全文HTML
--> --> -->Alq3有两种异构体[16]. 本文选用B3LYP/6-31G*方法和基组对8-羟基喹啉铝的经式结构的基态分子进行优化, 计算出该分子的红外与拉曼光谱, 分析了分子轨道成分. 继而采用含时密度泛函理论(TD-DFT)方法研究了mer-Alq3分子的电子光谱和前26个激发态的激发特性, 以此来分析该分子发光的电子跃迁机理, 从而对提高该分子的发光效率, 调控发光范围, 以及实验合成提供一定的理论指导.




从(1)式—(4)式可知, 可根据空穴与电子质心之间的距离、分布之间重叠的函数、总体平均分布广度、分离程度共同表征分子激发特征, 判断激发类型, 并进一步分析原子或片段对电子激发的贡献.
本文采用B3LYP方法和6-31G*基组对mer-Alq3的分子结构进行计算, 考虑方法和基组带来的误差, 将计算的频率乘以校正因子0.9613[19]进行修正. 优化时收敛指标为默认值, 相邻两次迭代的均方根密度矩阵元差值小于10–8, 迭代次数为128, 最大的相邻两次迭代的密度矩阵元绝对差值小于10–6, 相邻两次迭代的体系能量绝对差值小于10–6 Hartree.
3.1.基态几何结构
采用B3LYP/6-31G*方法和基组对mer-Alq3分子进行优化, 其振动频率无虚频, 即为稳定构型, 如图1所示. 结果还显示该分子对称性为C1对称性, 同文献[20]一致. 并且优化后的结构与实验[21]相符, 主要键长列于表1. 图 1 mer-Alq3的分子结构
图 1 mer-Alq3的分子结构Figure1. Structure of the mer-Alq3 molecule.
| Bond | B3LYP/6-31G*/? | Experimental results/?[21] |
| Al-Na | 2.08377 | 2.0502 |
| Al-Nb | 2.12565 | 2.0872 |
| Al-Nc | 2.06431 | 2.0172 |
| Al-Oa | 1.85545 | 1.8502 |
| Al-Ob | 1.88106 | 1.8602 |
| Al-Oc | 1.88398 | 1.8572 |
表1mer-Alq3分子的键长
Table1.Bond lengths of the mer-Alq3.
2
3.2.红外光谱与拉曼光谱
红外光谱是红外光子与分子振动、转动的量子化能级共振产生吸收而形成的特征吸收光谱曲线. 计算得到的红外光谱如图2所示. mer-Alq3的最强吸收峰、次强峰、第三强峰分别位于1512, 1422, 1550 cm–1处. 实验值[22]分别为1468, 1383, 1499 cm–1. 对比可知, 谱峰位置分别向高频移动了44, 39, 51 cm–1. 第一强峰归属于C—N与C—C的伸缩振动, 伴随C—H平面摇摆振动. 次强峰归属于C—C的伸缩振动, 伴随C—H的平面摇摆振动. 第三强峰归属于C—C伸缩振动, C—H平面摇摆振动. 红外光谱吸收峰所对应的振动模式指认列于表2中. 图 2 mer-Alq3分子的红外光谱
图 2 mer-Alq3分子的红外光谱Figure2. Infrared absorption spectrum of mer-Alq3.
| Vexperiment/cm–1 | Vtheory/cm–1 | Vibration analysis | Vexperiment/cm–1 | Vtheory/cm–1 | Vibration analysis |
| 398 | 408 | 分子骨架扭曲变形 | 1228 | 1268 | C—N伸缩振动, C—H平面摇摆振动, 剪式振动 |
| 416 | 422 | 分子骨架扭曲变形, 环1环2环5 环6上C—H平面摇摆振动, 环3环4上C—H扭曲振动 | 1280 | 1334 | C—O, C—C伸缩振动, C—H平面摇摆振动 |
| 457 | 470 | C—H扭曲振动 | 1328 | 1376 | C—H平面摇摆振动, 剪式振动 |
| 548 | 554 | Al-O50伸缩振动, 环1环2呼吸振动 | 1383 | 1422 | C—C伸缩振动, C—H平面摇摆振动 |
| 642 | 662 | Al-O50伸缩振动, C—H扭曲振动 | 1424 | 1438 | C—N、C—C伸缩振动, C—H 平面摇摆振动, 剪式振动 |
| 746 | 768 | Al-O面外弯曲振动, 环3环4呼吸 振动 | 1468 | 1512 | C—N伸缩振动, C—C伸缩振动, C—H平面摇摆振动 |
| 787 | 796 | C—H扭曲振动 | 1499 | 1550 | C—C伸缩振动, C—H平面摇摆振动 |
| 803 | 820 | 苯环和吡啶环变形振动 | 1579 | 1636 | C—N伸缩振动, C—C伸缩振动, C—H平面摇摆振动, 剪式振动 |
| 823 | 836 | C—H面外摇摆振动 | 1606 | 1658 | C—N伸缩振动, C—C伸缩振动, C—H平面摇摆振动, 剪式振动 |
| 1114 | 1140 | C—H剪式振动 | 3039 | 3202 | 苯环上C—H伸缩振动 |
表2mer-Alq3分子中部分振动模式指认
Table2.Identification of partial vibration modes of mer-Alq3.
图2与文献[22]谱图进行比较可知, 理论计算和实验测量的谱线整体比较一致, 但理论计算的谱线整体有一定程度蓝移. 本文计算的是单个气相分子, 但在实际晶体中存在分子间相互作用, 导致单个分子振转受范德瓦斯力作用而被轻微地束缚, 致使晶体光谱出现红移.
拉曼光谱作为红外光谱的重要补充, 可通过拉曼光谱测定分子的基态参数, 得到分子振动能级与转动能级结构的信息. 计算得到的拉曼光谱如图3所示. 拉曼振动主要分布在2个频区: 1330—400 cm–1(指纹区)、4000—1330 cm–1(特征频率区). 最强拉曼峰位于3216 cm–1处, 归属于C—H伸缩振动. 次强峰位于1422 cm–1, 实验值[23]为1393 cm–1, 向高频移动了29 cm–1, 归属于C—O, C—C伸缩振动, 伴随着C—H平面摇摆振动、剪式振动. 第三强峰在1438 cm–1处, 归属于C—N, C—C伸缩振动, 伴随着C—H平面摇摆振动、剪式振动. 将图3与文献[23]比较可以看出, 在波数小于1700 cm–1时, 理论计算和实验测量的谱线整体比较一致, 理论计算的拉曼光谱整体向短波方向有轻微的位移, 可能原因与上述红外光谱蓝移现象类似. 分子的拉曼峰的部分振动模式指认列于表3中.
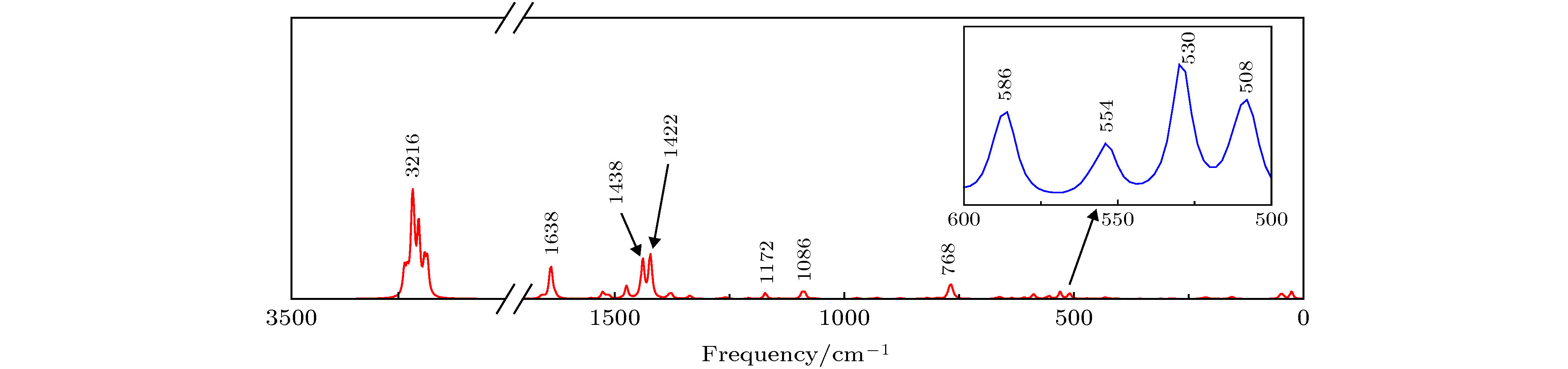 图 3 mer-Alq3的拉曼光谱
图 3 mer-Alq3的拉曼光谱Figure3. Raman spectrum of mer-Alq3.
| Vexperiment/cm–1 | Vtheory/cm–1 | Vibration analysis |
| 507 | 508 | Al—O扭曲振动, 苯环和吡啶环变形振动 |
| 529 | 530 | Al—O伸缩振动, 苯环和吡啶环呼吸振动 |
| 545 | 554 | Al—O伸缩振动, 苯环和吡啶环呼吸振动 |
| 581 | 586 | Al—O扭曲振动, 苯环和吡啶环变形振动 |
| 760 | 768 | Al—O伸缩振动, 苯环和吡啶环呼吸振动 |
| 1062 | 1086 | C—H平面摇摆振动, 剪式振动 |
| 1177 | 1172 | C—H平面摇摆振动, 剪式振动 |
| 1393 | 1422 | C—O, C—C伸缩振动, C—H平面摇摆振动, 剪式振动 |
| — | 1438 | C—N、C—C伸缩振动, C—H平面摇摆振动, 剪式振动 |
| 1593 | 1638 | C—C伸缩振动, C—H平面摇摆振动, 剪式振动 |
| — | 3216 | C—H伸缩振动 |
表3mer-Alq3分子中部分振动模式指认
Table3.Identification of partial vibration modes of mer-Alq3.
2
3.3.分子前线轨道与激发特性
电致发光器件中有机金属配合物的发光效率取决于电子跃迁和能量转移等机理, 配合物前线分子轨道上的电子受核束缚最小, 与电子传输性能关系密切, 所以研究其前线分子轨道非常重要[24]. 为探讨发光金属配合物mer-Alq3分子的电子跃迁实质, 以B3LYP/6-31G*优化得到的稳定几何构型为基准, 采用Hirshfeld方法[25]分析原子对前线分子轨道的贡献. 由表4可知, HOMO的电子云主要集中在a配体的苯酚环, 其中O占比19.25%, 苯占比57.52%. HOMO-1主要分布在c配体的苯酚环, 其中O占比17.19%, 苯占比58.30%. HOMO-2主要分布在b配体的苯酚环, 其中O占比20.36%, 苯占比64.59%. LUMO的电子云主要集中在b配体的吡啶环, 占比64.81%. LUMO+1主要分布在c配体的吡啶环, 占比47.98%. LUMO+2主要分布在a配体的吡啶环, 占比56.03%. 图4采用0.05等值面绘制. 说明有机金属配合物的配体对前线分子轨道的贡献是主要的, 中心离子贡献很小, 电子跃迁主要是含氧的苯酚环和含氮的吡啶环的电荷转移, 是一种配体发光的配合物.| 分子轨道 | 能级 | Al | a | b | c | ||||||||
| O | 苯 | 吡啶 | O | 苯 | 吡啶 | O | 苯 | 吡啶 | |||||
| H-2 | –0.19584 | 1.51 | 0.16 | 0.85 | 0.93 | 20.36 | 64.59 | 19.94 | 0.24 | 0.74 | 1.15 | ||
| H-1 | –0.19204 | 1.57 | 2.05 | 8.50 | 2.56 | 0.36 | 1.18 | 1.17 | 17.19 | 58.30 | 17.72 | ||
| H | –0.18397 | 1.47 | 19.25 | 57.52 | 17.84 | 0.36 | 0.20 | 0.24 | 3.58 | 7.26 | 2.42 | ||
| L | –0.06363 | 1.60 | 0.06 | 0.18 | 0.91 | 1.66 | 25.28 | 64.81 | 0.29 | 4.55 | 12.18 | ||
| L+1 | –0.05496 | 1.36 | 0.62 | 8.41 | 20.90 | 0.24 | 3.24 | 7.25 | 1.33 | 19.64 | 47.98 | ||
| L+2 | –0.05218 | 1.28 | 1.72 | 21.57 | 56.03 | 0.54 | 1.83 | 5.13 | 0.81 | 6.32 | 16.59 |
表4mer-Alq3的前线分子轨道能级(单位: arb.units)及分布(单位: %)
Table4.Frontier molecular orbital energy levels (in arb.units) and distribution (in %) of mer-Alq3.
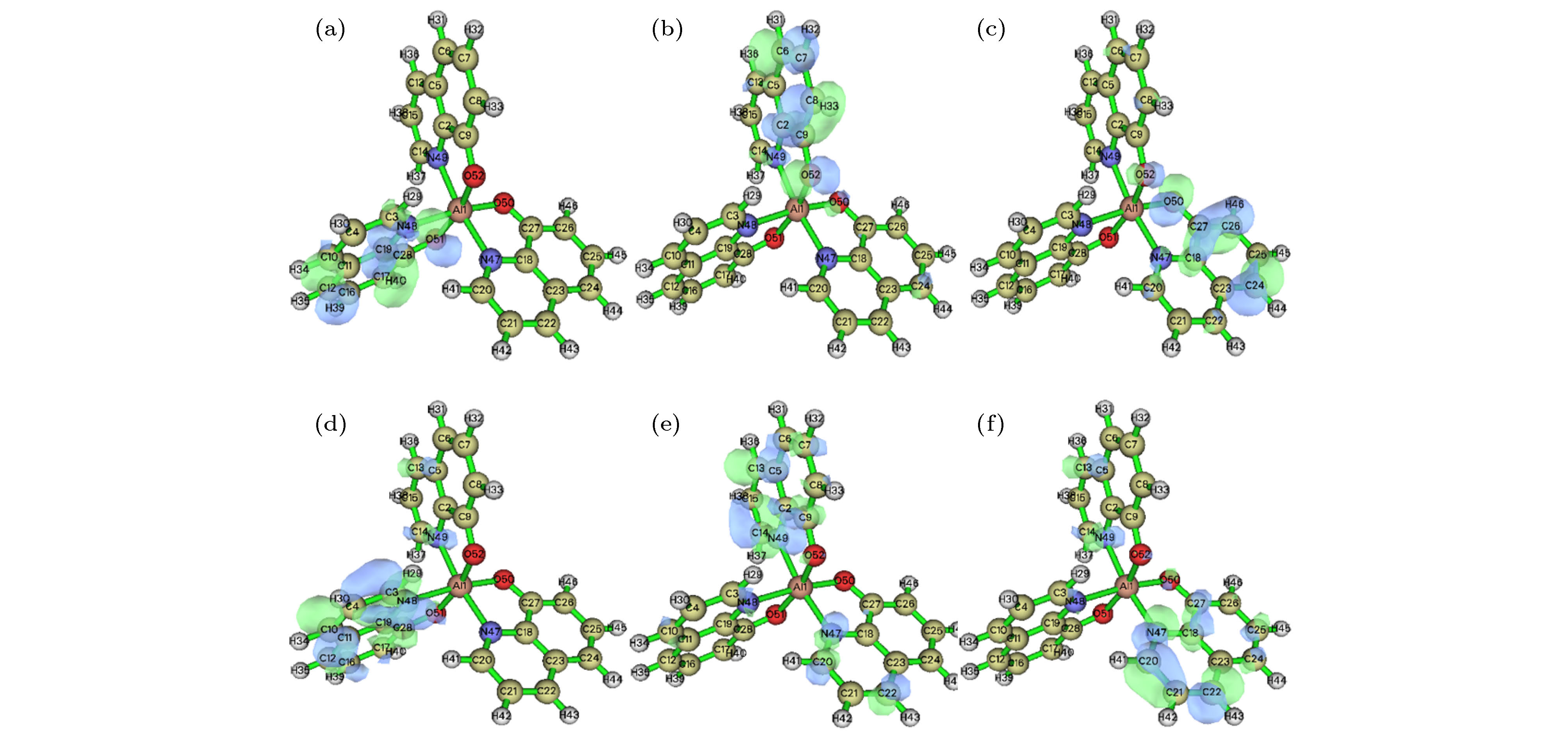 图 4 mer-Alq3前线分子轨道分布图 (a) HOMO-2轨道分布图; (b) HOMO-1轨道分布图; (c) HOMO轨道分布图; (d) LUMO轨道分布图; (e) LUMO+1轨道分布图; (f) LUMO+2轨道分布图
图 4 mer-Alq3前线分子轨道分布图 (a) HOMO-2轨道分布图; (b) HOMO-1轨道分布图; (c) HOMO轨道分布图; (d) LUMO轨道分布图; (e) LUMO+1轨道分布图; (f) LUMO+2轨道分布图Figure4. Frontier molecular orbits of mer-Alq3: (a) HOMO-2 distribution; (b) HOMO-1 distribution; (c) HOMO distribution; (d) LUMO distribution; (e) LUMO+1 distribution; (f) LUMO+2 distribution.
采用含时密度泛函理论(TD-DFT) B3LYP/6-31G*方法和基组计算了mer-Alq3分子的紫外-可见吸收光谱. 由图5可知, 四个最强吸收峰中427.15 和417.31 nm位于可见光区, 304.03 和302.87 nm位于紫外光区. 本节分析这四个贡献较大的激发态性质. 主要吸收峰跃迁波长、振子强度、相应的跃迁方式、跃迁轨道贡献率、跃迁能量分别列于表5.
 图 5 mer-Alq3分子的紫外-可见吸收光谱
图 5 mer-Alq3分子的紫外-可见吸收光谱Figure5. UV-Vis absorption spectrum of mer-Alq3.
| Excited state | λ/nm | f | Transition nature (contribution > 10%) | Transition energy/eV |
| 2 | 427.15 | 0.0672 | 119→121 (45.9956%); 119→122 (23.0683%); 118→120 (21.1263%) | 2.9026 |
| 4 | 417.31 | 0.0425 | 117→120 (88.1022%) | 2.9710 |
| 11 | 304.03 | 0.0151 | 119→124 (38.2445%); 119→125 (23.0208%) | 4.0781 |
| 12 | 302.87 | 0.0214 | 117→123 (66.2078%); 114→120 (20.1638%) | 4.0937 |
表5mer-Alq3分子的电子激发分析表
Table5.The analysis of electron excitation of mer-Alq3.
由表5可知, 以基态到第2激发态的跃迁为例, 119号轨道(HOMO)向121号轨道(LUMO+1)跃迁的贡献率为46%, 与119号轨道(HOMO)到122号轨道(LUMO+2)和118号轨道(HOMO-1)到120号轨道(LUMO)的贡献率之和44.19%相当. 同理, 由基态向第11, 12激发态跃迁中, 均表现出多组轨道跃迁共同的贡献. 由上述可知用单一的轨道跃迁模式描述电子跃迁性质不能更好地诠释电子激发特性. 因此本文选用空穴-电子分析法[26,27]研究电子激发. 从(1)式—(4)式可知, 表征电子激发性质可根据D, Sr, H, t四个参数来描述. 体系的4个激发态的D, Sr, H, t数值列于表6. 同时绘制了空穴-电子图、Chole-Cele图[17]以及Sr函数图, 如图6所示, 绿色和蓝色分别表示电子和空穴分布, 绘制均采用0.001等值面.
| D/? | Sr/arb.units | H/? | t/? | |
| S0 → S2 | 0.18 | 0.61 | 3.57 | 0.12 |
| S0 → S4 | 0.99 | 0.59 | 2.95 | 0.41 |
| S0 → S11 | 0.88 | 0.79 | 3.84 | –1.38 |
| S0 → S14 | 0.68 | 0.43 | 3.47 | 2.00 |
表6mer-Alq3分子的激发态参数
Table6.Excited state parameters of mer-Alq3.
 图 6 mer-Alq3的空穴-电子、Chole-Cele、Sr示意图 (a)?(c) S2的空穴-电子, Chole-Cele, Sr图; (d)?(f) S4的空穴-电子, Chole-Cele, Sr图; (g)?(i) S11的空穴-电子, Chole-Cele, Sr图; (j)?(l) S14的空穴-电子, Chole-Cele, Sr图
图 6 mer-Alq3的空穴-电子、Chole-Cele、Sr示意图 (a)?(c) S2的空穴-电子, Chole-Cele, Sr图; (d)?(f) S4的空穴-电子, Chole-Cele, Sr图; (g)?(i) S11的空穴-电子, Chole-Cele, Sr图; (j)?(l) S14的空穴-电子, Chole-Cele, Sr图Figure6. Electron-hole, Chole-Cele and Sr
从表6可知, 对于S0 → S2激发, D为0.18?, 小于C—C键的核间距, 说明空穴和电子质心距离较近. Sr数值为0.61, 表明空穴和电子超过一半的分布特征重合. t的数值是0.12, 大于0, 意味着空穴和电子分布有细微的分离. 结合表6中各项参数以及图6中Sr函数图和空穴-电子图可知, S0 → S2激发类型为局域激发. 根据空穴-电子图可得, 空穴由配体a和c上苯环的π轨道和氧原子的孤对电子轨道构成. 电子在吡啶环的平面上有个节面, 所以电子分布由π*轨道构成, 由上述可知S0→S2是苯环到吡啶环方向上的π-π*和氧原子到吡啶环方向上的n-π*局域激发(LE)的叠加.
S0→S4激发中, D, Sr, H, t值分别为0.99, 0.59, 2.95, 0.41. 表明空穴和电子质心距离较远, 总体平均分布广度小, 空穴和电子重叠程度超过一半, 但空穴和电子分离程度较大. 结合表6与图6中空穴-电子图可以看出, S0→S4激发的空穴和电子主体都分布在配体b上, 空穴是由苯环上的π轨道构成的, 电子在吡啶环的平面上有个节面, 因此电子分布是由π*轨道构成的. 即S0 → S4为苯环到吡啶环方向上的π-π*局域激发.
S0 → S11激发, Sr和H数值都是4个激发态中最大的, 空穴和电子的分布范围都比较广. t的数值为–1.38, 为负值, 表明空穴和电子分布没有显著分离. 空穴是由配体a和c上苯环的π轨道和氧原子的孤对电子轨道构成, 电子是由碳原子的孤对电子轨道构成. Chole-Cele图中蓝色和绿色等值面(分别对应空穴和电子的质心位置)几乎重合. 所以S0→S11激发是苯环到碳原子方向上的π-n和氧原子到碳原子方向上的n-n局域激发的叠加.
在S0 → S14激发中, Sr和t指数分别为0.43, 2.00, 空穴和电子分布有显著的分离. Chole-Cele图中蓝色和绿色等值面的中心相距较远, S0 → S14激发属于电荷转移激发. 根据空穴-电子图可以看出, 空穴由配体a和c上苯环的π轨道和氧原子的孤对电子轨道构成, 电子是由吡啶环上的π*轨道构成的, 因此, S0 → S14激发归属于苯环到吡啶环方向上的π-π*和氧原子到吡啶环方向上的n-π*电荷转移激发(CT)的叠加.
综上所述, 在四个主要激发态中, S0 → S2是苯环到吡啶环方向上的π-π*和氧原子到吡啶环方向上的n-π*局域激发的叠加. 苯环到吡啶环方向上的π-π*局域激发为S0 → S4激发. 苯环到碳原子方向上的π-n和氧原子到碳原子方向上的n-n局域激发的叠加由S0 → S11激发. S0 → S14激发由苯环到吡啶环方向上的π-π*和氧原子到吡啶环方向上的n-π*电荷转移激发的叠加所贡献. 这表明, mer-Alq3分子作为发光材料, 电子激发跃迁可看成空穴-电子对来完成, 不同电子激发态的激发有着不同激发性质, 本节对分子的发光机理做了系统的研究, 对进一步研究该材料的发光机制, 效率, 甚至调控发光范围有着重要意义.
采用TD-DFT计算了mer-Alq3分子的电子光谱, 结果表明: S0 → S2归属于苯环到吡啶环方向上的π-π*和氧原子到吡啶环方向上的n-π*局域激发的叠加; S0 → S4归属于苯环到吡啶环方向上的π-π*局域激发; S0 → S11归属于苯环到碳原子方向上的π-n和氧原子到碳原子方向上的n-n局域激发的叠加; S0 → S14归属于苯环到吡啶环方向上的π-π*和氧原子到吡啶环方向上的n-π*电荷转移激发的叠加.
