的数量变化曲线 (a)不同温度条件; (b)不同密度条件; (c)不同势能为0时的原子距离条件; (d)不同势能阱深度条件Figure9. The quantity curves of A and A—A: (a) Different temperatures; (b) different densities; (c) different distances between atoms when potential is 0; (d) different potential well depths.图9 可知, 在不同的物性条件下, A原子和A—A分子的数量变化均存在相同的规律, 随着时间的增长, A原子的数量不断增加并趋于一个定值, A—A分子的数量不断降低并趋于一个定值, A原子和A—A分子的转换过程趋于平衡状态; 当${\rho }^{*},\; \varepsilon ,\; \sigma$ 保持不变时(图9(a) ), 随着$ {T}^{*} $ 的上升, A—A分子的耗散速度和A原子的生成速度均增大; 当$ {T}^{*} $ , $\varepsilon ,\; \sigma$ 保持不变时(图9(b) ), 随着$ {\rho }^{*} $ 的增大, A—A分子的耗散速度和A原子的生成速度均减小; 当$ {T}^{*} $ , $ {\rho }^{*} $ 保持不变时(图9(c) 和图9(d) ), 无论$ \sigma $ 的增大还是$ \varepsilon $ 的增大, 均会导致A—A分子的耗散速度和A原子的生成速度减小.26 )式对所有条件下单原子L-J体内A原子数量随时间的变化进行拟合得到扩散和汇聚过程的弛豫时间即为本文提出的预测方法弛豫时间$ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^* $ , 结果示于图6 .$ {A}_{{\rm{a}}} $ 为指前因子, $ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^* $ 为预测方法弛豫时间.图6 可以看出, 在不同的物性参数下, 预测方法得到的弛豫时间$ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^* $ 与黏弹性特征弛豫时间$ {\tau }_{\rm{MD}}^{*} $ 具有相同的变化趋势; 当${\rho }^{*},\; \varepsilon ,\; \sigma$ 保持不变时(图6(a) ), 随着$ {T}^{*} $ 的下降, ${\tau }_{\rm{LC}}^{*},\; {\tau }_{\rm{Maxwell}}^{*}$ 与$ {\tau }_{\rm{MD}}^{*} $ 的差异均逐渐增大, 而$ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^* $ 与$ {\tau }_{\rm{MD}}^{*} $ 的差异无明显变化; 当$ {T}^{*} $ , $\varepsilon ,\; \sigma$ 保持不变时(图6(b) ), 随着$ {\rho }^{*} $ 的增大, $ {\tau }_{\rm{MD}}^{*} $ , $ {\tau }_{\rm{Maxwell}}^{*} $ , $ {\tau }_{\rm{LC}}^{*} $ , $ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^* $ 均呈现上升趋势, 但$ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^* $ 与$ {\tau }_{\rm{MD}}^{*} $ 十分接近, 而$ {\tau }_{\rm{Maxwell}}^{*} $ , $ {\tau }_{\rm{LC}}^{*} $ 与$ {\tau }_{\rm{MD}}^{*} $ 存在明显差异; 当$ {T}^{*} $ , ${\rho }^{*},\; \varepsilon$ 保持不变时(图6(c) ), 随着$ \sigma $ 的增大, $ {\tau }_{\rm{MD}}^{*} $ , $ {\tau }_{\rm{Maxwell}}^{*} $ , $ {\tau }_{\rm{LC}}^{*} $ , $ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^* $ 均不断增大, $ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^* $ 与$ {\tau }_{\rm{MD}}^{*} $ 的差异最小; 当$ {T}^{*} $ , ${\rho }^{*},\; \sigma$ 保持不变时(图6(d) ), 随着$ \varepsilon $ 的增大, $ {\tau }_{{\rm{m}}{\rm{o}}{\rm{d}}{\rm{e}}{\rm{l}}}^* $ 与$ {\tau }_{\rm{MD}}^{*} $ 与的结果最接近.4.结 论 本文应用平衡态分子模拟方法计算了${T}^{*}= 0.85—5$ , $ {\rho }^{*}= 0.85—1$ , $ \varepsilon = 0.97—1$ , $ \sigma = 0.8—1.3$ 范围内共22组液固共存态及液态的单原子L-J体的黏度(η * )、无限大频率的剪切模量($ G_∞^* $ )、储能模量($ G^{\prime *} $ )、损耗模量($ G^{\prime \prime *} $ )等黏弹性特性参量, 获得了黏弹性特征弛豫时间、Maxwell弛豫时间、原子连通弛豫时间, 在此基础上本文通过分析系统内的原子排布情况即径向分布函数曲线, 应用Kramers逃逸速率理论来描述单原子L-J体内原子的扩散和汇聚过程, 提出了单原子L-J体黏弹性弛豫时间的预测方法. 本文得到以下结论: 





























 图 1 L-J体计算系统
图 1 L-J体计算系统





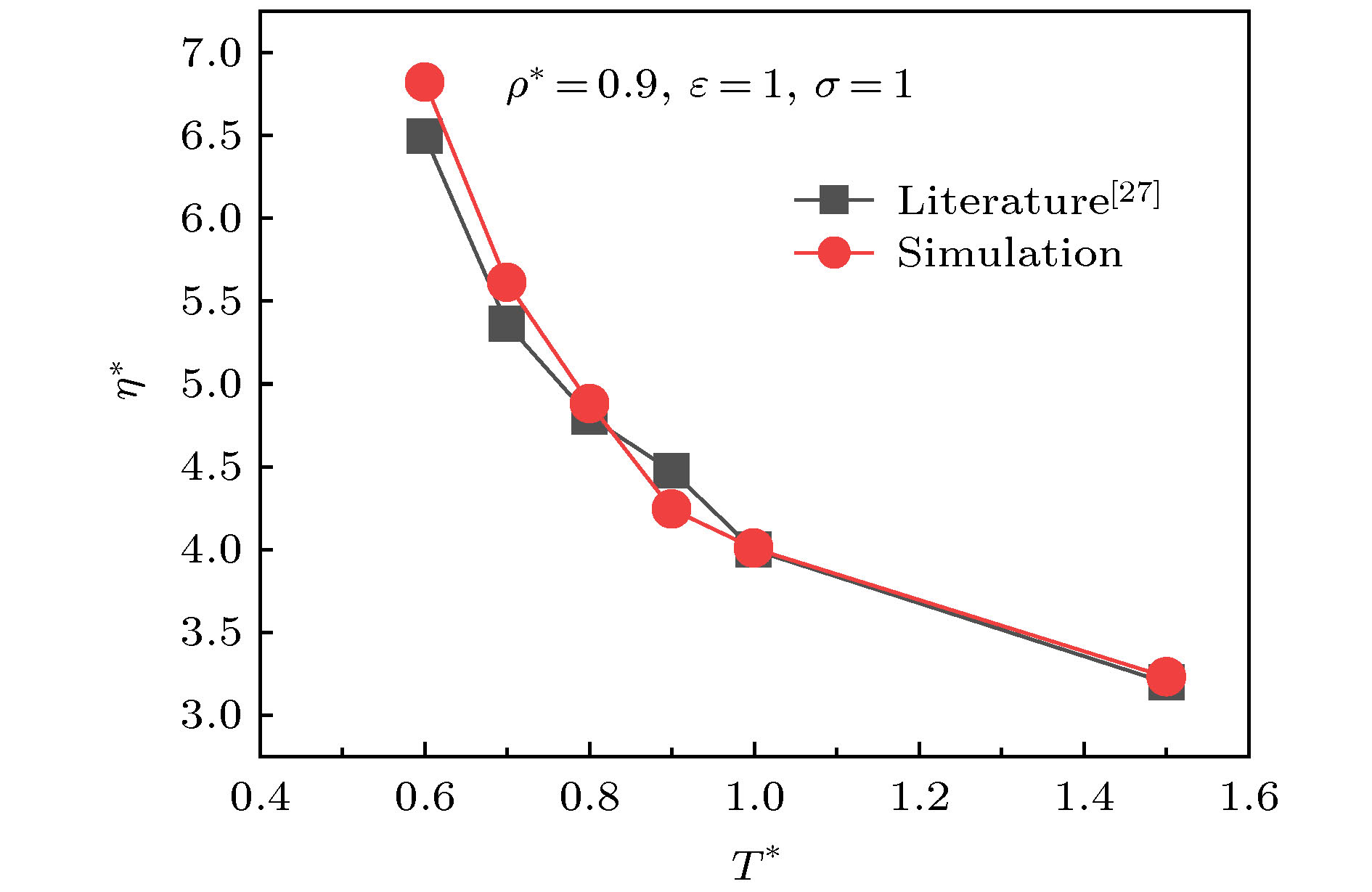 图 2 不同温度下L-J体的黏度验证
图 2 不同温度下L-J体的黏度验证
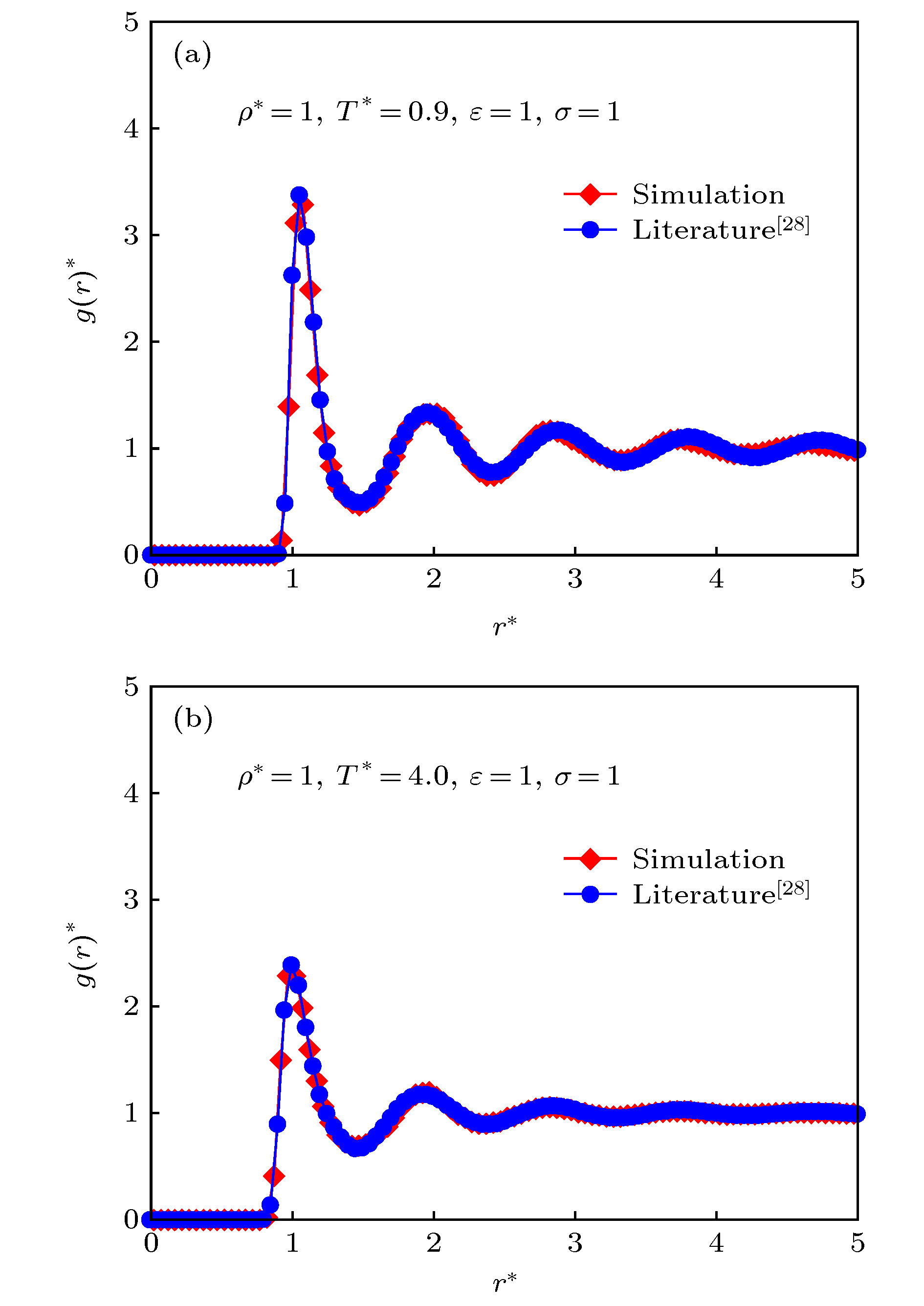 图 3 L-J体的径向分布函数验证 (a)
图 3 L-J体的径向分布函数验证 (a) 







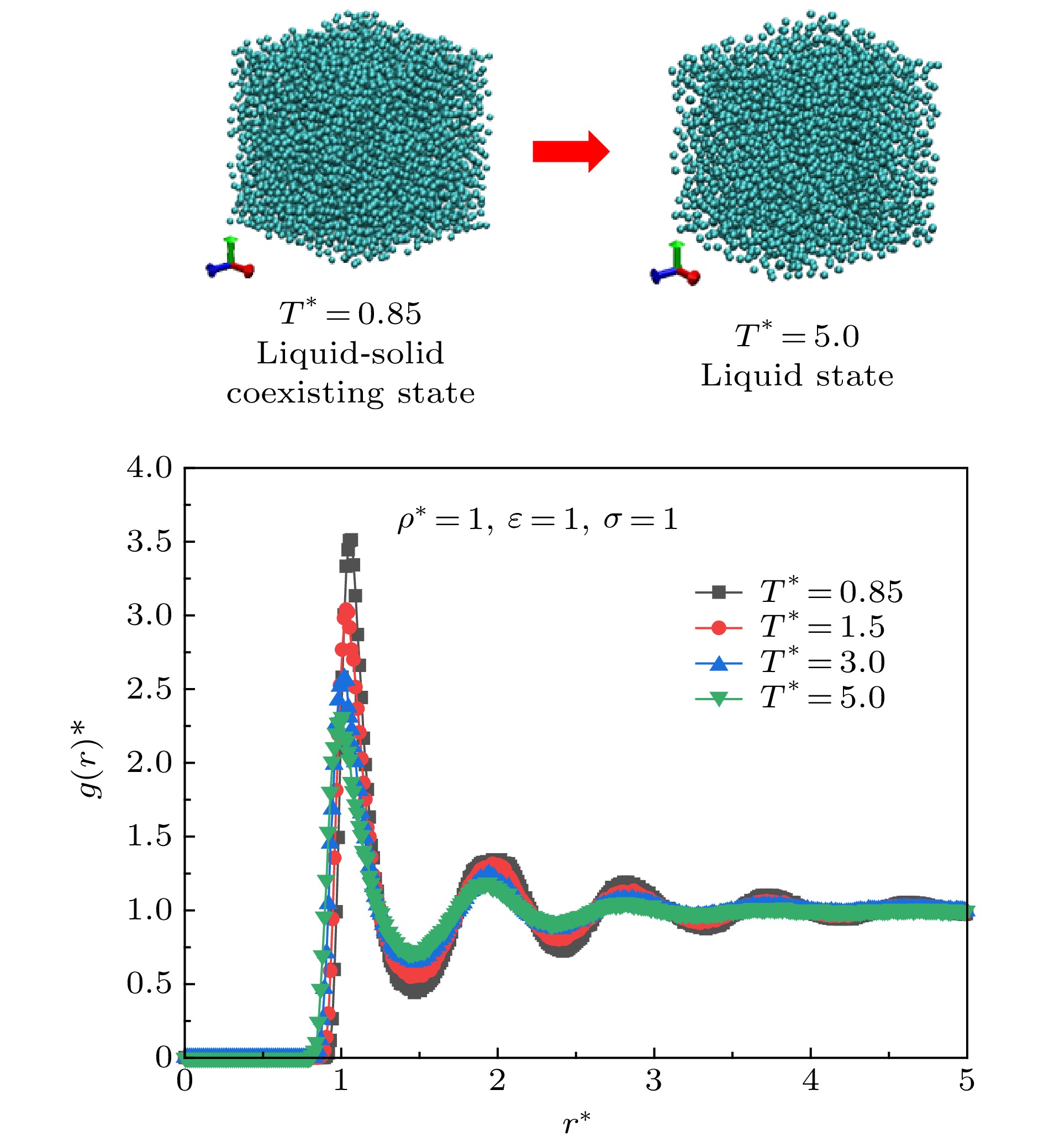 图 4
图 4 













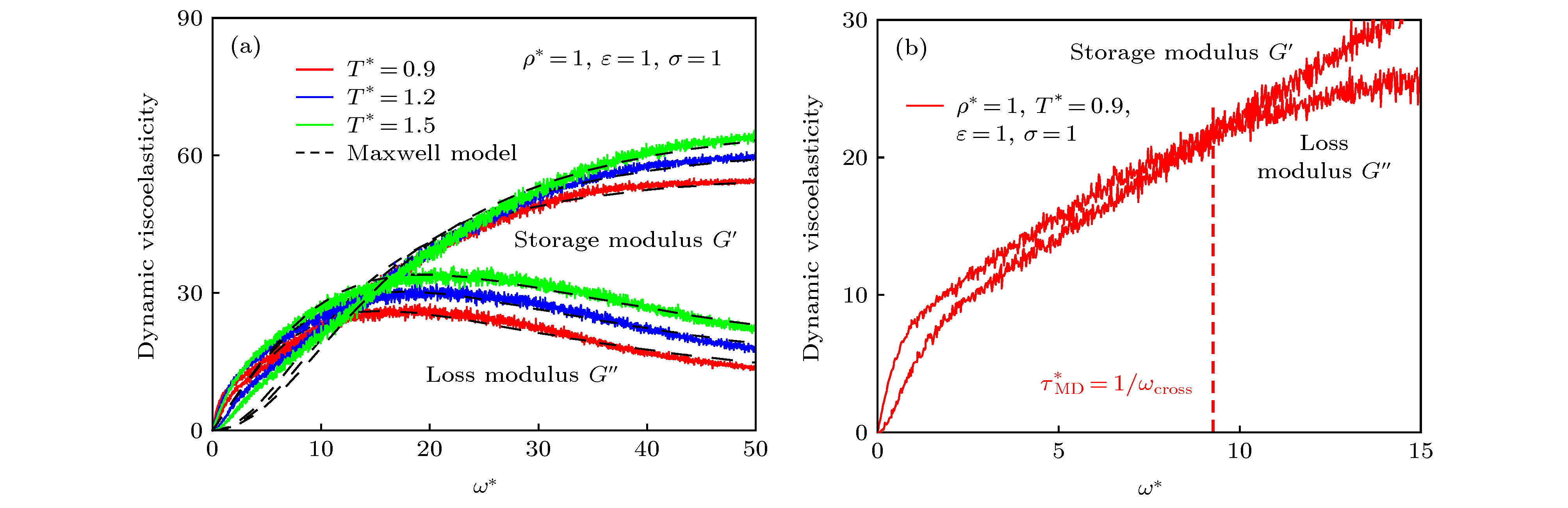 图 5 储能模量和损耗模量曲线 (a)不同温度条件写的储能模量和损耗模量曲线; (b)粘弹性特征弛豫时间
图 5 储能模量和损耗模量曲线 (a)不同温度条件写的储能模量和损耗模量曲线; (b)粘弹性特征弛豫时间














 图 6 不同温度、密度以及L-J势参数下
图 6 不同温度、密度以及L-J势参数下


















































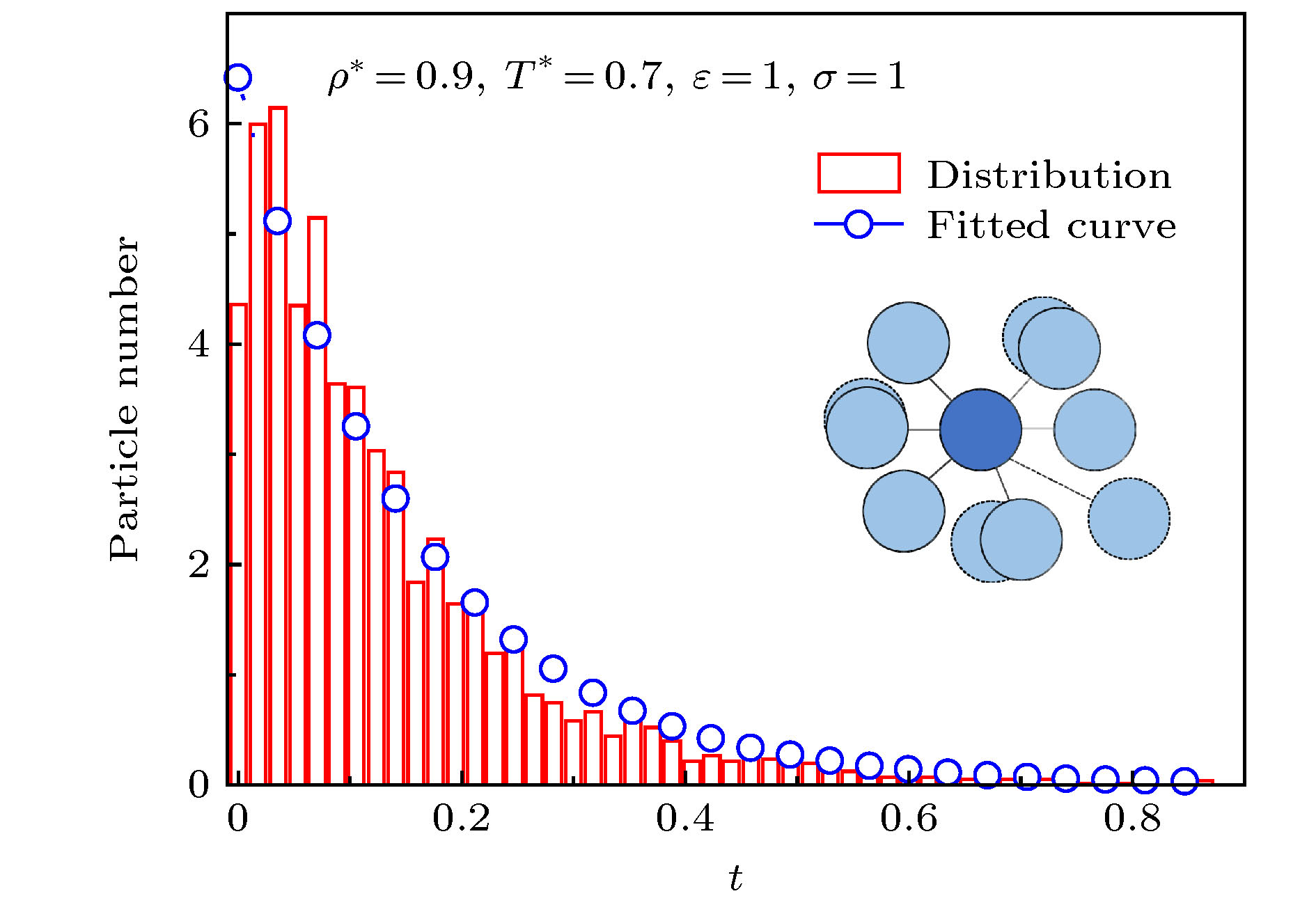 图 7
图 7 










































 图 8 Kramers逃逸速率理论示意图
图 8 Kramers逃逸速率理论示意图

 图 9 A原子和A—A分子的数量变化曲线 (a)不同温度条件; (b)不同密度条件; (c)不同势能为0时的原子距离条件; (d)不同势能阱深度条件
图 9 A原子和A—A分子的数量变化曲线 (a)不同温度条件; (b)不同密度条件; (c)不同势能为0时的原子距离条件; (d)不同势能阱深度条件




















































