摘要: 由维里定理导出的适用于均匀系统的平衡态压力张量表达式可以分成两部分: 动压力张量和位形压力张量. 人们进而对平衡的非均匀系统进行物理分析得到了局部平均压力张量表达式. 本文用更为简洁的方法推导出这一表达式. 给出以原子直径为长度单位的局部平均尺寸
L * > 8条件下均匀流体系统平均位形压力中的三部分贡献项(体贡献项、面贡献项和线贡献项)与
L * 的理论关系式(含有待定参数); 以氩原子气体为例, 在温度180 K、原子数密度0.8下, 对原子间采用林纳德-琼斯势进行了分子动力学模拟, 给出了0.4 ≤
L * ≤ 17条件下三项贡献及总位形压力的模拟曲线, 确定了
L * > 8条件下理论关系式中的待定系数, 并得到在
L * > 2时, 随着
L * 的增大, 体贡献项从正压单调下降并趋于负的总位形压力, 面贡献项和线贡献项都单调上升并趋于零, 但线贡献项趋向零最快. 从物理上解释了小尺寸
L * 下各项行为的复杂特点. 得出
L * 足够大, 才可以忽略面贡献项和线贡献项, 而在纳米尺度下, 忽略面贡献项和线贡献项, 也就是忽略边界效应会给计算带来明显的误差. 最后通过分子动力学模拟得出位形压力随着温度的升高而升高. 这些结论对于压力张量的分子动力学模拟计算时选项的最优化是有意义的.
关键词: 维里定理 /
分子动力学模拟 /
表面张力 /
局部压力张量 /
位形压力 English Abstract On the derivation of local mean pressure tensor for nonuniform systems and the analysis of uniform fluid Cui Shu-Wen 1,2 ,Liu Wei-Wei 1 ,Zhu Ru-Zeng 2,3 ,Qian Ping 4 1.Department of Physics and Information Engineering, Cangzhou Normal University, Cangzhou 061001, China Fund Project: Project supported by the National Key Research and Development Program of China (Grant No. 2016YFB0700500), the Key Sciencific Studies Program of Hebei Province Higher Education Institute, China (Grant No. ZD2018301), the Key Research and Development Program of Hebei Province, China (Grant No. 18211233), and the Cangzhou National Science Foundation, China (Grant No. 177000001).Received Date: 14 December 2018Accepted Date: 06 January 2019Available Online: 06 June 2019Published Online: 05 August 2019Abstract: The expression of equilibrium pressure tensor for uniform systems derived from Virial theorem can be divided into two parts: dynamic pressure tensor and configuration pressure tensor. The local mean pressure tensor is obtained by physical analysis of the equilibrium non-uniform system. In this paper, this expression is derived in a more concise way. The theoretical relationship, with undetermined parameters, between the three parts (the volume contribution, the surface contribution and the line contribution) of the average configuration pressure of a homogeneous fluid system and the local average size L * with the atomic diameter as length unit greater than 8 is given. Taking argon as an example, molecular dynamics simulation is carried out by using the Lennard-Jones potential between atoms at temperature 180 K and density 0.8 (atomic diameter)–3 . The simulation curves of configuration pressure and its three parts are given, and the undetermined coefficients in the theoretical relationship under the condition of L * > 8 are determined. It is found that for L * > 2, With the increase of L * , the volume contribution decreases monotonously from the positive pressure to the negative total configuration pressure, while the surface contribution and the line contribution both increase monotonously and tend to zero, but the line contribution tends to zero fastest. The complex characteristics of various behaviors are explained physically. It is concluded that the surface contribution and the line contribution can be neglected only if L * are large enough. In nanoscale, the neglect of the surface contribution and the line contribution, i.e., the neglect of the boundary effect, will bring obvious errors to the calculation. Finally, molecular dynamics simulation shows that the configuration pressure increases with the increase of temperature. These conclusions are significant for optimizing the selection of pressure tensor in molecular dynamics simulation.Keywords: Virial theorem /molecular dynamics simulation /surface tension /local pressure tensor /configuration pressure 全文HTML --> --> --> 1.引 言 对于平衡均匀无外场系统, Clausius[1 ] 和Maxwell[2 ] 基于维里定理, 给出了压力公式: 对于体积V 中包含N 个粒子的宏观体系, 平衡后的压力为i , j 表示粒子; ${m_i}$ 是粒子i 的质量; ${v_i}$ 是粒子i 的速度; $r_{ij}$ 是i , j 之间距离; ${f_{ij}}$ 表示i , j 之间的作用力, 吸引力为正; 〈 〉表示系综平均, 也即时间平均. (1 )式包含两部分, 第一项是原子运动所贡献的压力, 称为动压力, 第二项源于原子之间的相互作用力, 称为位形压力.${{{\sigma}} ^{\alpha,\beta }}({{r}})$ 需要定义, 使其满足连续介质的动量方程J 是动量密度, n 是粒子数密度, ${\phi ^{\rm ext}}$ 是外场势. Kirkwood和Buff[3 ] 最早就对势情况提出空间点R [4 ] 给予更简洁的表述: 作用在点R ${\rm{d}}A$ 上的力等于所有连线通过该面积元的分子对之间作用力之和, 加上由于分子通过面积元${\rm{d}}A$ 的运动在单位时间内交换的动量. Harasima[5 ] 最早认识到满足连续介质动量方程(2 )式的应力张量不唯一, 并提出了另一种针对液体表面层构造应力张量的方法. Schofield和Henderson[6 ] 证明将Irving和Kirkwood定义中的分子对之间的连线改为曲线, 其他不变, 也是一种应力张量的正确构成方法, 他们还给出了适合于多体势情况的应力张量的构成方法, 使Irving和Kirkwood (IK)方法和Harasima方法成为他们的特例. 显然, 对于对势情况而言, IK方法是最自然和最简单的, 因而也是后来对势情况下, 计算局部平均应力张量许多方案的基础[7 ,8 ] .N 个粒子组成的非均匀系统, 从Irving和Kirkwood的定义很容易得到作为空间点(R i , j 表示粒子; $\alpha $ , $\beta $ 表示方向; ${m_i}$ 是粒子i 的质量; $p_i^\alpha $ , $p_i^\beta $ 分别表示i 粒子的动量在$\alpha $ , $\beta $ 方向的分量; $r_{ij}^\beta $ 表示矢量${{{r}}_j} - {{{r}}_i}$ 在$\beta $ 方向上的分量; $\phi ({r_{ij}})$ 表示粒子i 与粒子j 之间的相互作用势.3 )式的理论意义是显然的. 但是由于其中含有δ函数, 不能直接用于实际测量和分子动力学模拟, 所以被称为微观压力张量[9 ] . Cormier等[9 ] 采用傅里叶变换方法得到了(3 )式的局部平均形式, 即对于由N 个粒子组成的非均匀流体中体积为V 的局部小区域, 平均压力张量为$v_i^\alpha $ , $v_i^\beta $ 表示i 粒子的速度在$\alpha $ , $\beta $ 方向的分量; $f_{ij}^\alpha $ 表示由j 施加在i 上的作用力矢量在$\alpha $ 方向的分量; 当粒子i 在平均体积内时${\varLambda _i}(V)$ 是1, 否则为0; ${l_{ij}}(V)$ 的范围是$0 \leqslant {l_{ij}} \leqslant 1$ , 它表示i -j 连线在体积V 中那部分长度的分数; ${l_{ij}}(V) = {l_{ji}}(V)$ ; 求和指标i , j 遍及整个流体而不仅仅是V .1 )和(4 )式可知, 就压力而言, 它们表示的都是体积V 中的平均值, 区别在于前者的V 是被刚性边界隔离着的均匀系统的总体积, 而后者的V 只是均匀或不均匀系统中的一小块的体积, 所以两者明显不是等价的. 一些研究者就非均匀系统中如果用前者代替后者将会引起的显著误差进行了讨论[10 -12 ] .4 )式适用于固体、液体、气体等各种系统, 因此应用价值十分广阔. 例如Li等[13 ] 采用巨正则蒙特卡罗方法模拟了超临界Lennard-Jones流体在纳米狭缝中的吸附行为, 他们在(4 )式中加进了壁-液势的影响项, 这也相当于对(1 )式做了修正, 获得了吸附流体的压力. 对于局部压力在原子量级的计算, 一直是人们研究的热点, 例如Torres-Sánchez等[14 ] 以及Chen[15 ] 对此进行了深入的研究. Yu和Jin[16 ] 用硬核双Yukawa流体混合物模型研究胶体的热力学和结构特性时, 在(4 )式的基础上计算了胶体体系的主体相压力.4 ). 鉴于在应用分子动力学计算(4 )式时需要考虑计算耗时的最优化问题, 但尚未见有关报道. 作为一个最简例子, 本文将给出均匀流体系统在以原子直径为长度单位的局部平均尺寸${L^*}$ 较大条件下, 局部平均位形压力中的三部分贡献项(体贡献项、面贡献项和线贡献项)与平均尺寸的关系式(含有待定参数), 这是计算最优化方案的依据; 以氩原子气体系统为例, 用分子动力学模拟方法确定待定参数, 并给出位形压力三部分贡献项在大尺寸和小尺寸$L^*$ 下各项行为的特点及温度的影响. 这将为压力张量的分子动力学模拟计算时选项的最优化方法提供一个范例.2.非均匀系统局部平均压力张量两种形式的简洁推导 22.1.第一种平均形式的推导 2.1.第一种平均形式的推导 第一种平均形式就是(4 )式. 推导如下: 将(3 )式对局部小体积V 求平均5 )式中6 )式代入(5 )式即可以得到(4 )式. (4 )式包含两部分: 第一部分是粒子的运动对压力的贡献, 称为动压力, 用$p_{{\rm{dong}}}^{\alpha \beta }$ 表示; 第二部分来源于粒子之间的相互作用, 称为位形压力, 用$p_{\rm{c}}^{\alpha \beta }$ 表示.7 )—(9 )式可以简化为$\left\langle {N} \right\rangle $ 为体积V 中的系综(即时间)平均粒子数; ${r_{ij}}$ 表示粒子对$i,j$ 之间的距离; ${f_{ij}}$ 表示$i,j$ 之间的作用力大小(吸引力取正).2.2.第二种平均形式的推导 -->2.2.第二种平均形式的推导 在由N 个分子组成的流体系统中, 取体积为V 的局部小长方体, 如图1 所示. 设流体系统中分子对的联线与长方体V 的交集, 所形成的非零长度的线段的总数为$M \leqslant \dfrac{{N(N - 1)}}{2}$ . 所有这些非零长度线段所构成的集合记为$\left\{ {i\left| {i = 1,2, \cdots,M} \right.} \right\}$ , 于是(9 )式可以改写为图 1 体积为V 的长方体系统示意图Figure1. Schematic figure of a rectangle with volume V .$r_i^\beta $ 表示第i 线段在$\beta $ 方向的投影(取正号), $f_i^\alpha $ 是提供该线段的分子对之间的作用力在$\alpha $ 方向上的分量(吸引力取正号).13 )式代入(7 )式, 可以得到平均压力张量,14 )式就是平均形式(4 )的第二种表示形式. 容易证明, (4 )和(14 )式中的体积V 可以取任何形状, 而不只限于长方体. 只是要注意, 此时一个分子对的联线与区域V 所交的线段可以超过一个, 求和时, 应遍及全部线段.3.均匀和非均匀系统位形压力张量局部平均形式的分析 23.1.理论分析 3.1.理论分析 在由N 个分子组成的系统(包括均匀系统和非均匀系统情况)中, 取体积为V 的小长方体, 如图1 所示. 在(13 )式中, 对长方体V 内的平均压力张量位形部分有贡献的粒子对中的两个粒子之间的距离必须小于分子间有效作用程长, 其相对位置有三种主要情况: 第一种是两个粒子都在长方体内, ${l_{ij}}(V)=1$ , 如图1 所示; 第二种是一个粒子在长方体内, 一个在长方体外, ${l_{ij}}(V) < 1$ , 如图2 所示; 第三种情况是两个粒子都不在长方体内, 但交长方体的侧面于两点, ${l_{ij}}(V) < 1$ , 如图3 所示. 三种情况的数量分别记为${M_1}$ , ${M_2}$ , ${M_3}$ .图 2 一个粒子在V 内, 一个粒子在V 外, 只有一个交点示意图Figure2. Geometry for calculating the contribution to the pressure from a pair of molecules i and j with only one intersection.图 3 两个粒子都在V 外有两个交点示意图Figure3. Geometry for calculating the contribution to the pressure from a pair of molecules i and j with two intersections.V 的边界面上或棱上的情况, 虽然不是完全不可能, 但是概率几乎为零, 对计算平均位形压力的贡献可以忽略, 故不计入. 于是(13 )式的求和上限M 满足关系式${M_1}$ , ${M_2}$ , ${M_3}$ 分别与长方体的体积、表面积及菱长近似成正比. 为方便计算, 无论长方体边长取多少, 这些粒子对对位形压力张量的贡献都分别简称为体贡献$\overline {p_1^{\alpha \beta }} $ 、面贡献$\overline {p_2^{\alpha \beta }} $ 和线贡献$\overline {p_3^{\alpha \beta }} $ ,V 中的平均位形压力张量总计为3.2.均匀流体系统位形压力三项贡献的分子动力学模拟 -->3.2.均匀流体系统位形压力三项贡献的分子动力学模拟 在平衡流体系统内部, 有$p_{\rm{c}}^{\alpha \beta } = {p_{\rm{c}}}{\delta _{\alpha \beta }}$ , $p_k^{\alpha \beta } = {p_k}{\delta _{\alpha \beta }}$ ($k = 1,2,3$ ), 故(19 )式成为3.2.1.分子动力学模拟的方案 -->3.2.1.分子动力学模拟的方案 采用氩原子的平衡系统作为研究对象进行模拟和分析. 氩的初始位型采用简立方的点阵结构, 原子间距$1.2\sigma $ . 模拟体系尺寸为: $ x \times y \times z =$ $ 18\sigma \times 18\sigma \times 18\sigma $ , $x,y,z$ 方向上均采用镜像边界条件. 氩原子之间采用截断距离为$3\sigma $ 的Lennard-Jones势能函数来描述$\varepsilon $ 为势能参数, $\sigma $ 为原子直径, r 为原子中心之间的距离. 对于氩原子$\sigma = $ 0.3405 nm, $\varepsilon = {k_{\rm{B}}} \times $ 120 K, 其中${k_{\rm{B}}} = 1.38 \times {10^{ - 23}}$ J/K.$\sigma $ , $\varepsilon $ 和氩原子的质量m = 6.63382 × 10–26 kg作为长度、能量和质量单位. 经过无量纲化之后, 给出了其他物理量的标度: 比如温度的无量纲量${T^*} = {k_{\rm{B}}}T/\varepsilon $ , 180 K相当于1.5; 时间的无量纲$\Delta {t^*} = \Delta t\sqrt {(\varepsilon /m{\sigma ^2})} $ . 体系的演化采用Velocity-Verlet方法, 截断长度${r_{{\rm{cutoff}}}} = 3.0\sigma $ , 弛豫过程采用温度为180 K的NVT系综, 时间步长取${\text{δ}}t = 5$ fs, 弛豫50万个时间步. 在达到迟豫平衡之后, 采用累积平均方法计算物理量的时间平均值${\overline {g(i \cdot {\text{δ}}t)} ^N}$ 称为累积平均值, 观察${\overline {g(i \cdot {\text{δ}}t)} ^N}$ 随N 的变化, 当累积平均值达到稳定的值时结束统计, 统计结果不少于100万步.3.2.2.模拟结果 -->3.2.2.模拟结果 在180 K下的氩系统弛豫平衡后的100万时步内, 在体系中取边长${L^*}$ 不同的立方体, 对相关物理量分别进行统计平均, 得到该温度下各个尺寸的$\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ 和$\overline {p_{\rm{c}}^*} $ , 列于表1 中. ${L^{*3}}$ 与立方体含有的粒子平均数成正比. $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ 和$\overline {p_{\rm{c}}^*} $ 与${L^*}$ 的关系如图4 所示.${L^*}$ $\overline {p_1^*} $ $\overline {p_2^*} $ $\overline {p_3^*} $ $\overline {p_c^*} $ 0.4 0 0.014 –0.082 –0.069 0.8 0 0.00814 –0.0773 –0.069 1.2 0.035 –0.04 –0.067 –0.0705 1.6 0.039 –0.071 –0.038 –0.07 2 0.034 –0.072 –0.031 –0.069 2.4 0.024 –0.072 –0.021 –0.069 2.8 0.018 –0.07 –0.015 –0.069 3.2 0.011 –0.068 –0.012 –0.069 3.6 0.0053 –0.067 –0.0089 –0.069 4 –3 × 10–4 –0.064 –0.007 –0.07 5 –0.01 –0.06 –0.0054 –0.07 6 –0.02 –0.05 –0.0038 –0.073 8 –0.035 –0.034 –0.0021 –0.071 10 –0.045 –0.024 –0.0014 –0.07 12 –0.051 –0.02 –9 × 10–4 –0.071 14 –0.055 –0.015 –5 × 10–4 –0.07 16 –0.06 –0.012 –2.6 × 10–4 –0.072 17 –0.06 –0.01 –1 × 10–4 –0.07
表1 $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ 和$\overline {p_{\rm{c}}^*} $ 的模拟值Table1. Values of $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ and $\overline {p_{\rm{c}}^*} $ given by simulation.图 4 $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ , $\overline {p_{\rm{c}}^*} $ 与${L^*}$ 的关系Figure4. Relation between $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ , $\overline {p_{\rm{c}}^*} $ and ${L^*}$ .图4 可以看出, $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ 随${L^*}$ 的变化显著, 但$\overline {p_{\rm{c}}^*} $ 是常数(当${L^*}$ 很小时, 由于系统的误差较大, 出现小波动), 这说明本文模拟是可靠的. 三种贡献随${L^*}$ 的变化行为将在下面做理论分析. 为了节省机时, 我们一般关心$\overline {p_2^*} $ 和$\overline {p_3^*} $ 的可忽略性. 图4 显示, 随着${L^*}$ 的增加, 当${L^*} \geqslant 2$ 时, $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ 的变化是单调的, 其中, $\overline {p_2^*} $ 和$\overline {p_3^*} $ 总是负的, 并且对负压的贡献都不断衰减, 尤其$\overline {p_3^*} $ 衰减得最快. 而$\overline {p_1^*} $ 则在${{L^*}=4}$ 附近由正变负, 并且当${L^*} \geqslant 8$ 时, $\overline {p_1^*} $ 对位形负压的贡献超过$\overline {p_2^*} $ 和$\overline {p_3^*} $ 的贡献. 随着${L^*}$ 的继续增加, $\overline {p_1^*} $ 的负压贡献越来越远超过$\overline {p_2^*} $ 和$\overline {p_3^*} $ 的贡献, 这将有利于我们忽略后两者的贡献. 反过来, 当${L^*} < 16$ 时, $\overline {p_2^*} $ 对负压的影响明显不可忽略, 当${L^*} \leqslant 8$ 时, $\overline {p_2^*} $ 和$\overline {p_3^*} $ 对负压的影响都明显不可忽略. 因此, 在纳米尺度下, 忽略$\overline {p_2^*} $ 和$\overline {p_3^*} $ , 也就是忽略边界效应会给计算带来明显的误差.3.3.均匀流体位形压力三部分贡献的尺度分析 -->3.3.均匀流体位形压力三部分贡献的尺度分析 33.3.1.大${L^*}$ ![]()
![]()
-->3.3.1.大${L^*}$ 分析 随着所取平均体积的大小不同, (14 )式中的平均动压力保持不变, 平均位形压力包含的三项相对大小会有很大不同. 弄清楚它们与计算尺度${L^*}$ 的关系, 对于我们在实际应用中节省计算机时非常重要. 在${L^*}$ 较大, 即${L^*} > 8$ 的情况下, 首先分析位形压力中$\overline {p_2^*} $ 和$\overline {p_3^*} $ 与${L^*}$ 之间的依赖关系, 再分析$\overline {p_1^*} $ 与${L^*}$ 之间的依赖关系.$\overline {p_2^*} $ 与${L^*}$ 的依赖关系${L^*}$ 条件下, 作为近似, 可以先不考虑棱对面贡献的影响, 根据(17 )式, 面贡献与体积的乘积应与第二种粒子对数${M_2}$ 成正比, 故与V 的总面积, 即与${L^*}$ 的平方成正比. 由于此情况下计算的面贡献没有考虑棱对面贡献的影响, 计算的面贡献与实际的面贡献有差别, 因此应该校正这样计算的面贡献: 先不计棱两端的端点效应, 棱效应与体积的乘积应与棱长, 即边长${L^*}$ 成正比, 这就是(23 )式中的第二项. 依此类推, 还应加上顶角的影响, 即第三项它与${L{^*}}$ 无关, 是个待定常数. $k_1$ , $k_2$ , $k_3$ 是与具体的系统和条件有关的常数.$\overline {p_3^*} $ 与${L^*}$ 的依赖关系${L^*}$ 条件下, 作为近似, 可以先不考虑顶角对线贡献的影响, 依据(18 )式, 线贡献与体积的乘积, 与${M_3}$ 成正比, 故与V 的边长, 即与${L^*}$ 成正比. 这样计算的线贡献与实际的线贡献有差别, 因此要加上顶角端点的影响, 即第二项$k_5$ . $k_4$ , $k_5$ 是常数, 由具体的系统和条件决定.$\overline {p_1^*} $ 与${L^*}$ 的依赖关系${L^*}$ 无关, 由(11 )式可知, 平均动压力也与${L^*}$ 无关, 所以总平均位形压力$\overline {p_{\rm{c}}^*} $ 与所取的${L^*}$ 无关. 平均位形压力的体贡献可以表示为${L^*}$ 较大时对应的分子动力学模拟值, 进行拟合, 结果给出: ${k_1} = 0.24053$ , ${k_2} = - 0.01509$ , ${k_3} = 0.0345$ , ${k_4} = $ $0.10328 $ , ${k_5} = - 0.02699$ , 如图5 和图6 所示.图 5 $\overline {p_2^*} $ 的拟合曲线Figure5. Fitting curve of $\overline {p_2^*} $ .图 6 $\overline {p_3^*} $ 的拟合曲线Figure6. Fitting curve of $\overline {p_3^*} $ .23 ), (24 ), (25 )与分子动力学结果拟合的方差都很小, 这也证明了本文的尺度分析和推理是正确的.3.3.2.较小尺度分析 -->3.3.2.较小尺度分析 从氩系统的模拟结果图4 可以看出, 在${L^*} \leqslant 4$ 区域, 三种贡献的行为有些复杂, 其中特征及物理根源分析如下.$\overline {p_1^*} $ , 它来自两个粒子同时出现在体积V 中的粒子对的贡献. 当${L^*} \leqslant 0.8$ 时, 这个概率几乎为零, 所以$\overline {p_1^*} \approx 0$ ; 当$0.8 \leqslant {L^*} \leqslant 4$ 时, 这个概率不能忽略. 但是因为粒子之间的距离很近, 斥力平均占优势, 所以$p_1^* > 0$ ; 当${L^*} \geqslant 4$ 时, 引力平均占优势, 故$\overline {p_1^*} < 0$ , 并与大尺度行为相衔接.$\overline {p_2^*} $ , 它来自粒子对中一个粒子在体积V 内, 一个粒子在体积V 外时的贡献. 随着${L^*}$ 从0增大, ${M_2}$ 中粒子对间平均距离从小增大, 处于斥力状态的概率从大减少, 引力状态的概率从小增大. 当${L^*} \leqslant 0.8$ 时, 一直维持总的斥力状态, 所以$\overline {p_2^*} > 0$ . 当${L^*} > 0.8$ 时, 才转化为总的引力状态, 所以$p_2^* < 0$ . 随着${L^*}$ 继续增加, 引力优势增加, 所以在$0.8 \leqslant {L^*} \leqslant 2$ 时, ${\rm d}\overline {p_2^*} /{\rm d}{L^*} < 0$ . ${L^*}$ 超过2以后总的引力开始减小, ${\rm d}\overline {p_2^*} /{\rm d}{L^*} > 0$ . 以后的变化就与大尺度行为相衔接.$\overline {p_3^*} $ , 它来自粒子对中两个粒子都在体积V 外的贡献. 由于两个粒子都在V 外, 相距较远, 平均来说引力占优势, 所以$\overline {p_3^*} < 0$ . 随着${L^*}$ 的增大, ${M_3}$ 中粒子对的平均距离不断增加, 斥力迅速消失, 引力不断减小, 所以$\overline {p_3^*} $ 不断增加, 并与大尺度行为相衔接.${L^*} \leqslant 8$ )下, $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ 的变化非单调, 计算$\overline {p_{\rm{c}}^*} $ 时, 不可以忽略其中任何一项的贡献.${k_1}$ , ${k_2}$ , ${k_3}$ , ${k_4}$ , ${k_5}$ 及大小尺度${L^*}$ 分界线可能不同外, 所有定性性质对其他系统不会有实质改变.6 —8 , 12 ]直接将宏观大体积整体平均定理(1 )式用到局部小体积上是不准确的. 原因是: 1)遗漏了线贡献和面贡献; 2)即使对(1 )式做了修正, 没有遗漏面贡献, 对于面贡献项, 都用1/2代替了(4 )式右边的${l_{ij}}(V)$ , 即用分子对距离的一半在$\beta $ 方向的投影代替了(17 )式中的$r_i^\beta $ . 对于第1)点, 只有当${L^*}$ 远远大于分子有效作用距离时, 这种遗漏才不会带来明显的误差; 对于第2)点, 仅对分子有效作用距离范围内密度均匀的情况适用, 此时${l_{ij}}(V)$ 的统计平均值为1/2.3.4.温度对位形压力的影响 -->3.4.温度对位形压力的影响 在讨论界面特性及气液固相变过程中, 需要研究界面压力张量及表面张力随温度的变化情况[7 ,8 ] . 因此本节采用分子动力学模拟, 进一步研究了温度对氩系统位形压力的影响. 分子动力学模拟的细节与3.2 节相同, 取${L^*}= 6$ , 温度取值范围${T^*}=$ 1.4—2.3, 模拟结果如图7 所示.图 7 $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ , $\overline {p_{\rm{c}}^*} $ 与${T^*}$ 的关系Figure7. Relation between $\overline {p_1^*} $ , $\overline {p_2^*} $ , $\overline {p_3^*} $ , $\overline {p_{\rm{c}}^*} $ and ${T^*}$ .图7 可以看出, 位形压力随着温度的升高而升高. 这是由于温度升高, 分子平均动能增加, 使得粒子对处于高斥力区(${r_{ij}} < \sigma $ )的概率增加, 斥力增大, 从而位形压力增高.4.结 论 对于平衡的非均匀系统, 人们推导了局部平均压力张量的表达式. 本文用更为简洁的方法推导了这一表达式. 此表达式也适用于均匀流体系统. 本文给出在局部平均尺寸${L^*}>8$ 的条件下均匀流体系统平均位形压力中的三部分贡献项(体贡献项、面贡献项和线贡献项)与${L^*}$ 的理论关系式(含有待定参数), 并以氩原子气体系统为例, 在温度180 K, 原子数密度$0.8{\sigma ^{- 3}}$ 下, 对分子间采用林纳德-琼斯势进行了分子动力学模拟, 给出了$0.4 \leqslant $ ${L^*} \leqslant 17$ 条件下三项贡献及总位形压力的模拟曲线, 确定了${L^*}$ > 8条件下理论关系式中的待定系数, 并做了大$L^*$ 和小$L^*$ 下各项行为的特点分析和温度影响的模拟分析, 得到${L^*}$ 足够大, 才可以忽略面贡献项和线贡献项, 在纳米尺度下, 忽略面贡献项和线贡献项, 也就是忽略边界效应会给计算带来明显的误差. 这些结论对于压力张量的分子动力学模拟计算时选项的最优化是有意义的. 







































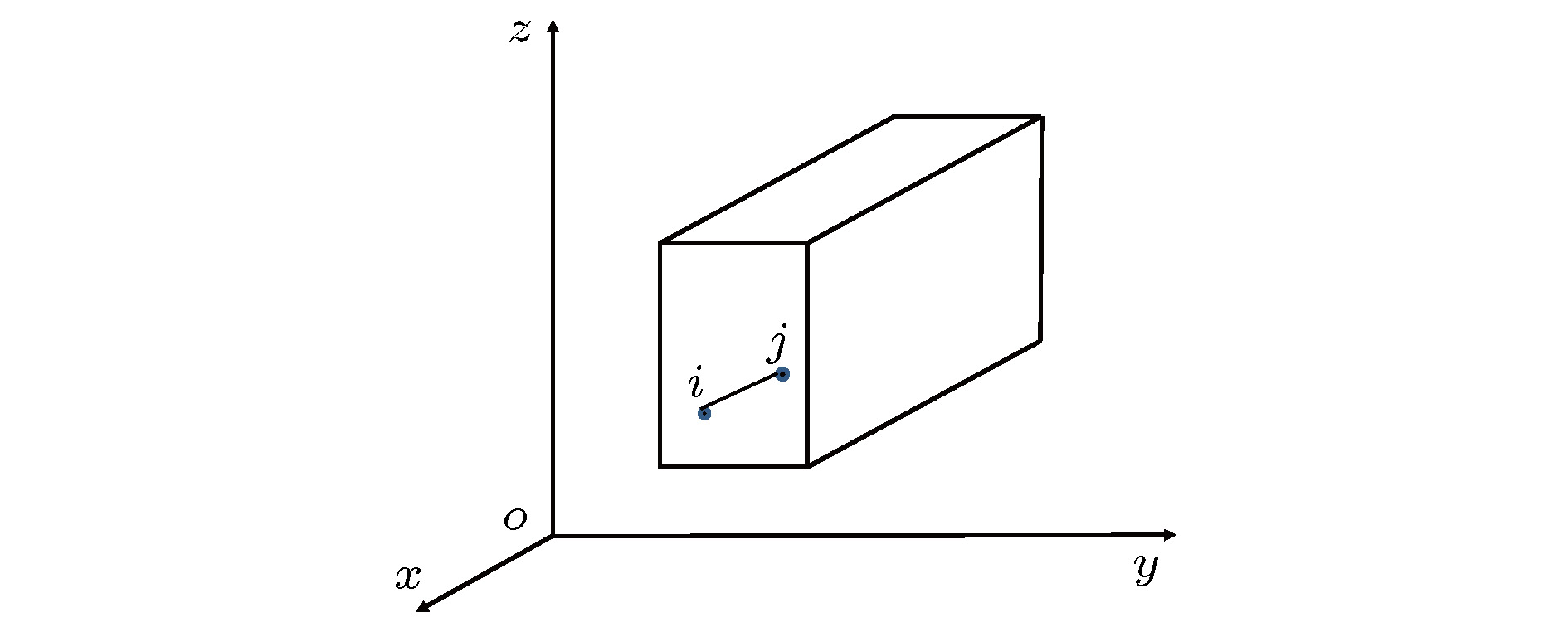 图 1 体积为V的长方体系统示意图
图 1 体积为V的长方体系统示意图









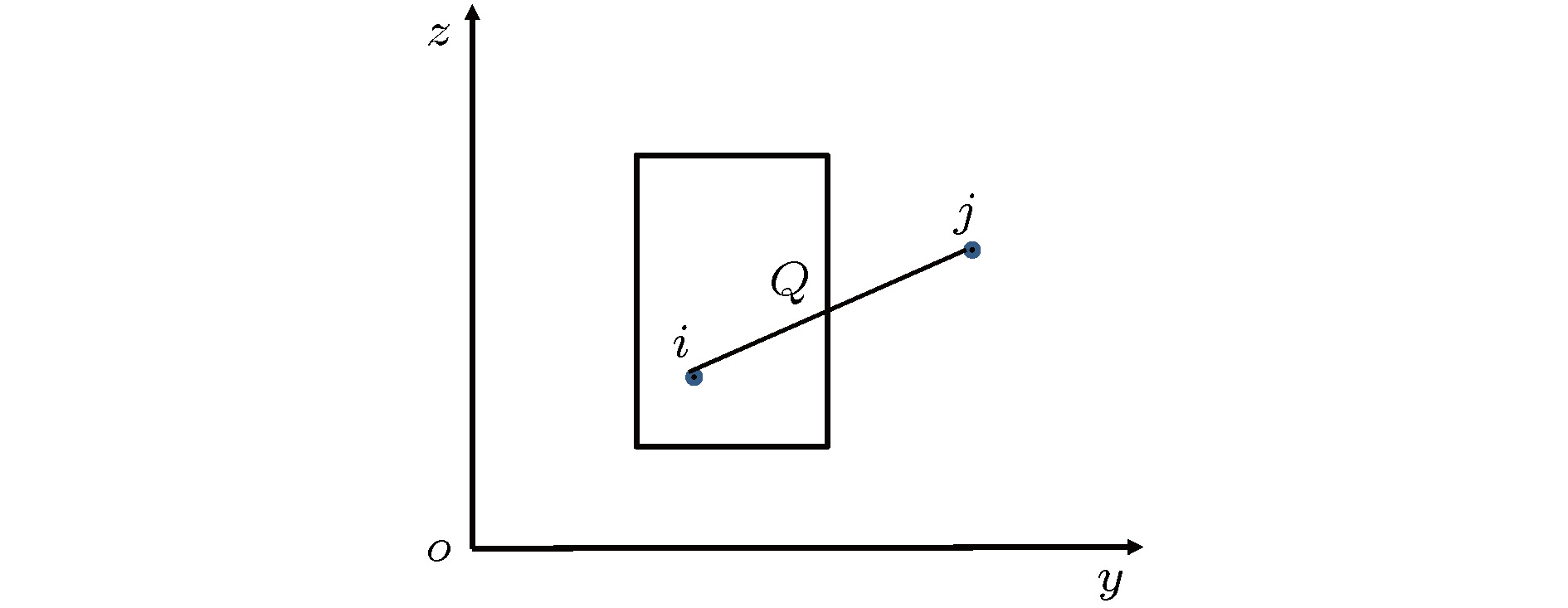 图 2 一个粒子在V内, 一个粒子在V外, 只有一个交点示意图
图 2 一个粒子在V内, 一个粒子在V外, 只有一个交点示意图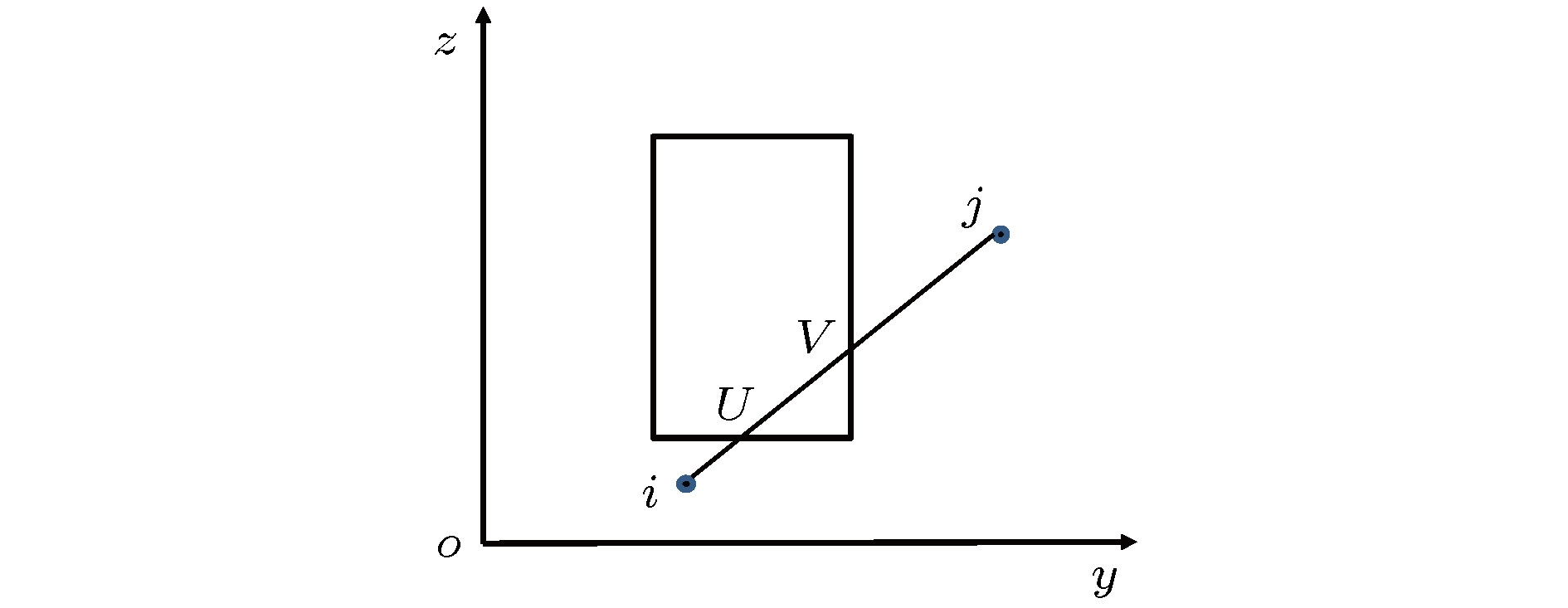 图 3 两个粒子都在V外有两个交点示意图
图 3 两个粒子都在V外有两个交点示意图













































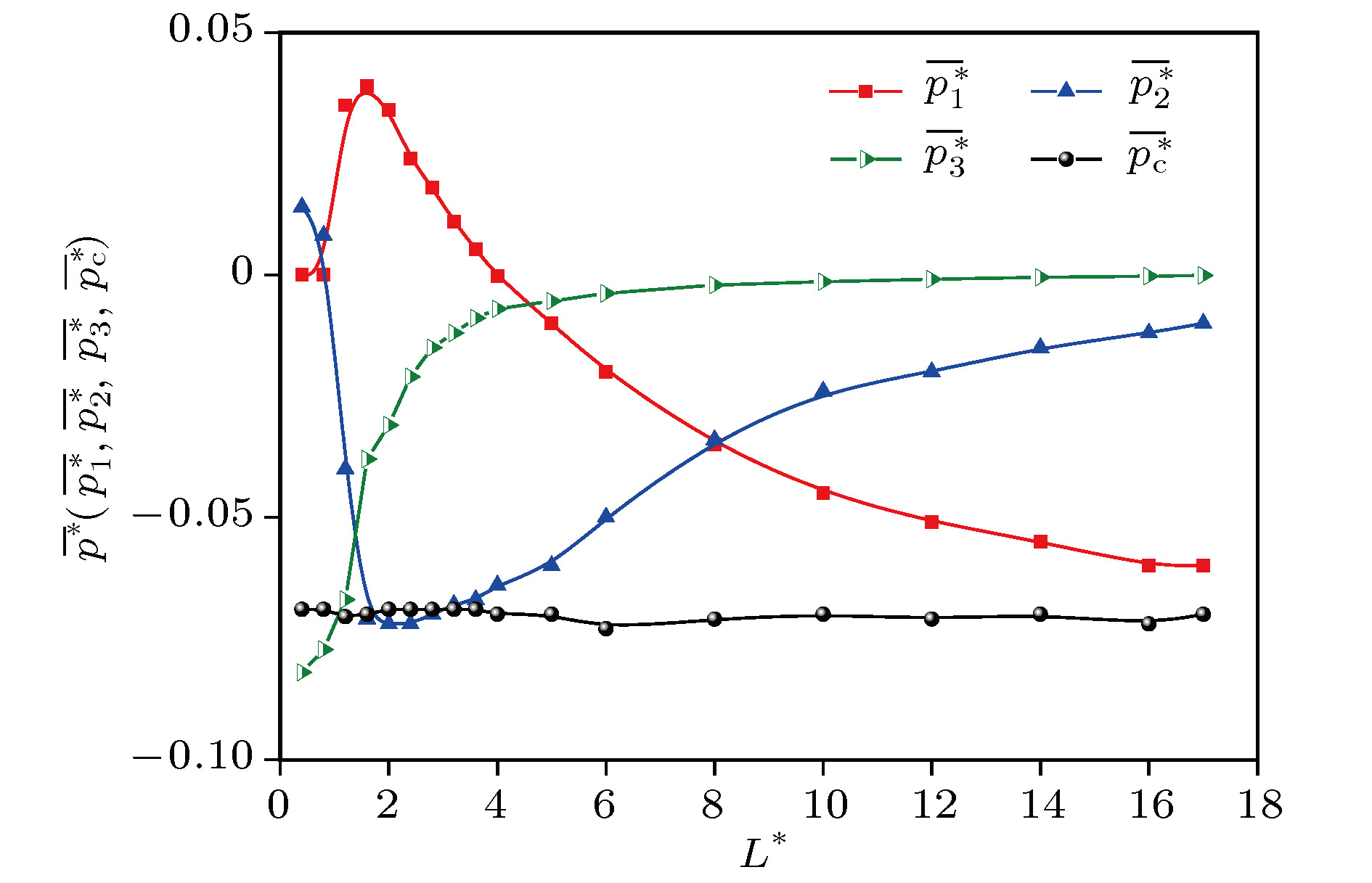 图 4
图 4 



















































































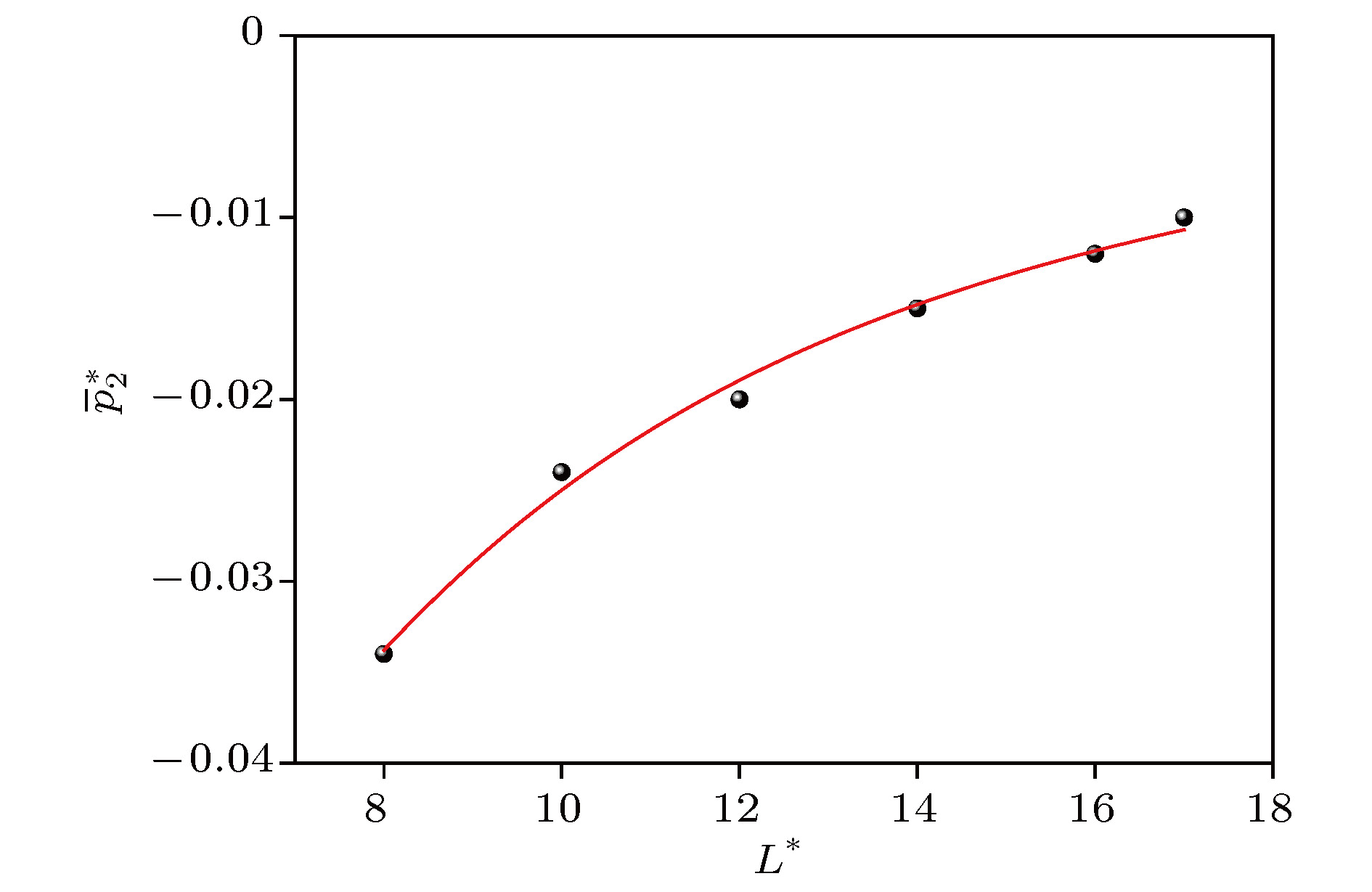 图 5
图 5 

 图 6
图 6 












































 图 7
图 7 



















