摘要: 基于一个最新的CH
2 $(\tilde{\rm X}{}^3 {\rm A''})$ 势能面, 运用切比雪夫波包方法对初始态为(
$\nu = 0{\rm{ }},j = 0$ )的
$ {\rm{C}}\left( {^3{\rm{P}}} \right) + $ $ {\rm H}_2 ({\rm X}^1\Sigma^+_{\rm g}) \to {\rm H} ({}^2{\rm S}) + {\rm CH}({}^2\Pi)$ 反应体系在1.0—2.0 eV 的碰撞能量范围内进行了动力学研究. 通过对角动量量子数
J = 60以下的所有分波进行计算, 得到了反应几率、积分散射截面和速率常数. 计算中用到了耦合态近似方法和考虑科里奥利耦合效应的精确量子方法. 通过对比发现, 随着角动量量子数以及能量的增加, 科里奥利耦合效应的影响越发显著, 因而对于该反应体系, 科里奥利耦合效应不可忽略. 本文计算所得的积分散射截面和速率常数尚无实验数据可以比较, 对该反应的后续研究有一定的参考价值.
关键词: CH2 体系 /
势能面 /
反应几率 /
积分散射截面 English Abstract Wave packet quantum dynamics of ${\bf{C}}{(^3}{\bf{P}}) + {{\bf{H}}_2}({{\bf{X}}^1} \Sigma _{\bf{g}}^ + ) $ $ \to {\bf{H}}{(^2}{\bf{S}}) + {\bf{CH}}{(^2} \Pi ) $ reaction based on new CH2 (${\tilde {\bf X}{}^3}\bf A''$ ) surface Zhao Wen-Li 1 ,Wang Yong-Gang 1 ,Zhang Lu-Lu 2 ,Yue Da-Guang 2 ,Meng Qing-Tian 3 1.School of Information Science and Engineering, Shandong Agricultural University, Taian 271018, China Received Date: 18 January 2020Accepted Date: 20 February 2020Published Online: 20 April 2020Abstract: The C(3 P) + H2 → CH+H reaction in a collision energy range of 1.0–2.0 eV with the initial state $\nu = 0{\rm{ }},j = 0$ is investigated based on the new potential energy surface (PES) by using the Chebyshev wave packet method. All partial wave contributions up to J = 60 are calculated explicitly by the coupled state (CS) approximation method and the Coriolis coupling (CC) effect. Dynamic properties such as reaction probabilities, integral cross sections, and state specific rate constants are calculated. The calculated probabilities and integral reaction cross sections display an increasing trend with the increase of the collision energy and an oscillatory structure due to the CH2 well on the reaction path. The thermal rate constants of the endoergic reaction with a temperature ranging from 1000 K to 2000 K are obtained also. The calculated rate constants increase in the entire temperature range, showing a sharp T dependence in a range of 1400–2000 K. The rate constants are sensitive to the temperature due to the high threshold of the title reaction. In addition, the results of the exact calculations including CC effect are compared with those from the CS approximation. For smaller J , the CS probabilities are larger than the CC results, while for larger J , they are smaller than the CC ones. For reaction cross sections and rate constants, the CS results and the CC ones are in good agreement with each other at lower energy. However, they turn different at higher energy. The comparison between the CC and CS results indicates that neglecting the Coriolis coupling leads the cross sections and the rate constants to be underestimated due to the formation of a CH2 complex supported by stationary point of CH2 (${\tilde{\rm X}}{}^3 \rm A''$ ) PES. It is suggested that the CH2 complex plays an important role in the process of the title reaction. However, it seems to overestimate the CS and CC rate constants because the barrier recrossing is neglected. Unfortunately, the results obtained in the present work have no corresponding theoretical or experimental data to be compared with, therefore these results provide simply a certain reference significance to the follow-up study of the title reaction.Keywords: CH2 system /potential energy surface /reaction probability /integral cross section 全文HTML --> --> --> 1.引 言 在燃烧化学[1 ] 和天体化学[2 ,3 ] 中, 碳原子和氢分子间的反应是一种非常重要的化学反应, 而CH2 自由基作为中间产物, 在这类碳氢反应中发挥着重要的作用[4 ] . 迄今为止, 人们在实验[5 -7 ] 和理论 [8 -19 ] 上对亚甲基分子CH2 进行了大量的研究, 且主要集中于CH2 的${\tilde {\rm a}}{}^1{\rm A'}$ , $1{}^1{\rm{A''}}$ 和${}^1{\rm{A'}}$ 等几个单态的势能面性质[8 -11 ] 和相关的${\rm{C}}{(^1}{\rm{D}}) + {{\rm{H}}_2}{(^1}\Sigma _{\rm{g}}^+) \Leftrightarrow $ $ {\rm{H}}{(^2}{\rm{S}}) + {\rm{CH}}{(^2}\Pi )$ 的动力学计算[12 -19 ] , 而对于三态CH2 (X3 A'')的势能面和相关的${\rm{C}}\left( {^3{\rm{P}}} \right) + {{\rm{H}}_2}\left( {{{\rm{X}}^1}\Sigma_{\rm{g}}^ + } \right)$ $ \Leftrightarrow {\rm{H}}\left( {^2{\rm{S}}} \right) + {\rm{CH}}\left( {^2\Pi } \right) $ 反应动力学研究相对较少, 特别是正向反应少有涉及. 本文的工作就是基于一个最新的CH2 (X3 A'')势能面并对反应${\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2}({{\rm{X}}^1}\Sigma _{\rm{g}}^ + )$ $ \to {\rm{H}}{(^2}{\rm{S}}) + {\rm{CH}}{(^2}\Pi ) $ 开展量子波包动力学研究.[20 ] 在1983年给出了一个${\rm{C}}{{\rm{H}}_{\rm{2}}}({}^1{{\rm{B}}_3})$ 态的势能面, 该势能面能较好地反映体系在所有渐近区的性质. 基于该势能面, Murrell和Dunne[21 ] 用准经典轨线(QCT)的方法研究了${\rm{H}}{(^2}{\rm{S}}) + {\rm{CH}}{{\rm{(}}^2}\Pi ) \to {\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2}({{\rm{X}}^1}\Sigma _{\rm{g}}^+)$ 并计算了该反应的速率常数. 1993年, Harding等[22 ] 应用多参考组态相互作用(MRCI)[23 ,24 ] 结合Dunning相关一致基组的方法得到了CH2 基态$({\tilde {\rm X}}{}^3{\rm{A''}})$ 的全维势能面(HGS). 随后, 该小组应用HGS计算了反应${\rm{H}}{(^2}{\rm{S}}) + {\rm{CH}}{{\rm{(}}^2}\Pi ) \to {\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2}({{\rm{X}}^1}\Sigma _{\rm{g}}^+)$ 和${\rm{H}}{(^2}{\rm{S}}) + {\rm{CH}}{{\rm{(}}^2}\Pi ) \to {\rm{C}}{{\rm{H}}_2}$ 的速率常数, 在高温区域(1500—2000 K), 理论结果和实验符合得很好. 基于HGS, van Harrevelt等[25 ] 应用量子波包和QCT的方法计算了${\rm{CH}} + {\rm{H}} \to {\rm{C}} + {{\rm{H}}_2}$ 的散射截面和速率常数. 另外, Gamallo等[26 ] 使用玻恩-奥本海默(BO)近似的量子力学方法对${\rm{H}}{(^2}{\rm{S}}) + $ $ {\rm{CH}}{{\rm{(}}^2}\Pi ) \to {\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2}({{\rm{X}}^1}\Sigma _{\rm{g}}^+)$ 反应进行了动力学计算, 并指出该反应只发生在解耦合的基态$({\tilde {\rm X}}{}^3{\rm{A''}})$ 上. 1996年, Guadagnini和Schatz[27 ] 使用QCT方法研究了${\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2} \to {\rm{H}} + {\rm{CH}}$ 反应的产物分布, 并指出产物${\rm{CH}} + {\rm{H}}$ 的形成完全取决于C原子靠近H2 的插入机制.[28 ] 使用激光烧蚀石墨的方法获得${\rm{C}}{(^3}{\rm{P}})$ 粒子束, 研究了${\rm{C}}{(^3}{\rm{P}})$ 与H2 , HCl, HBr和CH3 OH的反应, 并指出产物CH的生成与反应物的转动态呈现明显依赖关系. 1997年, Ehbrecht等[29 ] 使用高能量(10 —180 eV)的C原子束与H2 发生光化学反应, 观测到了生成CH产物不同电子态辐射带(${}^{\rm{2}}\Delta - {\rm{X}}{}^{\rm{2}}\Pi $ , ${}^{\rm{2}}{\Sigma^ - }{\rm{ - X}}{}^{\rm{2}}\Pi $ 和${}^{\rm{2}}{\Sigma^+}{\rm{ - X}}{}^{\rm{2}}\Pi $ ), 并指出三种态可观测的能量范围不同.[30 ] 应用多参考组态相互作用方法(MRCI), 采用包含Davidson修正的aug-cc-pVX Z(X = Q和5)基组计算得到的5285个能量点, 将其外推到完备基组极限(CBS)并利用多体展开(MBE)的方法拟合构建了一个关于${\rm{C}}{{\rm{H}}_2}({{\tilde {\rm X}{}^3}}{\rm{A''}})$ 的全维解析势能面, 其中势能最大值与最小值之间的跨度为17 eV, 势能面的总方均根偏差为0.0349 eV. 基于该势能面, 该团队运用QCT方法, 计算了${\rm{H}}{(^2}{\rm{S}}) + {\rm{CH}}{{\rm{(}}^2}\Pi ) \to {\rm{C}}{(^3}{\rm{P}}) + $ ${{\rm{H}}_2}({{\rm{X}}^1}\Sigma_{\rm{g}}^+) $ 和${\rm{H}}{(^2}{\rm{S}})\!+\!{\rm{CH}}{{\rm{(}}^2}{{\rm{X}}^1}\Pi ) \!\to\! {\rm{CH}}({{\rm{X}}^1}\Pi) +\!$ $ {{\rm{H}}_2}{(^2}{\rm{S}}) $ 反应的积分散射截面和速率常数, 与基于HGS势能面的动力学计算结果和实验结果符合得很好, 从而验证了该势能面的精确性.2 自由基电子态性质复杂, 相关的动力学研究涉及到BO近似和非绝热过程, 不过对该体系性质进行全面的研究并不是本文的目的. 本工作主要是基于Zhang等[30 ] 构建的${\rm{C}}{{\rm{H}}_2}({{\tilde{\rm X}{}^3}}{\rm{A''}})$ 势能面, 用切比雪夫量子波包方法对反应${\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2}({{\rm{X}}^1}\Sigma _{\rm{g}}^ + )$ $ \to {\rm{H}}{(^2}{\rm{S}}) + {\rm{CH}}{{\rm{(}}^2}\Pi ) $ 的反应几率和散射截面等性质进行研究. 本文所采用的方法, 相对于传统的含时方法有着更优越的标度律, 只需要波包的一次传播就能够给出所有能量点处的反应几率, 并且波包在实空间的传播没有近似, 因而在计算上更具有优势[31 -33 ] . 本文主要包含以下几部分: 第二部分介绍所采用的理论方法以及相应的数值计算方法. 第三部分给出理论计算结果和讨论. 第四部分总结得出相关的结论. 除特别说明之外, 文中的所有公式和计算都使用原子单位.2.理 论 对于反应物满足交换对称的体系(C + H2 ), 使用雅克比坐标($R, r, \gamma $ )能够减少计算基组个数, 在雅克比坐标下C + H2 体系的哈密顿量的表达式为$r$ 表示双原子(H-H)之间的距离, $R$ 表示碳原子与双原子质心之间的距离(C-H2 ), $ {\mu _r} = {m_{\rm{H}}} /2 $ 和${\mu _R} \!=\! {{2{m_{\rm{C}}}{m_{\rm{H}}}} / {\left( {{m_{\rm{C}}} \!+\! 2{m_{\rm{H}}}} \right)}}$ 表示约化质量, $V(R, r, \gamma )$ 为体系的势能面, $\hat j$ 和$\hat l$ 是双原子转动角动量算符和轨道角动量算符. 轨道角动量算符的平方${\hat l^2}$ 可以表示为$\hat J$ 表示总角动量算符, ${\hat J_ + }$ (${\hat J_ - }$ )和${\hat j_ + }$ (${\hat j_ - }$ )分别表示总角动量升(降)算符和转动角动量升(降)算符, 其中${\hat J_z}$ 和${\hat j_z}$ 分别是$\hat J$ 和$\hat j$ 在定体坐标系中(body-fixed, BF) z 轴的投影(BF z 轴为H-H质心指向C原子的方向). (2 )式中的最后两项对应于科里奥利耦合(CC)效应, 在耦合态(CS)近似中, 这两项会被忽略掉.[34 ,35 ] ${H_{{\rm{norm}}}} = {{\left( {H - {H^ + }} \right)} / {{H^ - }}}$ 是归一化的哈密顿量, 且${H^ \pm } = {{\left( {{H_{\max }} \pm {H_{\min }}} \right)} / 2}$ , ${H_{\max }}$ (${H_{\min }}$ )通过光谱的上(下)限计算获得[36 ] . 在$R$ (或$r$ )格点边缘, 用高斯型函数D 来描述输出波的边界条件${d_x}$ 决定阻尼范围, ${x_{\rm{d}}}$ 为阻尼起始点. 初始波包$\left| {{\psi _0}} \right\rangle $ 表示为$N$ 是归一化常数, ${k_0}$ 为初动量, ${R_0}$ 与$\delta $ 分别为初始波包的中心位置和宽度, $\left| {{\varphi _i}} \right\rangle $ 为一个具体的振转本征函数.S -散射矩阵元计算耗时较长[37 ] , 为了避免这种情况, 本文采用流计算法, 应用这种方法获得的初始态总反应几率为[38 ] ${r_{\rm{f}}}$ 决定了产物通道的分界面, $\theta \equiv $ $ \arccos \left[ {{{\left( {E - {H^ + }} \right)}/ {{H^ - }}}} \right]$ 是切比雪夫角, ${a_i}(E)$ 是初始波包的振幅[39 ] ${{\rm h}}_{li}^{(2)}({k_i}R)$ 为第二类汉克尔函数. 应用这种汉克尔函数代替平面波函数的好处在于, 初始波包能够被放在一个R 足够小的范围内, 进而无需考虑长程区离心势能的影响, 这种近似方法已经被成功地应用到N + H2 等反应体系中[40 ] .J 的反应几率进行求和${E_{\rm{c}}}$ 为碰撞能量, $k_{{\upsilon _i}{j_i}}^2 = 2{\mu _R}{E_{\rm{c}}}$ , $P_{{\upsilon _i}{j_i}{l_i}}^{Jp}({E_{\rm{c}}})$ 是宇称为p 、角动量量子数为$J$ 时, 初始态$\left( {{\upsilon _i}{j_i}{l_i}} \right)$ 的反应几率. 将相应的积分散射截面${\sigma _{{\upsilon _i}{j_i}}}({E_{\rm{c}}})$ 对碰撞能量${E_{\rm{c}}}$ 结合玻尔兹曼权重进行积分, 得到初态$\left( {{\upsilon _i}{j_i}{l_i}} \right)$ 的速率常数为f 是电子的简并因子, ${k_{\rm{B}}}$ 和T 分别为玻尔兹曼常数和热力学温度.3.结果和讨论 23.1.势能面和最小能量路径 3.1.势能面和最小能量路径 图1 2 $(\tilde {\rm X}{}^3\rm A'')$ 的等势线. 图1(a) 为C原子沿着H-H连线方向靠近${{\rm{H}}_2}$ 时的等势线, 该势能面最显著的特征是有一个鞍点(${R_{{\rm{HH}}}} \approx 2.601{a_0}$ , ${R_{{\rm{CH}}}} \approx 2.201{a_0}$ ). 图1(b) 为C原子沿着T构型插入${{\rm{H}}_2}$ 分子时的等势线, 很显然, 该曲线有一个局部极小值(${R_{{\rm{HH}}}} \approx 3.760{a_0}$ , ${R_{{\rm{C}}{{\rm{H}}_{\rm{2}}}}} \approx 0.805{a_0}$ 处)和一个过渡态 (${R_{{\rm{HH}}}} \approx 1.595{a_0}$ , $ {R_{{\rm{C}}{{\rm{H}}_{\rm{2}}}}} \approx 2.370 a_0$ ), 这意味着势能面的几何结构中有一个势阱.图 1 CH2 等势线, 图中等势线间隔为0.1 eV (a) C沿着共线构型靠近H2 分子; (b) C 沿着T构型插入H2 Figure1. Equipotential contour plot for CH2 , the contour increments are 0.1 eV: (a) For bond stretching in C-H-H linear geometry; (b) for T-shaped insertion of C into H2 diatoms.图2 给出了${\rm{C}}{(^3}{\rm{P}}) \!+\! {{\rm{H}}_2}({{\rm{X}}^1}\Sigma _{\rm{g}}^+) \!\to\! {\rm{H}}{(^2}{\rm{S}}) \!+$ $ {\rm{CH}}{{\rm{(}}^2}\Pi ) $ 反应最小能量路径(MEP), 它描述了C原子从不同角度插入时, CH2 体系的势能(角度$\angle {\rm{CHH}}$ 固定)随着反应坐标${R_{{\rm{CH}}}} - {R_{{\rm{HH}}}}$ 的变化关系. 红线对应于T构型的情况, 黑线对应于共线结构. 从反应物C+H2 生成产物CH+H吸热为1.10908 eV, 即25.8 kcal·mol–1 , 该数值比文献[41 ]中的理论值小3.7 kcal·mol–1 , 比其中的实验值大2.6 kcal·mol–1 . 从图2 中可以看出, 当$\angle {\rm{CHH}} = 9{0^\circ }$ 时, 能量曲线有一个深度为0.2 eV 的浅势阱和一个1.1 eV的势垒. 当$\angle {\rm{CHH}} = {180^ \circ }$ 时, 曲线仅仅有一个高度为1.3 eV的势垒. 势能面的这些特征对于动力学起着决定性的作用, 下文将会详细讨论这一点.图 2 90°和180°的最小能量路径Figure2. The minimum energy paths as a function of R CH -R HH at 90° and 180°.3.2.计算细节 -->3.2.计算细节 本文采用切比雪夫量子波包方法研究${\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2} \to {\rm{H}} + {\rm{CH}}$ 反应. 由上述分析可知, 该反应是一个有较大的势垒和较小的势阱的典型吸热反应, 为了计算得到精确的ICS和速率常数, 需要较高的碰撞能量、大量的分波以及较大的基组数, 尤其是考虑完全的CC效应时. 对于$J = 0$ 的情况, 经过反复的收敛性测试, 本文选取了最优的计算参数列于表1 . 应用这些计算参数, 对于J = 0, 5, 15, ···, 60的分波进行了CS 近似和完全的CC效应的计算, 其他反应几率通过上述反应几率用三次样条插值法插值得出[42 ] , 进而得出碰撞能量范围在1.0—2.0 eV 的ICS, 最后给出速率常数.坐标取值范围 $R \in ({10^{{\rm{ - }}16}}, \, 16)$, $({N_R} = 203)$ $r \in (0.5, \, 12)$, $({N_r} = 99)$ $\gamma \in ({90^ \circ }, \, {180^ \circ })$, $({N_\gamma } = 50)$ 吸收势 ${R_{\rm{d}}} = 11.0$, ${d_R} = 0.0006$ ${r_{\rm{d}}} = 7.5$, ${d_r} = 0.001$ 初始波包 ${R_0} = 8.0$, ${E_0} = 1.55\;{\rm{ eV}}$, $\delta = 0.3$ 光谱控制 0.5 流计算的位置 ${r_{\rm{f}}} = 7.4$ 传播步数 100000
表1 波包计算中的数值参量(除特殊说明, 均采用原子单位a.u.)Table1. Parameters used in wave packet calculation (The atomic unit is used in the calculation unless otherwise stated).3.3.反应几率 -->3.3.反应几率 图3 给出了J = 0, J = 10, J = 30和J = 50的精确量子计算(考虑完全CC效应)反应几率随碰撞能量的变化. 从图中可以看出, 整体上反应几率数值较小(< 0.22), 这是因为C原子无论从哪个角度碰撞H2 , 过程中都有可能形成中间络合物CH2 , 当能量较低、J 较小时, 络合物还可能比较“长寿”, 并且CH2 也有很大的几率衰变回反应物C+H2 [27 ] . 图3 中不同分波的反应几率显示范围是0—0.22, 碰撞能量显示范围是1.0—2.0 eV. 对于J = 0, 反应几率逐渐从0开始增大, 约为1.10 eV时, 反应几率大于0.1, 这与前面MEP讨论的${\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2} \to {\rm{H}} + {\rm{CH}}$ 反应是一个吸热反应是一致的, 与该反应需要吸热1.109 eV也是一致的. 但是同时要注意到, 在1.0—1.1 eV的范围内, 碰撞能量不足以越过该体系的势垒, 而反应几率仍然不为0, 这只能用量子隧穿效应解释. 随着总角动量J 的增加, 离心势垒逐渐增大, 反应阈能逐渐增加, J = 30的阈能约在1.2 eV, J = 50的阈能约在1.65 eV. 从整体上看, 反应几率随着碰撞能量的增加而增加, 这也符合一般的吸热反应的反应几率变化规律. 在低能区域反应几率呈现强烈的振荡, 这是由体系的势阱(0.2 eV)所产生的共振引起的. 当碰撞能量增加时, 反应几率的振荡减弱, 逐渐变得不明显.图 3 不同分波的反应几率随着碰撞能量的变化Figure3. The reaction probabilities vs. collision energy at different J .[43 ] . 但是, CS近似中忽略了科里奥利效应. 实际上, 有动力学研究表明在一些化学反应中, 这种效应会产生非常重要的影响因而不能忽略[44 ,45 ] . 例如 Meijer和Goldfield[44 ] 发现CC效应在H+O2 反应中扮演着重要的角色, 如果忽略CC效应将会使J > 0时的计算变得不可靠, 这种偏差在高能区越发明显.图4 画出了J = 5, 10, 20的CS近似和CC两种方法下的反应几率. 两种计算结果都表现出共振特性, 随着J 的增大, 一些共振会消失, 这是因为J 越大, 波函数计算中包含的分波更多. 此外, CS的振荡幅度比CC的大得多, 这是反应中科里奥利耦合效应受到长程力支配的结果. 整体上来讲, 在角动量J 比较小的情况下(J = 5), CC计算的反应几率小于CS近似的数值; 当J = 10时, 两种方法的反应几率基本符合; 当J = 20时CC计算的反应几率大于CS近似的结果, J 越大, 二者差别越明显. 从J 较大时的单一分波来看(J = 20), 在低能区域(小于1.3 eV), CS的计算结果比CC的大, 在1.3—2.0 eV的范围内, CC计算的结果大于CS近似的数值, 并且碰撞能量越大偏差越大. 理论认为, 中间络合物CH2 的形成对于反应机制起着主导作用. 当J 较小时, CS近似下由于H2 分子态的限制, C更容易与其发生碰撞, 从而促进产物生成, 所以CS的几率较大. 随着J 的增大, 更多分波贡献被考虑其中. 一方面, CS近似下长程相互作用影响较大, 长程势会使得碰撞时间变长, 原子的重新组合变慢从而使得反应几率变小; 另一方面, 由于科里奥利耦合效应激发的振动模式更有利于破坏H-H键从而促进产物的形成. 这种情况下, CC效应就会显著影响反应过程, 所以CC的几率较大.图 4 CC与CS反应几率比较Figure4. Comparisons between the CC and CS probability.3.4.积分散射截面和速率常数 -->3.4.积分散射截面和速率常数 根据(8 )式, ICS是把所有分波的反应几率乘以各自权重, 然后再对所有的分波求和得到的. 在1.0—2.0 eV的碰撞能量范围下, 我们对所有J < 60分波进行加权求和得到了${\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2} \to {\rm{H}} + {\rm{CH}}$ 反应的ICS. 图5 给出了分别用CS近似和CC方法得到的ICS随着碰撞能量的变化. 从图中可以看出, 随着碰撞能量的增加, 两种计算方法的ICS都会增加, 与一般吸热反应的ICS的变化趋势一致. 相比于CS近似的ICS, CC计算的结果振荡得更加剧烈, 但幅度比CS的小, 并且CC的ICS随碰撞能量的增加而增加得更快. 当E C < 1.27 eV时, 两种方法的计算结果基本符合. 当E C > 1.27 eV时, CC的ICS迅速增加, CS的ICS则缓慢增加, 碰撞能量越高, 二者的差距越大. 例如, 当E C = 1.21 eV时, ${\sigma _{{\rm{CC}}}}$ = 0.135 ?2 , ${\sigma _{{\rm{CS}}}}$ = 0.130 ?2 , 二者数值几乎相当; 当E C = 1.61 eV时, ${\sigma _{{\rm{CC}}}}$ = 0.647 ?2 , ${\sigma _{{\rm{CS}}}}$ = 0.256 ?2 , 前者是后者的2.53倍; 当E C = 1.99 eV时, ${\sigma _{{\rm{CC}}}}$ = 1.022 ?2 , ${\sigma _{{\rm{CS}}}}$ = 0.305 ?2 , 前者是后者的3.35倍. 考察势能面的特征就会发现, 势能面的势阱、鞍点和过渡态的位置对于中间络合物的形成是非常有利的, 即不利于产物的形成, 这就导致了CS近似下ICS随能量增加而缓慢攀升, 然而, 正如前文所述, 科里奥利耦合效应就可以克服这一点, 所以CC的ICS随能量增加上升得非常快.图 5 C+H2 反应的积分散射截面Figure5. The integral cross section of the C+H2 reaction.9 )式, 我们计算出了${\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2} \to {\rm{H}} + {\rm{CH}}$ 反应的速率常数, 如图6 所示. 温度的取值范围为1000—2000 K, 对应ICS碰撞能量的取值范围1.0—2.0 eV. 图中红色实线表示CC计算的速率常数, 黑色虚线表示CS近似的结果. 从图6 中可以看出, 在1000—1400 K, 速率常数增加比较平缓, CC和CS的结果基本相一致; 当T > 1400 K时, 随着温度的增加, 速率常数迅速增加, 我们认为速率常数对于温度如此敏感归因于该反应为吸热反应, 有较高的反应阈能. 另一方面, 我们注意到文献[46 ]报道的类似电子结构O(3 P ) 与CH4 反应的速率常数, 该文献认为计算过程中忽略势垒交叉的影响会导致速率常数计算结果偏大. 基于同样的原因, 我们预测本文速率常数计算结果可能比实际偏大. 此外, 类似于ICS的结果, 随着温度的增加, CC速率常数比CS攀升得更快, 例如在T = 1400 K时, CC计算和CS近似的速率常数分别为5.52 × 10–15 和5.07 × 10–15 cm3 ·molecule–1 ·s–1 , 二者的差值为4.50 × 10–16 cm3 ·molecule–1 ·s–1 , CS比CC结果小8.25%; 在T =1700 K时, CC计算和CS近似的速率常数分别为2.91×10–14 和2.37×10–14 cm3 ·molecule–1 ·s–1 , CS比CC结果小18.58%; 在T = 2000 K时, CC计算和CS近似的速率常数分别为9.43 × 10–14 和6.98 × 10–14 cm3 ·molecule–1 ·s–1 , 二者的差值为2.46 × 10–14 cm3 · molecule–1 · s–1 , CS比CC结果小25.95%. 可见, 温度越高, CC与CS的计算结果差别越大.图 6 C+H2 反应的速率常数Figure6. The rate constant of the C+H2 reaction.4.结 论 基于最新CH2 $({\tilde {\rm X}}{}^3{\rm{A''}})$ 的势能面, 本文应用切比雪夫量子波包方法对${\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2}({{\rm{X}}^1}\Sigma _{\rm{g}}^ + ) \to $ ${\rm{H}}{(^2}{\rm{S}}) + {\rm{CH}}{{\rm{(}}^2}\Pi ) $ 反应进行了动力学研究, 对碰撞能量1.0—2.0 eV范围内的大量分波(J < 60)进行了计算. 为了考察科里奥利耦合的影响, 计算中分别采用了CS近似和考虑完全CC效应精确量子方法. 通过对计算数据的分析发现, J 比较小的情况下(J < 10), CC反应几率小于CS近似; J > 10的情况下, CC计算的反应几率远大于CS近似的结果, 并且总角动量J 越大, 二者差值越大. 基于反应几率, 计算得到了ICS和速率常数, 对比可知, 随着能量增加和温度的升高, CS与CC的结果差别越来越大, 这种差别应该归因于该反应中间络合物的产生机制与势能面之间的密切关系. 所以, 对于该反应, CS近似是一种比较粗糙的方法, 科里奥利效应在${\rm{C}}{(^3}{\rm{P}}) + {{\rm{H}}_2} \to {\rm{H}} + {\rm{CH}}$ 反应中的影响是不可忽略的. 
































































































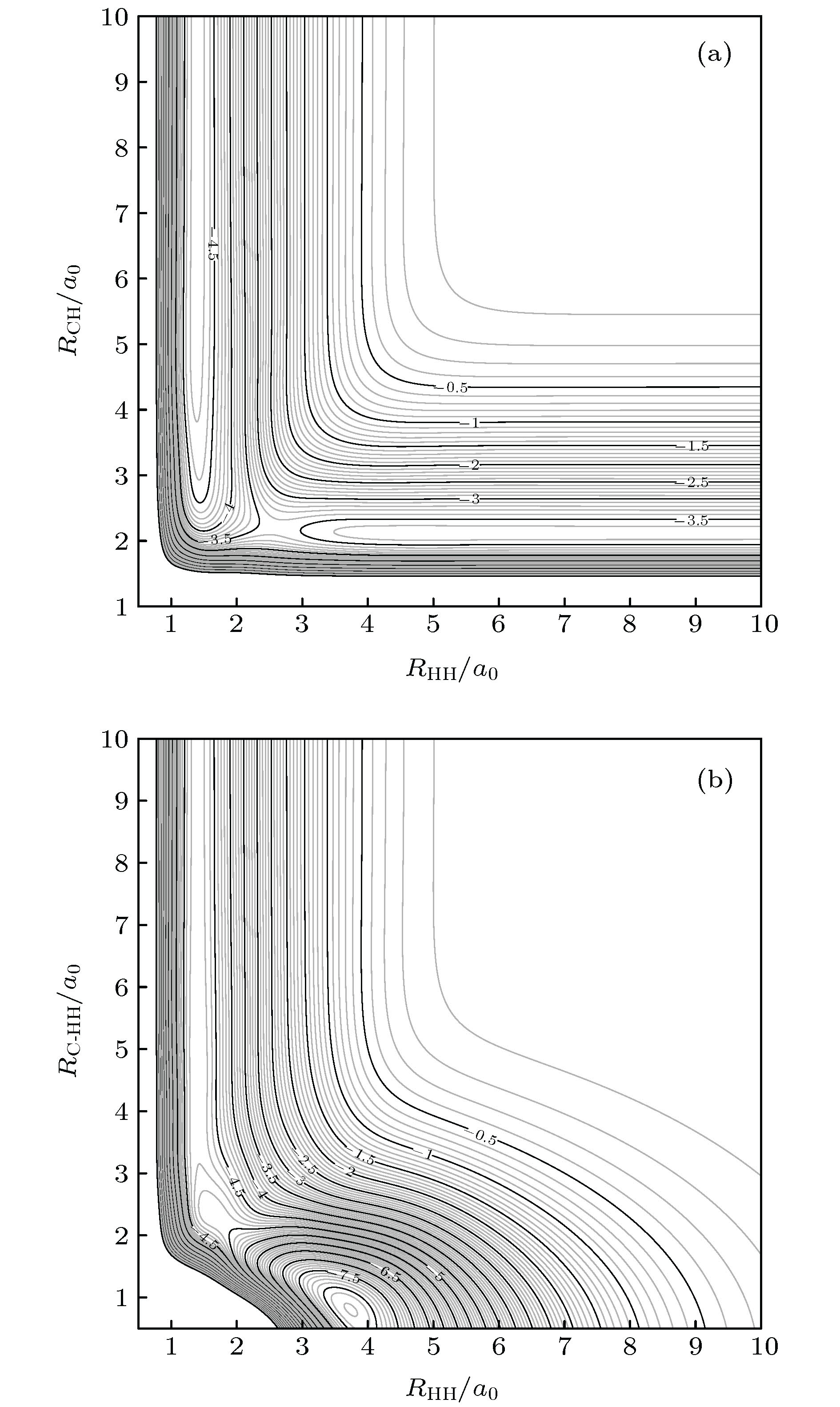 图 1 CH2等势线, 图中等势线间隔为0.1 eV (a) C沿着共线构型靠近H2分子; (b) C 沿着T构型插入H2
图 1 CH2等势线, 图中等势线间隔为0.1 eV (a) C沿着共线构型靠近H2分子; (b) C 沿着T构型插入H2





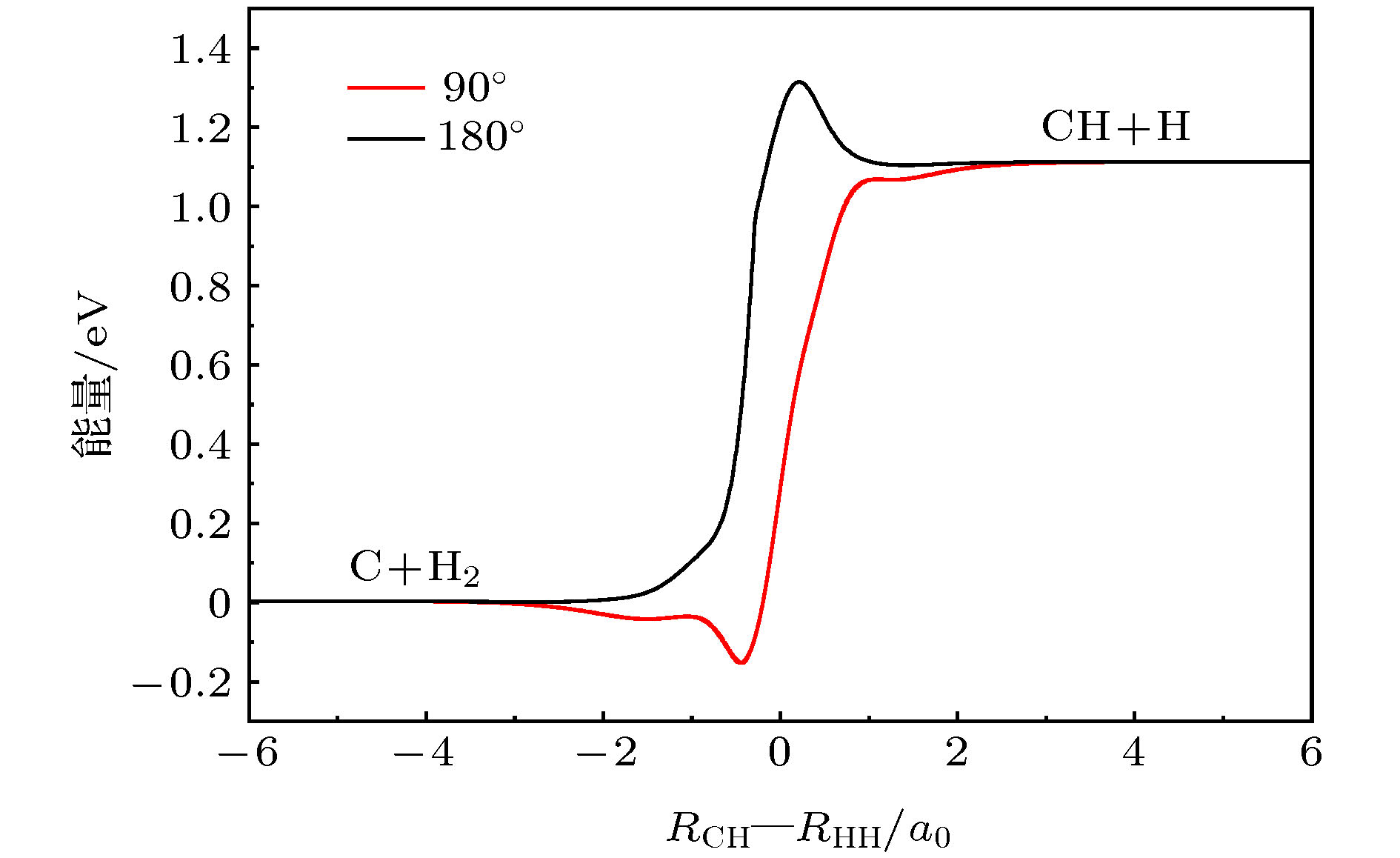 图 2 90°和180°的最小能量路径
图 2 90°和180°的最小能量路径


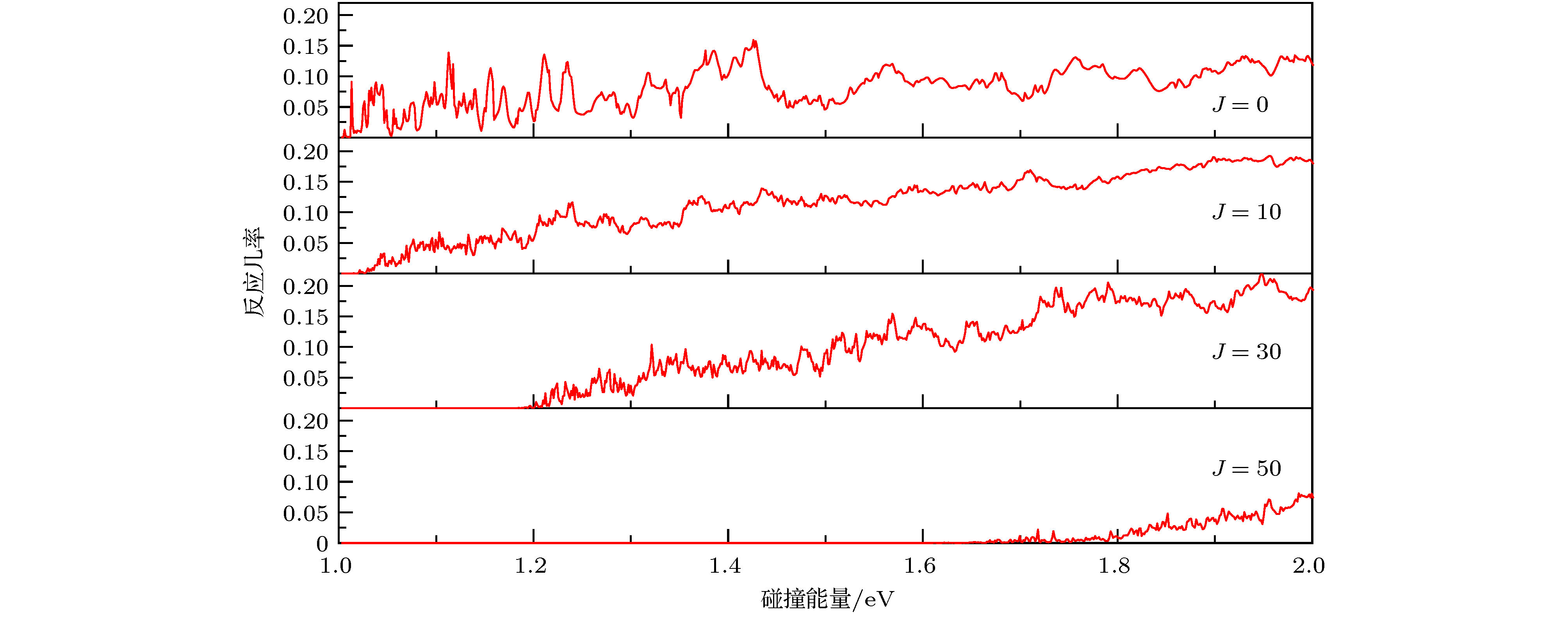 图 3 不同分波的反应几率随着碰撞能量的变化
图 3 不同分波的反应几率随着碰撞能量的变化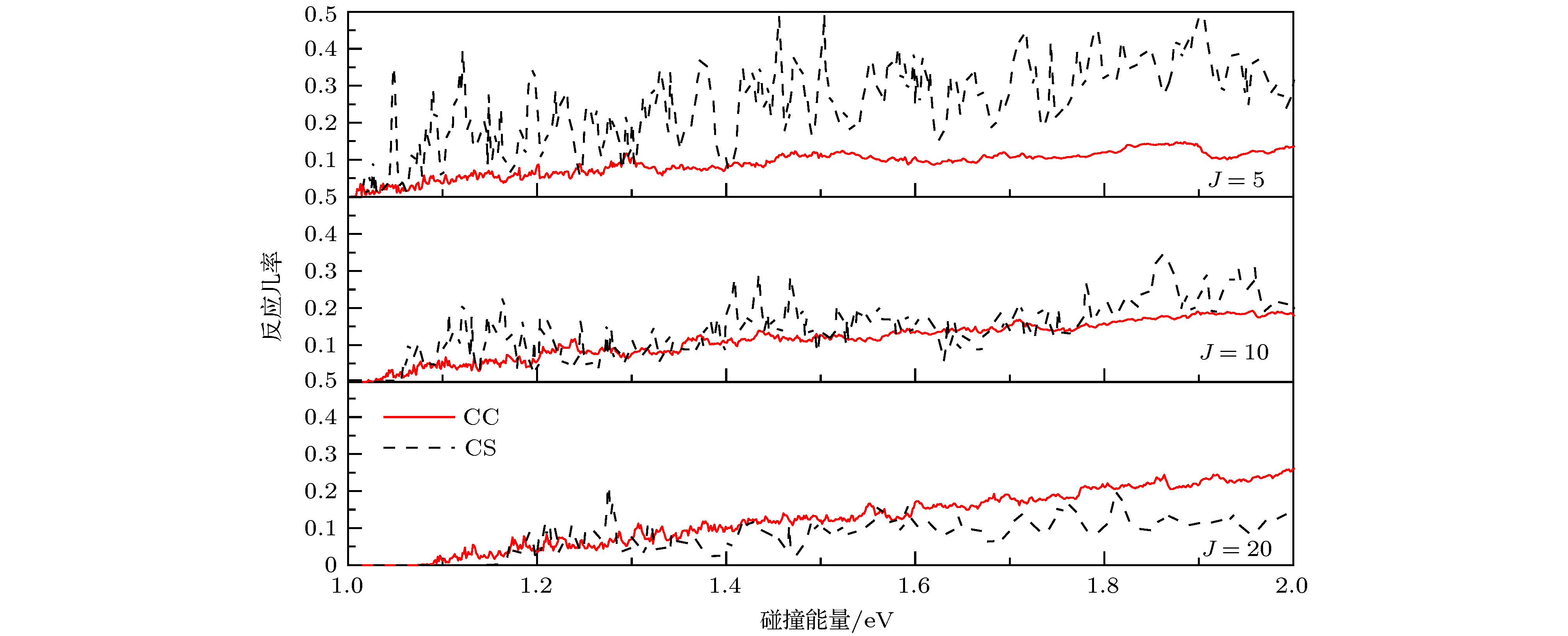 图 4 CC与CS反应几率比较
图 4 CC与CS反应几率比较






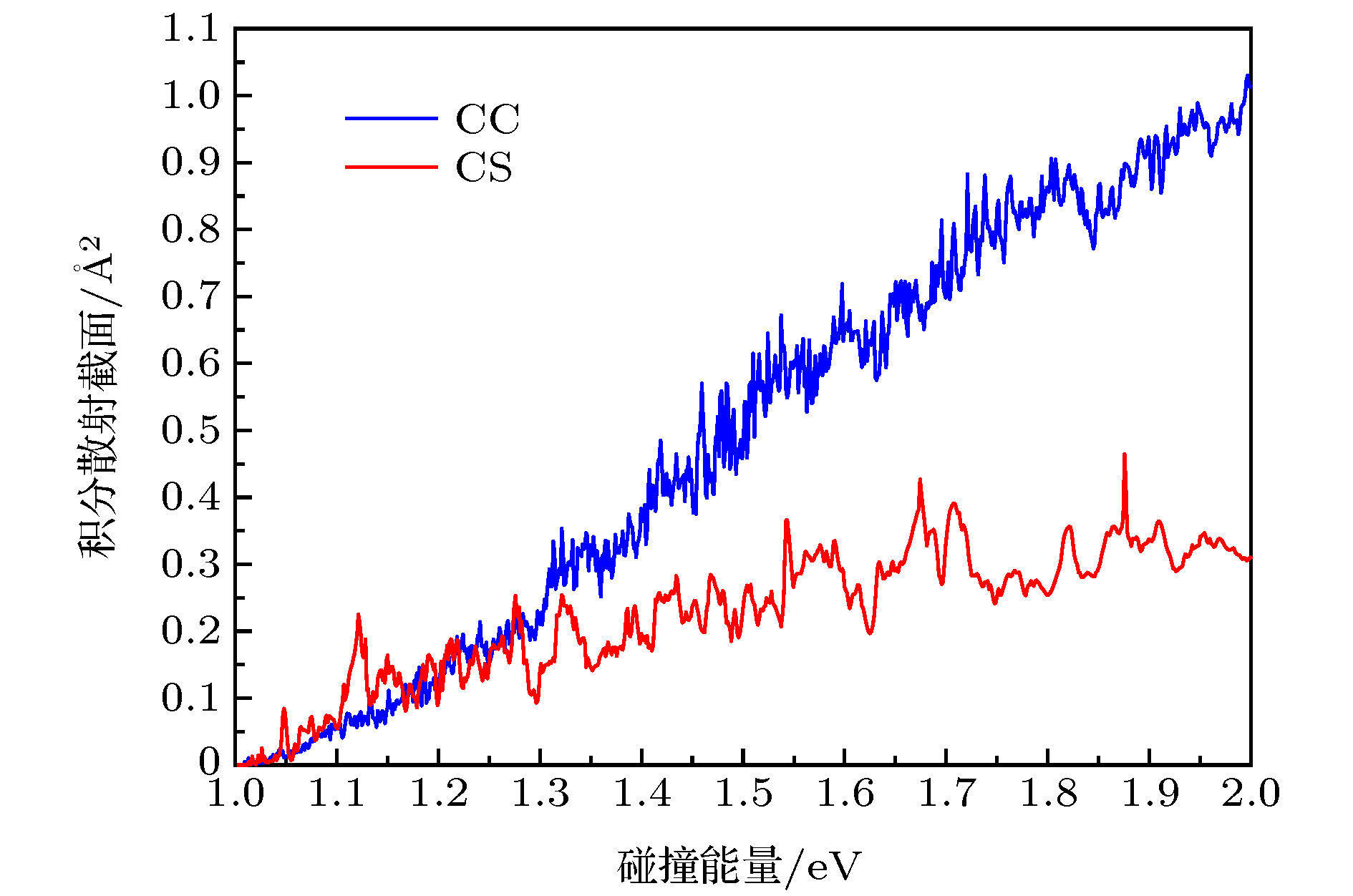 图 5 C+H2反应的积分散射截面
图 5 C+H2反应的积分散射截面
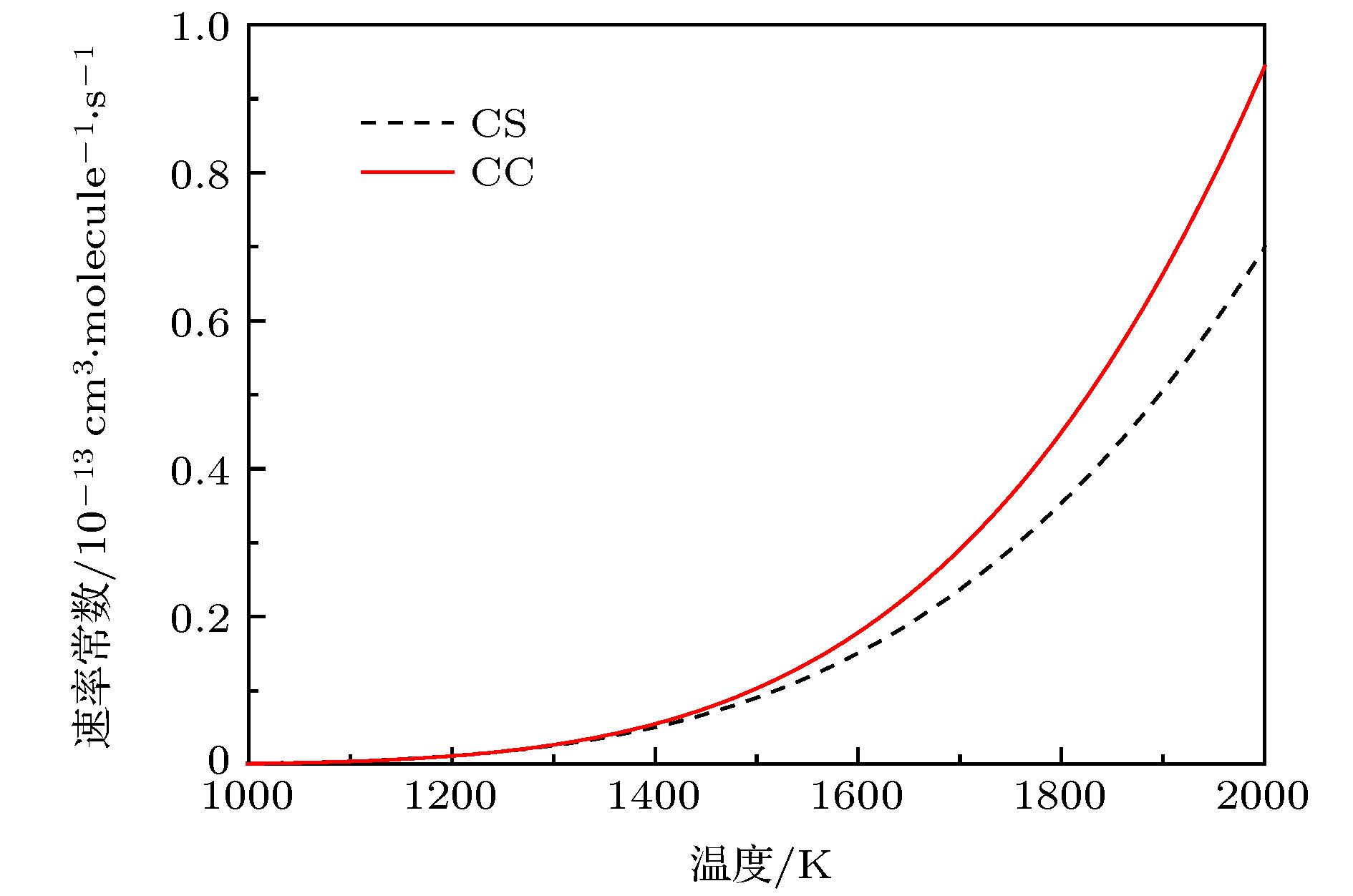 图 6 C+H2反应的速率常数
图 6 C+H2反应的速率常数



