全文HTML
--> --> -->在纳米尺寸上同时进行形貌设计和组份控制是改良Pt基催化剂性能的一种重要策略. 一方面, 通过添加其他过渡金属, 利用不同元素间的协同作用和电子转移, 调控Pt的电子结构, 从而改善Pt基催化剂的催化性能[11-13]. 另一方面, 对双金属合金Pt基(Pt-M, M是非贵过渡金属Fe, Co, Ni, Mn等)纳米催化剂进行形貌设计, 通常也被认为是增强电催化性能的一种重要手段[14,15]. 一般来说, 催化剂与电解液的接触面积越大, 催化反应过程中能够提供更大的比表面积和更多的活性位点, 催化活性也会随之越强[16]. 目前已有很多研究表明, 尺寸较小且分散性良好的零维Pt-M纳米颗粒催化剂具有优异的催化活性. 例如, 呈单分散的中空PtNi纳米球(直径约32 nm)对MOR的电催化活性比商业Pt/C提高了3倍[17]; 超薄PtNi空心纳米球(球壁只有2 nm)的电化学活性比表面积是商业Pt/C的约2倍之高[18]; 超小粒径(1.9 nm)的PtRu纳米颗粒在催化MOR时的电化学比表面和质量活性分别高达相同原子比的商业PtRu/C的1.8和2.5倍[19]. 但是, 由于尺寸效应[20]的存在, 具有小尺寸的零维Pt-M纳米颗粒虽然可以提供更大的活性比表面积, 但随着尺寸的减小, 将会受到结构溶解和奥斯特瓦尔德(Ostwald)熟化的影响, 导致稳定性变差[21,22]. 近期的研究表明, 以纳米棒[23,24]、纳米线[25,26]或纳米管[27]等形式存在的一维Pt基金属纳米材料, 由于在酸性电催化条件下较难发生溶解和奥斯瓦德熟化等现象, 在催化MOR时能表现出优异的催化稳定性[26,28]. 但一维Pt基纳米结构通常因具有较小的比表面积而使其催化活性受到限制. 因此, 迫切需要设计合成出一种新颖的双金属Pt基合金纳米结构, 以确保拥有较高催化活性的同时实现较高的稳定性. 和传统的具有平整表面或由纳米球无间隙组装在一起的一维纳米结构不同, 将具有一定长度的一维纳米结构与零维小尺寸纳米颗粒组装在一起, 纳米颗粒间由极细的纳米线串联而成的纳米结构, 能够同时拥有二者的结构优越性, 在保持高稳定性的同时, 催化活性达到最大化. 近期, 华中科技大学的Tian等[29]利用油浴热法合成了串状PtNi纳米球, 然后通过酸刻蚀法处理得到一维PtNi纳米笼结构同时形成富Pt表面, 将一维结构和中空结构结合在一起, 证明这种一维组装结构的催化剂对质子交换膜燃料电池阴极反应(ORR)表现出了优异的电催化性能.
本工作利用非常简单的一步溶剂热法直接合成具有富Pt表面的链状Pt-Ni合金纳米颗粒(chain-like Pt-Ni nanoparticles, Pt-Ni CNPs), 将一维纳米线和零维小尺寸纳米颗粒完美地组合在一起, 使其同时具备一维纳米线高稳定性和零维小尺寸纳米颗粒高活性的优点. 通过X射线衍射、电镜等表征手段, 发现Pt-Ni CNPs具有巧妙且独特的微观形貌、合金化结构以及富Pt表面, 非常有利于催化反应过程中电子的传递以及在反应过程中提供更多的活性位点, 从而增强催化活性. 与Tian等[29]的研究内容不同的是, 本工作是在反应釜中直接合成具有富Pt表面的Pt-Ni CNPs, 并且在反应中添加了葡萄糖作为还原剂, 适量的葡萄糖和表面活性剂共同控制一维纳米晶体的成核和生长动力学[30], 其中葡萄糖对纳米颗粒自组装成一维纳米结构并保持稳定起关键作用[31,32]. 此外, 本工作是对DMFC的阳极(MOR)进行催化研究, 华中科大的工作主要讨论的是催化阴极反应(ORR). 与商业Pt/C相比, 合成的Pt-Ni CNPs对MOR具有显著增强的催化活性和稳定性, 这能够为制备在工业生产中大规模应用的高效催化剂提供新思路和新方法.
2.1.实验材料准备
乙酰丙酮镍(Ni(acac)2)、乙酰丙酮铂(Pt(acac)2)和标准商业Pt/C(40 wt.%)均从阿法埃莎(Alfa Aesar)公司购买. 十六烷基三甲基氯化铵(CTAC)、油胺(C18H37N)和葡萄糖(C6H12O6)在阿拉丁(Aladdin)试剂公司购买. 甲醇(CH3OH)、异丙醇((CH3)2CHOH)、硫酸(H2SO4)、无水乙醇(CH3CH2OH)和环己烷(C6H12)购买于国药集团化学试剂有限公司. 所有试剂均为分析纯, 没有进一步提纯和处理.2
2.2.样品制备
将20 mg Pt(acac)2, 7 mg Ni(acac)2, 32 mg CTAC和90 mg葡萄糖添加到25 mL的样品瓶中, 加入20 mL油胺, 超声30 min使之形成均匀的混合溶液. 将混合溶液转移至25 mL的反应釜中, 密封后放入鼓风干燥箱中由室温加热至160 ℃, 在160 ℃下保持20 h后自然冷却至室温. 将得到的黑色产物离心并用乙醇/环己烷(v∶v, 9∶1)的混合溶液洗涤数次.2
2.3.样品形貌、结构、成分、电子结构表征
通过透射电子显微镜(transmission electron microscopy, TEM; 200 kV, JEOL, JEM-2200F)表征所制备样品的形态和晶体结构. 样品的化学组成通过配置有能谱仪(energy dispersive spectroscopy, EDS)的扫描电子显微镜(scanning electron microscopy, FIB-SEM, Zeiss/Auriga)和电感耦合等离子体光谱仪(inductively coupled plasma atomic emission spectroscopy, ICP-AES, 213DF0G/ICPE-9820)测试得到. 通过粉末X射线衍射(X-ray diffraction, XRD, PANalytical Empyrean, Cu Kα)研究样品的晶体学信息. 通过X射线光电子能谱(X-ray photoelectron spectroscopy, XPS, AXIS Supra)测量不同元素的电子结构.2
2.4.电化学测量
电化学表征在室温下采用三电极测试系统在电化学工作站(CHI660E, 上海辰华仪器有限公司)中进行. 工作电极是玻碳电极(glassy carbon electrode, GCE, 几何面积为0.071 cm2), 对电极和参比电极分别是铂丝和饱和甘汞电极(saturated calomel electrode, SCE). 工作电极制备过程如下: 适量的催化剂(Pt-Ni CNPs或商业Pt/C)通过超声处理分散在异丙醇/去离子水(3∶1)的混合溶液中, 形成匀相溶液. 将上述混合溶液滴加到预处理过的GCE上, 并在室温下自然干燥, 保证工作电极上负载Pt的质量均为16 μg. 最后, 取5 μL Nafion溶液(0.1 wt.%)滴在工作电极上, 保证完全覆盖样品并再次干燥.CV测试是在N2饱和的0.5 M H2SO4溶液中完成的, 扫描电压范围是0—1.25 V (vs. RHE), 扫描速率为50 mV/s. MOR测试是在0.5 M H2SO4和1 M CH3OH的混合溶液中进行的, 扫描电压范围是0.25—1.25 V (vs. RHE), 扫描速率为50 mV/s. 线性扫描伏安(linear sweep voltammetry, LSV)测试也是在0.5 M H2SO4和1 M CH3OH的混合溶液中进行的, 扫描电压范围是0.25—0.85 V (vs. RHE), 扫描速率为5 mV/s. CO溶出曲线是在0.5 M H2SO4溶液中测试的: 向0.5 M H2SO4溶液中持续通入CO气体, 工作电极在低电位0.01 V(vs. SCE)下吸附CO 30 min, 之后持续通入N2 2 h以去除溶液中的CO气体, 然后再以50 mV/s的扫速在0—1.25 V (vs. RHE)范围内进行CV测试.
3.1.Pt-Ni CNPs的形貌及结构表征
一维组装Pt-Ni CNPs独特的微观结构用TEM来表征, 如图1所示. 图1(a)清楚地表明, 制备得到的样品是纳米链状结构, 单根纳米链长度可达几百纳米. 从图中可以明显看出, 每个纳米链都包含直径约为3 nm的超细纳米线主干(图1(b)中箭头处所示)和由该主干串起的纳米颗粒结构. 这与传统的一维纳米球链有明显不同, 传统一维纳米球链一般由数个纳米球组成, 纳米球间彼此没有间隙, 而本工作中制备得到的纳米链的各组成单元(纳米颗粒结构)由纳米线连接, 这样能够将一维纳米线和零维小尺寸纳米颗粒完美地组合在一起, 更有利于催化反应过程中电子的传递以及在反应过程中提供更多的活性位点. 图1(b)是高放大倍数下单根Pt-Ni CNPs的TEM图像, 可以清楚地看到每个组成单元(小尺寸纳米颗粒)的平均直径约为10 nm, 与图1(a)插图中的统计结果一致. 图 1 (a) Pt-Ni CNPs的TEM图像, 插图是纳米颗粒的直径分布统计图; (b) 高放大倍数下单根Pt-Ni CNPs的TEM图像; (c) Pt-Ni CNPs的HAADF-STEM图像; (d)和(e)分别为(c)中Pt和Ni的EDS元素分布
图 1 (a) Pt-Ni CNPs的TEM图像, 插图是纳米颗粒的直径分布统计图; (b) 高放大倍数下单根Pt-Ni CNPs的TEM图像; (c) Pt-Ni CNPs的HAADF-STEM图像; (d)和(e)分别为(c)中Pt和Ni的EDS元素分布Figure1. (a) TEM image of Pt-Ni CNPs. Inset: graph of the diameter distribution of nanoparticles; (b) TEM image of a single Pt-Ni CNPs at a higher magnification; (c) HAADF-STEM images of Pt-Ni CNPs; (d) and (e) are EDS element distribution images of Pt and Ni in Pt-Ni CNPs corresponding to (c), respectively.
图1(c)是Pt-Ni CNPs的HAADF-STEM图像, 对应的EDS元素面扫图展示在图1(d) (Pt元素) 和图1(e) (Ni元素) 中. 从图中可以明显看出, Pt元素分布均匀, 而Ni的分布相对集中在中心, 这证明了材料具有Ni稀缺/Pt富集的表面, 这种表面元素分布能够使更多的Pt和外界电解液接触, 从而在催化过程中提供更多的活性位点, 有利于催化活性的提高.
图2(a)显示的是Pt-Ni CNPs的XRD图谱, 其中, 位于40°, 46°, 67°, 81°和86°的衍射峰分别对应于Pt标准卡片(JCPDS No. 04-0802)的(111), (200), (220), (311)和(222)平面, 属于典型的fcc结构. 与纯Pt相比, Pt-Ni CNPs的衍射峰向大角度有微小偏移, 这是由于引入金属Ni后, 合金化导致了Pt的晶格间距减小[33,34]. 图2(b)所示的SEM-EDS结果显示了Pt-Ni CNPs的元素组成, 从图中通过计算峰的面积比可以得出Pt/Ni原子比为77.2/22.8, 该结果与ICP-AES估计的Pt∶Ni = 76.5∶23.5一致. 为了进一步分析Pt和Ni的电子结构, 用XPS对样品进行了表征, 如图2(c)和图2(d)所示. Pt-Ni CNPs的XPS图谱由C的1 s峰在284.6 eV校准得到. 在图2(c)中, 位于74.64和71.34 eV的峰分别对应于Pt 4f5/2和Pt 4f7/2. 经过分析处理后发现, 这两个峰均由Pt0和Pt2+两组峰组成. 金属态Pt0的特征峰位于74.64 和71.24 eV, 与纯Pt的Pt 4f5/2和Pt 4f7/2峰(74.25和70.9 eV)相比[35,36], 结合能向正向偏移, 这主要是因为与Ni合金化后, Pt向Ni提供电子, 导致Pt原子上产生了更多的电子缺陷[37]. 而大量的Pt原子失去电子也会减弱O与Pt的结合能力, 从而暴露出更多的活性位点来提高催化性能[33]. 而另一组峰Pt2+ 4f5/2和Pt2+ 4f7/2被认为是表面Pt原子由于暴露在空气中形成了少量的Pt-O[9,38]. 类似方法分析Ni的2p峰, 如图2(d)所示, 金属态的Ni0 2p1/2和2p3/2分别位于870.24和852.74 eV, 氧化态的Nix+是因为样品表面部分Ni被氧化失去电子[39,40]. 同时图中可以看到在879.64和861.84 eV存在两个振荡峰, 这是由Nix+2p1/2和Nix+2p3/2导致. 此外, Ni0的特征峰信号较弱, 归因于XPS仅检测到样品表面上几纳米的元素信号, 而Ni在样品Pt-Ni CNPs的表面含量很少[33].
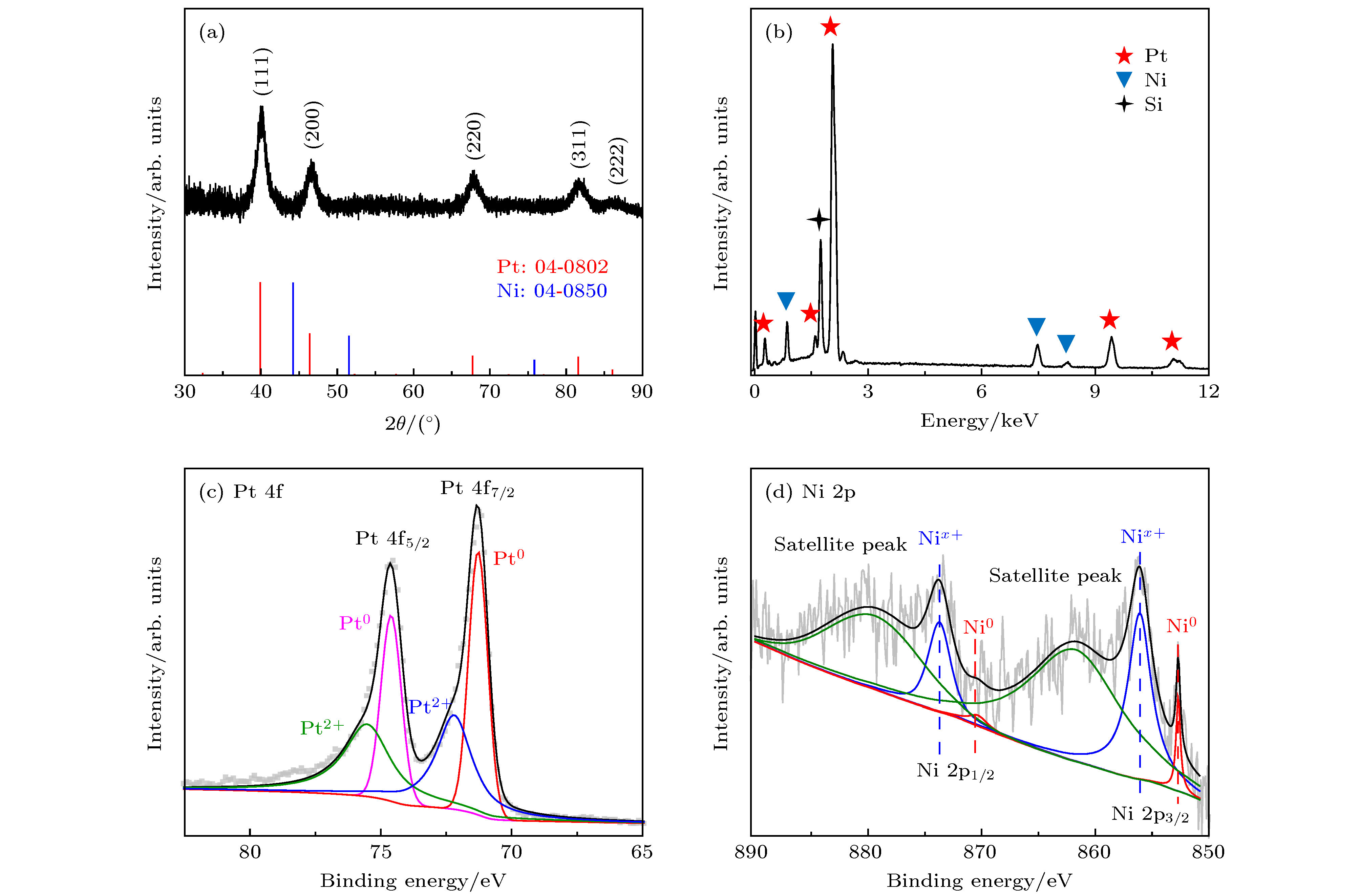 图 2 (a) Pt-Ni CNPs的 XRD 谱图及Pt 和 Ni 的标准卡片峰(分别对应红色和蓝色); (b) Pt-Ni CNPs的 EDS能谱图; (c)和(d)为Pt-Ni CNPs的XPS谱图, 分别对应Pt的4f峰和Ni的2p峰
图 2 (a) Pt-Ni CNPs的 XRD 谱图及Pt 和 Ni 的标准卡片峰(分别对应红色和蓝色); (b) Pt-Ni CNPs的 EDS能谱图; (c)和(d)为Pt-Ni CNPs的XPS谱图, 分别对应Pt的4f峰和Ni的2p峰Figure2. (a) XRD patterns of Pt-Ni CNPs and standard card peaks of Pt and Ni (corresponding to red and blue respectively); (b) EDS spectrum of Pt-Ni CNPs; (c) and (d) are XPS spectra of Pt-Ni CNPs, corresponding to the 4f peak of Pt and the 2p peak of Ni, respectively.
2
3.2.电催化性能测试及分析
图3(a)是在N2饱和的0.5 M H2SO4溶液中测得的Pt-Ni CNPs和商业Pt/C的CV曲线. 可以明显看出曲线可划分为四个区域: 其一是电压范围为0—0.35 V的氢区(Hupd, 氢的欠电位沉积区间), 上半部分是氢脱附曲线, 这个过程会使得Pt表面更好地暴露出来, 下半部分是氢吸附曲线; 其二是电压范围为0.35—0.55 V的双电层区; 其三是电压范围为0.55—0.95 V的氧脱附曲线; 最后是电压范围为0.95—1.25 V的氧吸附区[41]. 氢的吸附/脱附峰得到的催化剂电化学活性表面积(electrochemically activity surface area, ECSA)可以用来评估电极表面的催化活性[42]. 通过欠电位沉积的氢吸附[43], 可以计算得到Pt-Ni CNPs和商业Pt/C的ECSA分别为53.4和70.8 m2·gPt–1. 结果表明, Pt-Ni CNPs的ECSA略低于商业Pt/C, 归因于以下两点因素: i) ECSA与颗粒大小密切相关, 商业Pt/C具有更小的尺寸(2—5 nm); ii) Pt的Hupd行为对其化学环境非常敏感, 尤其是与Pt具有强电子相互作用的非贵过渡金属合金化时, 会改变氢与Pt之间的吸附强度[41]. 通常, 由于合金化抑制了样品对Hupd的吸附/脱附, 因此Hupd这种测试方法会略低估Pt基合金催化剂的ECSA[30].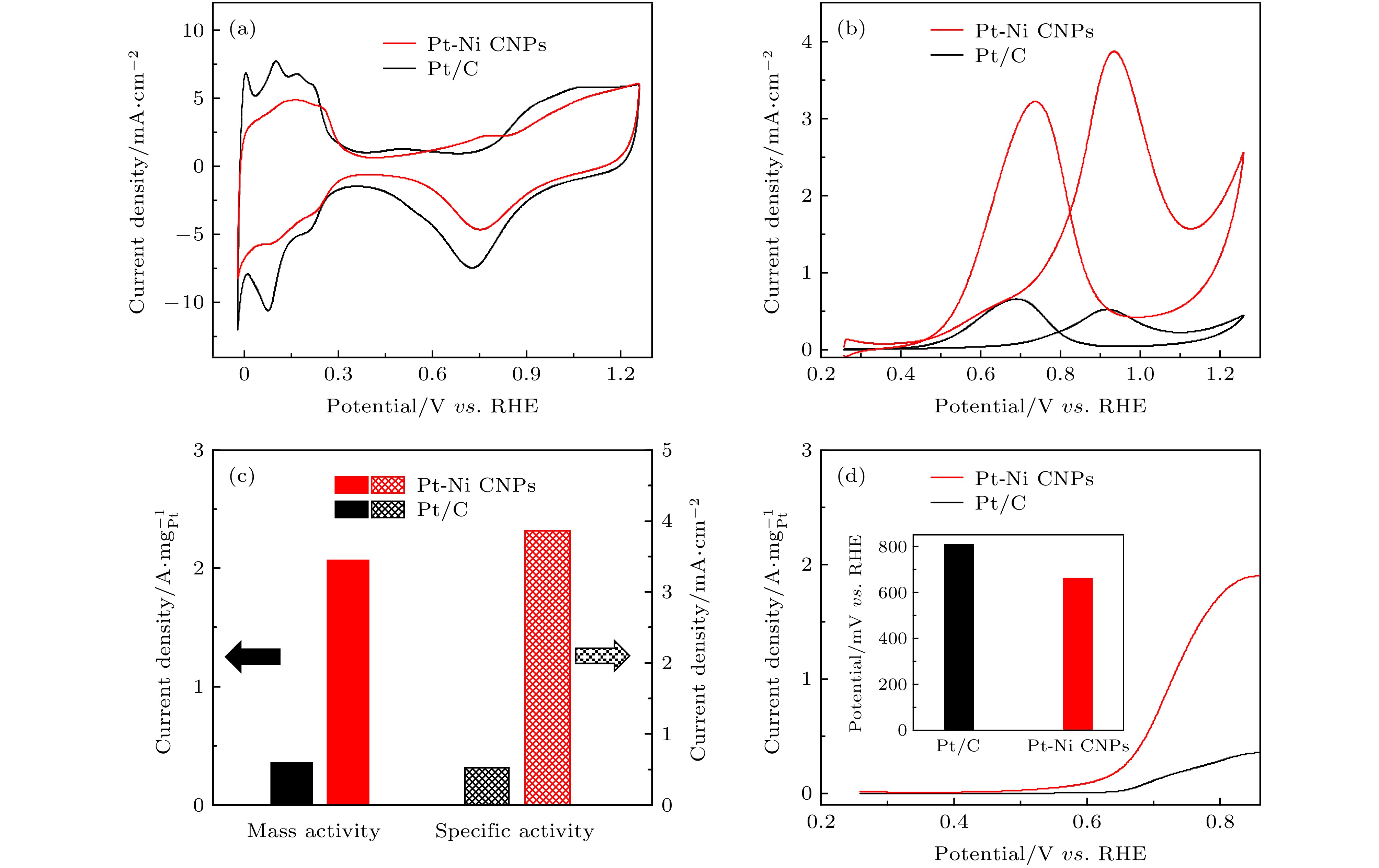 图 3 Pt-Ni CNPs (红色) 和商业Pt/C (黑色) 的MOR性能对比 (a), (b)两种催化剂的CV曲线, 分别是ECSA和MOR; (c) 两种样品相对应的质量活性和比活性; (d) 以5 mV/s的扫描速率测得的LSV曲线, 插图是固定电流密度所需提供的电位值
图 3 Pt-Ni CNPs (红色) 和商业Pt/C (黑色) 的MOR性能对比 (a), (b)两种催化剂的CV曲线, 分别是ECSA和MOR; (c) 两种样品相对应的质量活性和比活性; (d) 以5 mV/s的扫描速率测得的LSV曲线, 插图是固定电流密度所需提供的电位值Figure3. MOR performance comparison for Pt-Ni CNPs (red) and commercial Pt/C (black): (a) CV of the above catalysts for ECSAs; (b) CV of the above catalysts for MOR; (c) corresponding mass and specific activities of different catalysts for MOR; (d) LSV curves of the above electrocatalysts with a low scan rate of 5 mV/s. Inset:the potential required for fixed current density.
Pt-Ni CNPs和商业Pt/C的MOR测试是在0.5 M H2SO4和1 M CH3OH的混合溶液中进行的, 图3(b) 是经过ECSA归一化后的CV曲线图. 其中可以明显观察到两个分离良好的峰: 其一是在正扫描过程中, 在约0.93 V处出现了第一氧化峰, 是因为在酸性环境中催化剂能够将CH3OH一部分完全氧化为CO2, 另一部分氧化为CHO等中间产物; 其二是负扫描过程中, 在约0.73 V处出现的第二氧化峰, 这时催化剂将一次氧化中产生的中间产物完全氧化为CO2[31]. 第一氧化峰的高低反映了催化剂的催化能力, 由测试结果可知, Pt-Ni CNPs的第一氧化峰的峰值电流密度为3.86 mA·cm–2, 是商业Pt/C (0.51 mA·cm–2)的7.6倍. 为了能更加明显的地比较两种催化剂的催化活性, 通常可以用两种方法对第一氧化峰的数值进行标准化处理, 其一是用电极表面负载催化剂的电化学活性表面积归一化处理, 得到该催化剂的比活性, 比活性反映的是催化剂表面的固有反应活性. 其二是用电极表面负载Pt的质量归一化处理, 得到其质量活性, 质量活性反映的是催化剂材料的利用效率[20,44]. 如图3(c) 中所示, Pt-Ni CNPs的比活性(3.86 mA·cm–2)和质量活性(2.06 A·mgPt–1)分别是商业Pt/C(0.51 mA·cm–2和0.36 A·mgPt–1)的7.6倍和5.7倍. 图3(d)中所示的LSV曲线是在0.5 M H2SO4和1 M CH3OH的混合溶液中以5 mV /s的低扫描速率测量得到的. Pt-Ni CNPs和商业Pt/C的起始电位分别为0.54和0.65 V. Pt-Ni CNPs显著降低的起始电位能够明确表明在MOR中Pt-Ni CNPs具有增强的动力学行为, 更容易触发反应开始和去除未完全氧化所产生的中间产物[45,46]. 例如, 若想使氧化电流密度达到0.3 A·mgPt–1, Pt-Ni CNPs的电势要求仅为0.66 V, 比商业Pt/C(0.81 V)低150 mV(如图3(d)的插图所示). 显著的低触发电压需求意味着Pt-Ni CNPs更容易催化MOR[47].
众所周知, Pt基催化剂容易被催化反应过程中形成的中间产物CO吸附, 导致催化剂失去活性, 通常被称为催化剂中毒. 为了检测催化剂抗CO中毒的能力, 我们在CO饱和溶液中进行了CO溶出实验, 如图4(a)所示. 在CO溶解曲线中, 起始电位和峰值电位可以作为评估催化剂抗CO中毒能力的重要参数[48]. 如图4(a)所示, Pt-Ni CNPs的CO溶解曲线表现出较低的起始电位(0.64 V)和峰值电位(0.8 V), 而商业Pt/C对应的两个数值分别为0.78和0.88 V. 这意味着与商业Pt/C相比, 在催化反应过程中Pt-Ni CNPs表面仅需提供较低的电势即可将CO等有害中间产物完全氧化[49]. 这是因为Pt和Ni之间的协同作用能够更好地调控Pt的电子结构[50], 减弱Pt对CO的吸附能力[51], 从而增强催化剂的抗CO中毒能力.
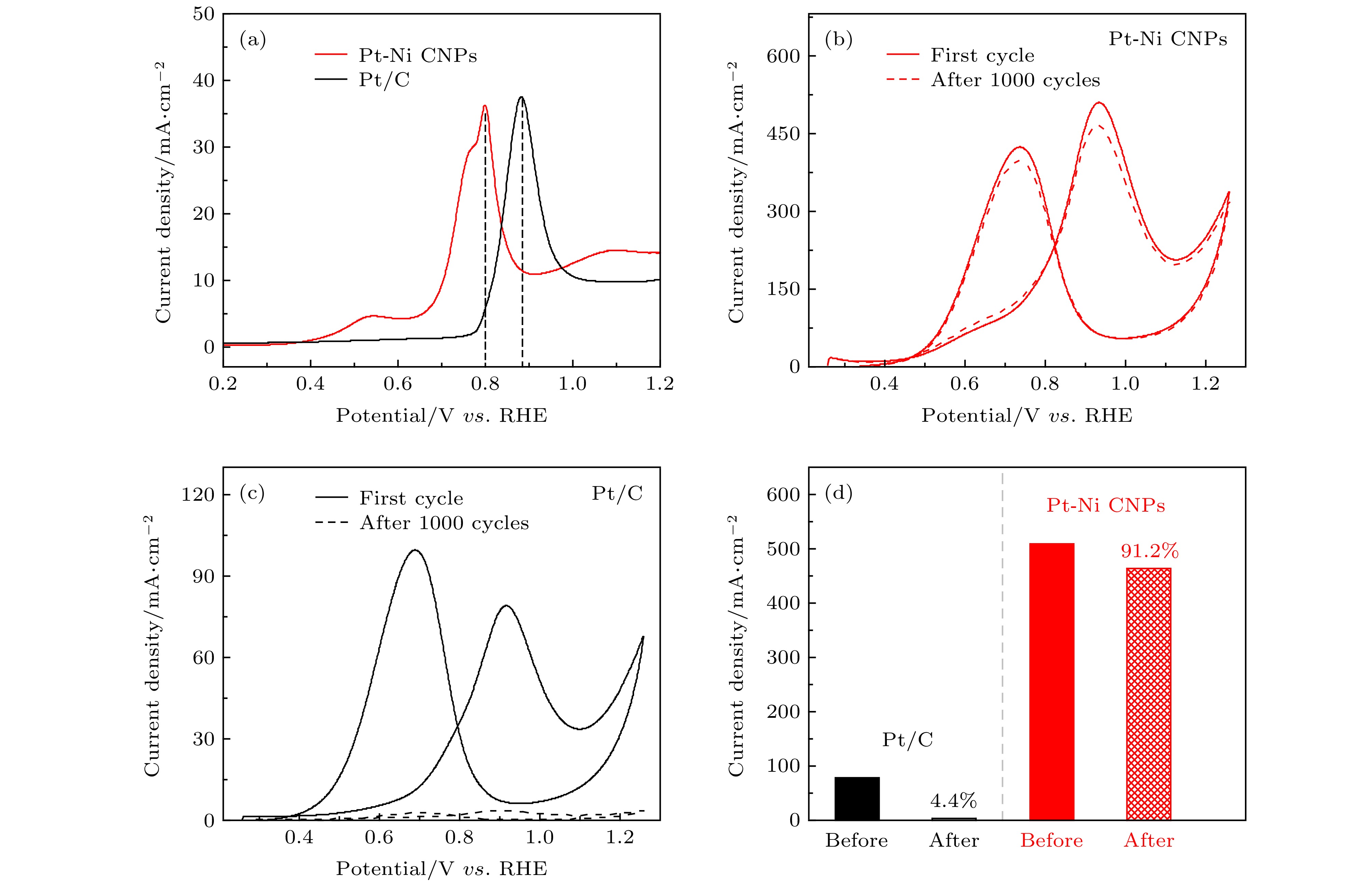 图 4 (a) CO溶解曲线; (b)和(c)分别为Pt-Ni CNPs (红色)和商业Pt/C (黑色)在0.5 M H2SO4和1 M CH3OH混合溶液中的稳定性测试: 实线为第一圈CV循环曲线, 虚线为第1000圈CV循环曲线; (d) 两种样品1000圈CV循环前后比活性对比
图 4 (a) CO溶解曲线; (b)和(c)分别为Pt-Ni CNPs (红色)和商业Pt/C (黑色)在0.5 M H2SO4和1 M CH3OH混合溶液中的稳定性测试: 实线为第一圈CV循环曲线, 虚线为第1000圈CV循环曲线; (d) 两种样品1000圈CV循环前后比活性对比Figure4. (a) The electrode area-normalized CO stripping curves; Stability test in 0.5 M H2SO4 and 1 M CH3OH solutions: (b) Pt-Ni CNPs (red) and (c) commercial Pt/C (black) with solid line as the first cycle and dashed line as the 1000th cycle; (d) specific activities of two samples before and after 1000 cycles.
为评估催化剂的电化学稳定性, 我们在酸性环境中对催化剂进行了连续1000圈的CV测试, 扫描速率为50 mV/s, 测试结果用电极面积进行归一化处理. 如图4(b)所示, 实线为Pt-Ni CNPs在催化MOR性能稳定后的CV曲线, 虚线是连续循环1000圈后的结果. 可以看出在经过长达1000圈的CV循环后, Pt-Ni CNPs的电流密度几乎没有损失, 保持了绝大部分的催化活性. 相反的是, 商业Pt/C在MOR反应循环1000圈之后, 其电流密度损失极其严重(如图4(c)所示). 为更直观对比两种催化剂的电化学稳定性, 图4(d)中分别给出了它们在连续1000圈CV循环前后的第一氧化峰值电流密度. 可以清楚地看出, 在经过1000次循环之后Pt-Ni CNPs保留了初始峰值电流密度的91.2%, 而商业Pt/C仅剩余初始电流密度的4.4%. 由上述结果分析可知, 与商业Pt/C相比, Pt-Ni CNPs具有显著的电化学稳定性, 这种优势意味着在对MOR催化过程中Pt-Ni CNPs具有更好的抗CO中毒能力, 其大部分的活性位点在催化过程中不会因为CO中毒而失活.
基于上述讨论, 与商业Pt/C相比, Pt-Ni CNPs增强的电催化活性和稳定性可归因于以下因素: 1) Pt-Ni CNPs独特的一维结构, 将一维纳米结构和零维小尺寸纳米颗粒的优势结合在了一起: a) Pt-Ni CNPs整体呈现出较长的一维链状结构(长度为几百纳米), 不仅有利于减小高指数晶面的表面能[32], 更能减弱催化过程中溶解和奥斯特瓦尔德(Ostwald)熟化对催化剂的影响[21,22], 有助于保持良好的稳定性; 同时该链状结构固有的各向异性和高的导电率, 将会更加有利于电子的转移和传递[29,52]; b) 组成Pt-Ni CNPs的零维纳米颗粒具有较小的尺寸, 能够增大催化剂的电化学比表面积, 向外界提供更多的活性位点, 提高催化活性. 2) Pt-Ni CNPs具有Ni稀缺/ Pt富集的表面: 其一, 这增大了催化剂中Pt与反应物的接触概率, 有利于增加催化剂的活性位点; 其二, 富Pt表面可以有效地保护有益的Ni原子在长期的电催化过程中免于溶解, 有利于保持催化剂优异的活性和稳定性[53,54].
