Key Laboratory of Radiation Physics and Technology, Ministry of Education, Institute of Nuclear Science and Technology, Sichuan University, Chengdu 610064, China
Fund Project:Project supported by the National Magnetic Confinement Fusion Program of China (Grant No. 2013GB109000), the National Natural Science Foundation of China (Grant No. 11205107), and the Open Research Fund of Key Laboratory of Radiation and Technology of Education Ministry of China (Grant No. 2018SCURPT12).
Received Date:05 March 2019
Accepted Date:08 April 2019
Available Online:01 June 2019
Published Online:20 June 2019
Abstract:Tungsten, due to its desirable properties (high melting point, low sputtering coefficient, good irradiation resistance etc.), is considered as a promising candidate for the plasma facing materials in future nuclear fusion reactors. Therefore, it will work in extremely harsh environments because it is subjected to the bombadement of high-flux plasma particles and the irradiation of high energy neutrons, resulting in vacancies and interstitials. The migration behavior of self-interstitial atoms is one of the most important factors determining the microstructure evolution in irradiated metals because it will greatly affect the mechanical properties of materials. The study of the diffusion behavior of di-interstitials with different configurations contributes to a better understanding of the self-interstitial atom behavior in tungsten. Despite the inherent difficulty in experimental approaches, atomistic simulation provides an effective means of investigating the defect evolution in materials. In this paper, based on the newly developed interatomic potential for W-W interaction, the diffusion behavior of self-interstitial atoms in tungsten is studied by molecular dynamics simulation. This work focuses on the investigation of the diffusion behavior of di-interstitials with different configurations at different temperatures. The obtained results show that the di-interstitials with the first nearest neighbor configuration presents the one-dimensional migration in the $\left\langle 111 \right\rangle $ direction at temperatures below 1400 K. As the temperature increases, it makes rotations from one $ \left\langle 111 \right\rangle$- to other $\left\langle 111 \right\rangle $-directions. Thus migration of di-interstitial atoms with the first nearest neighbor configuration exhibits a change in mechanism from one-dimensional to three-dimensional migration, keeping the stable $\left\langle 111 \right\rangle $ configuration in the whole investigated temperature range. The migration of di-interstitial atoms with the second nearest neighbor configuration is one-dimensional along the $\left\langle 111 \right\rangle$ direction within a certain temperature range. When the temperature is above 600 K, the di-interstitial atoms will dissociate into two individual self-interstitial atoms and move independently. However, the migration of di-interstitial atoms with the third nearest neighbor configuration dissociates at a temperature just above 300 K. The non-parallel self-interstitial atoms form a sessile configuration within a certain temperature range. Once the sessile cluster is formed it can hardly move. Interestingly, it will transform into mobile defect when the temperature is higher than 1000 K. By comparing the migration energy values of these configurations obtained by nudged elastic band method with those of the Arrhenius fits, we find that the diffusivity for each of single- and di-interstitial atoms in tungsten is a linear function of temperature rather than Arrhenius as usually assumed. Keywords:self-interstitial atoms/ diffusion behavior/ irradiation damage/ molecular dynamics simulation
式中$\phi $为两体作用函数; $\rho $为有效电子密度; F为嵌入函数; $\theta (x)$是Heaviside阶跃函数, 当$x \geqslant 0$时, $\theta (x)=1$, 其他情况下$\theta (x)=0$. 上述参数的具体取值见文献[21]. 在进行分子动力学模拟之前, 首先构建一个BCC结构的钨晶体, 基底共20层, 每层10 × 10个原子排列在(001)面. 根据研究的具体需要, 引入SIAs. 本文所涉及的SIAs结构包括1-SIA (图1)和多种构型的2-SIAs (图2), 2-SIAs结构包括最近邻(2-SIAs-1st)、次近邻(2-SIAs-2nd)、三近邻结构(2-SIAs-3rd)以及非平行结构. 为了将体系扩展到无限大情形, 分别沿着[100], [010], [001]方向加上周期性边界条件. 值得一提的是, 当SIAs运动出基底盒子时, 由于周期性边界条件的作用, SIAs将会从另外一面进入基底, 这将会丢失掉真实的扩散信息. 为了获得SIAs在无限大体系中的运动轨迹, 在SIAs扩散出基底时修正了其位置信息, 具体的修正方式见文献[22, 23]. 本文所有的模拟都在NVT (固定粒子数、体积、温度)系综下进行, 为了将系统升至所需温度, 将系统中的每个原子按Maxwell随机分配速度, 然后弛豫. 在演化过程中利用Wigner-Seitz cell方法判断SIAs的位置, 并按一定的时间间隔保存位置信息, 从而得到SIAs运动的轨迹. 演化的时间步长设为10–15 s (1 fs), 每一种结构配置演化时间为10 ns. 图 1 钨中1-SIA的结构图(紫色球为SIAs结构, 蓝色球为格点原子) Figure1. 1-SIA configuration in W. The purple sphere represents the SIA; the blue one represents the lattice atom.
图 2 2-SIAs的不同构型图 (a), (b), (c), (d)分别代表最近邻、次近邻、三近邻以及非平行结构的结构示意图; 右上方的插图分别代表这几种结构$ \left\langle 111 \right\rangle$方向的视图; 紫色球为SIAs, 蓝色球为格点原子 Figure2. Schematic illustrations of the 2-SIAs with different configurations: (a), (b), (c), (d) Represent the configuration of the 2-SIAs-1st, 2-SIAs-2nd, 2-SIAs-3rd and the non-parallel SIAs, respectively. Insets represent the views corresponding to their $ \left\langle 111 \right\rangle$ orientations; the purple sphere stands for the SIA and the blue one stands for the lattice atom.
式中${{{r}}_i}(0)$, ${{{r}}_i}(t)$分别代表粒子初末位置矢量; $\langle {\left| {{{{r}}_i}(t) - {{{r}}_i}(0)} \right|^2}\rangle $代表粒子的系综均方位移; t是演化时间; d是空间维数. 图6给出了各种结构在不同温度下的扩散系数. 通常钨中SIAs的扩散规律(扩散系数与温度的依赖关系)都被认为满足Arrhenius关系[5,22]: 图 6 不同结构的扩散系数(图中实线是根据Arrhenius关系拟合的结果) (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd Figure6. Arrhenius plots of diffusion coefficients of single SIA and di-interstitial atoms in tungsten, which is determined using MD simulations and plotted as a function of the absolute temperature T: (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd.
式中, $k_{\rm B}$是玻尔兹曼常数, kB = 1.38 × 10–23 J·K–1; T是温度, 单位是K; B和A为拟合常数. 如图8所示, 这些间隙结构的扩散系数与温度形成良好的线性关系. 从图8可以看出2-SIAs结构的扩散能力明显低于1-SIA, 在一定温度范围内2-SIAs-1st和2-SIAs-2nd的扩散能力接近. 图 8 不同缺陷的扩散系数 Figure8. Diffusion coefficient for self-interstitials of different configuration in tungsten determined by molecular dynamics simulations and plotted as a function of the absolute temperature T (the solid lines are linear fits).













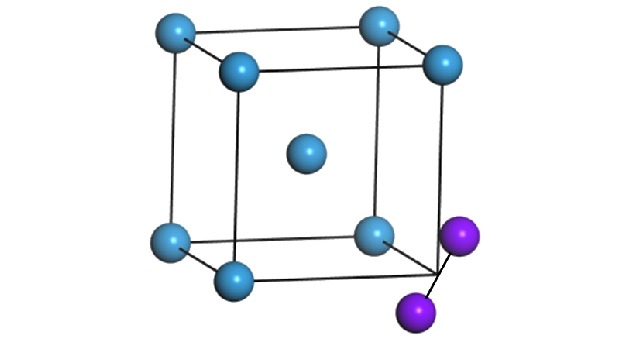 图 1 钨中1-SIA的结构图(紫色球为SIAs结构, 蓝色球为格点原子)
图 1 钨中1-SIA的结构图(紫色球为SIAs结构, 蓝色球为格点原子)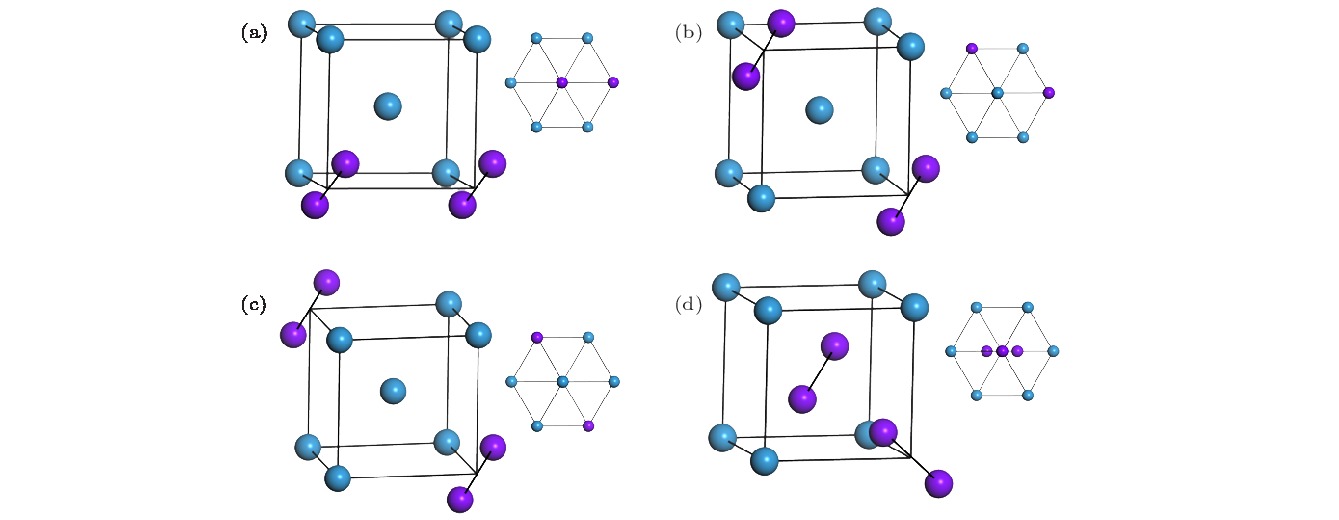 图 2 2-SIAs的不同构型图 (a), (b), (c), (d)分别代表最近邻、次近邻、三近邻以及非平行结构的结构示意图; 右上方的插图分别代表这几种结构
图 2 2-SIAs的不同构型图 (a), (b), (c), (d)分别代表最近邻、次近邻、三近邻以及非平行结构的结构示意图; 右上方的插图分别代表这几种结构












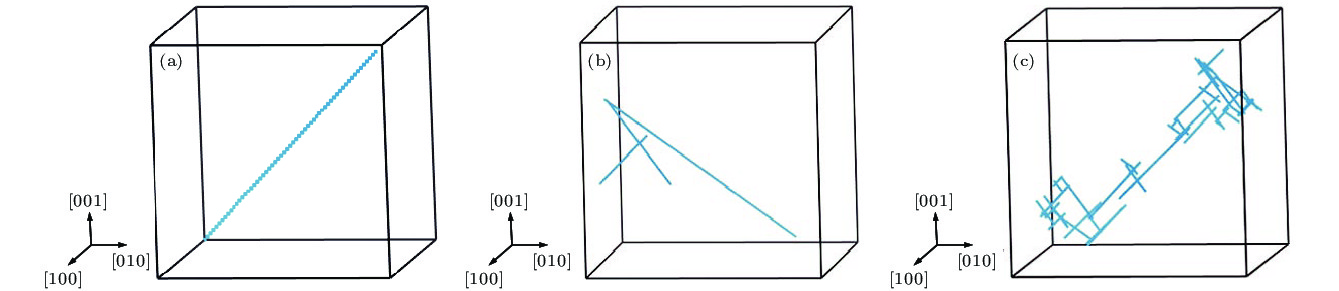 图 3 1-SIAs在不同温度下演化10 ns的扩散径迹图 (a) T = 100 K; (b) T = 700 K; (c) T = 1000 K
图 3 1-SIAs在不同温度下演化10 ns的扩散径迹图 (a) T = 100 K; (b) T = 700 K; (c) T = 1000 K

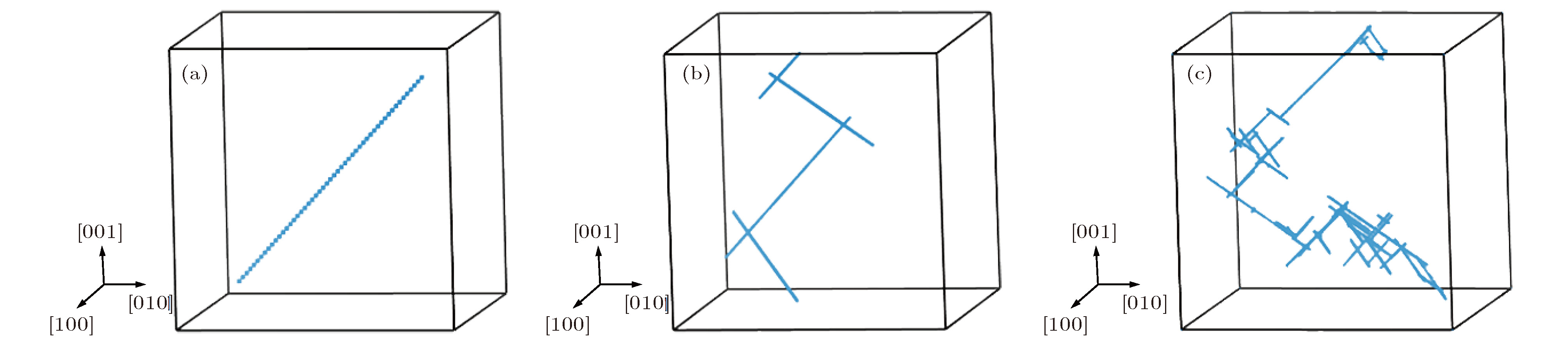 图 4 最近邻结构在不同温度下演化10 ns的扩散径迹图 (a) T = 100 K; (b) T = 1400 K; (c) T = 2000 K
图 4 最近邻结构在不同温度下演化10 ns的扩散径迹图 (a) T = 100 K; (b) T = 1400 K; (c) T = 2000 K



 图 5 T = 500 K时sessile结构在不同时间的结构图 (a) t = 0.5 ns; (b) t = 2 ns; (c) t = 5 ns
图 5 T = 500 K时sessile结构在不同时间的结构图 (a) t = 0.5 ns; (b) t = 2 ns; (c) t = 5 ns


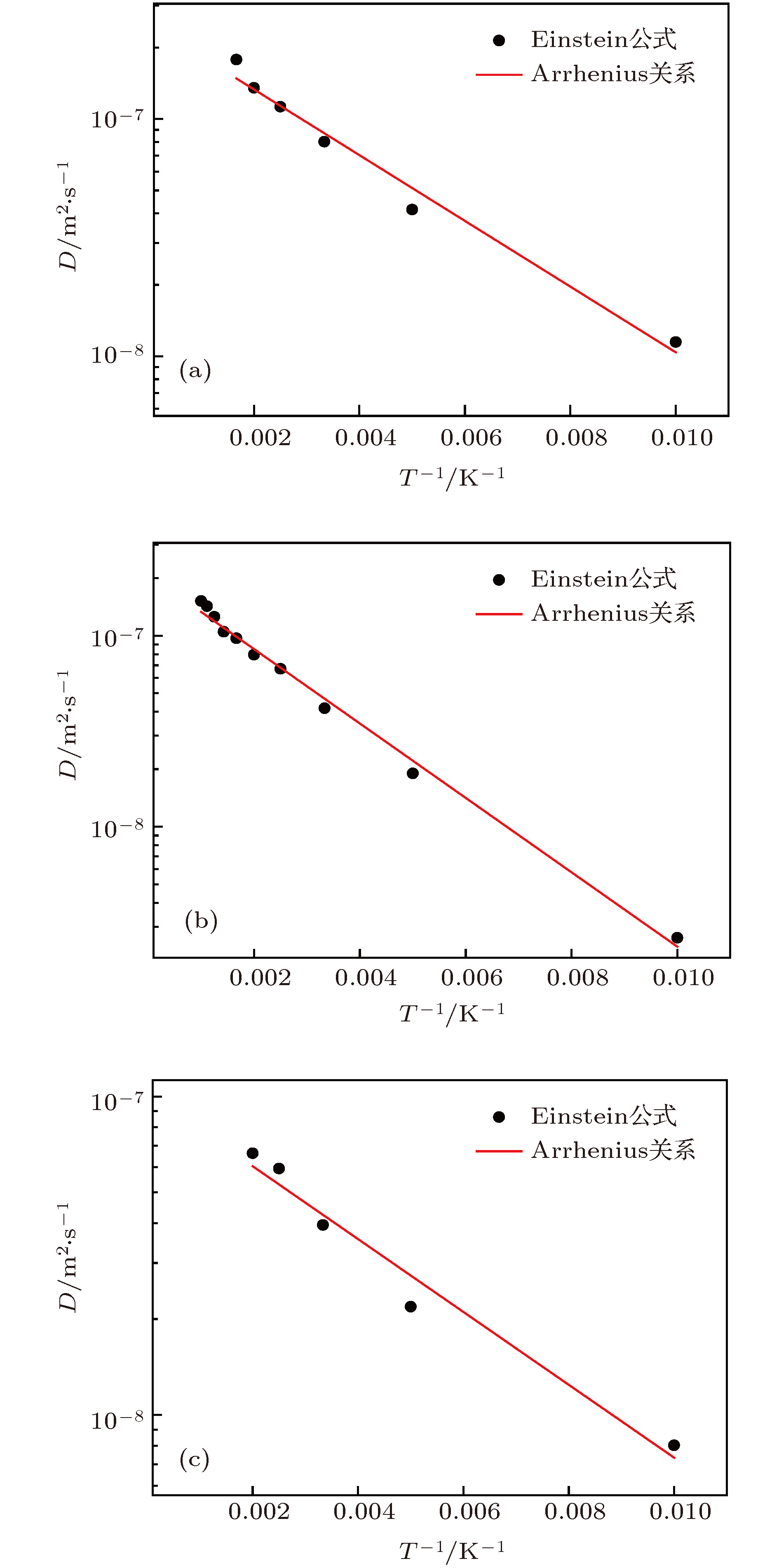 图 6 不同结构的扩散系数(图中实线是根据Arrhenius关系拟合的结果) (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd
图 6 不同结构的扩散系数(图中实线是根据Arrhenius关系拟合的结果) (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd








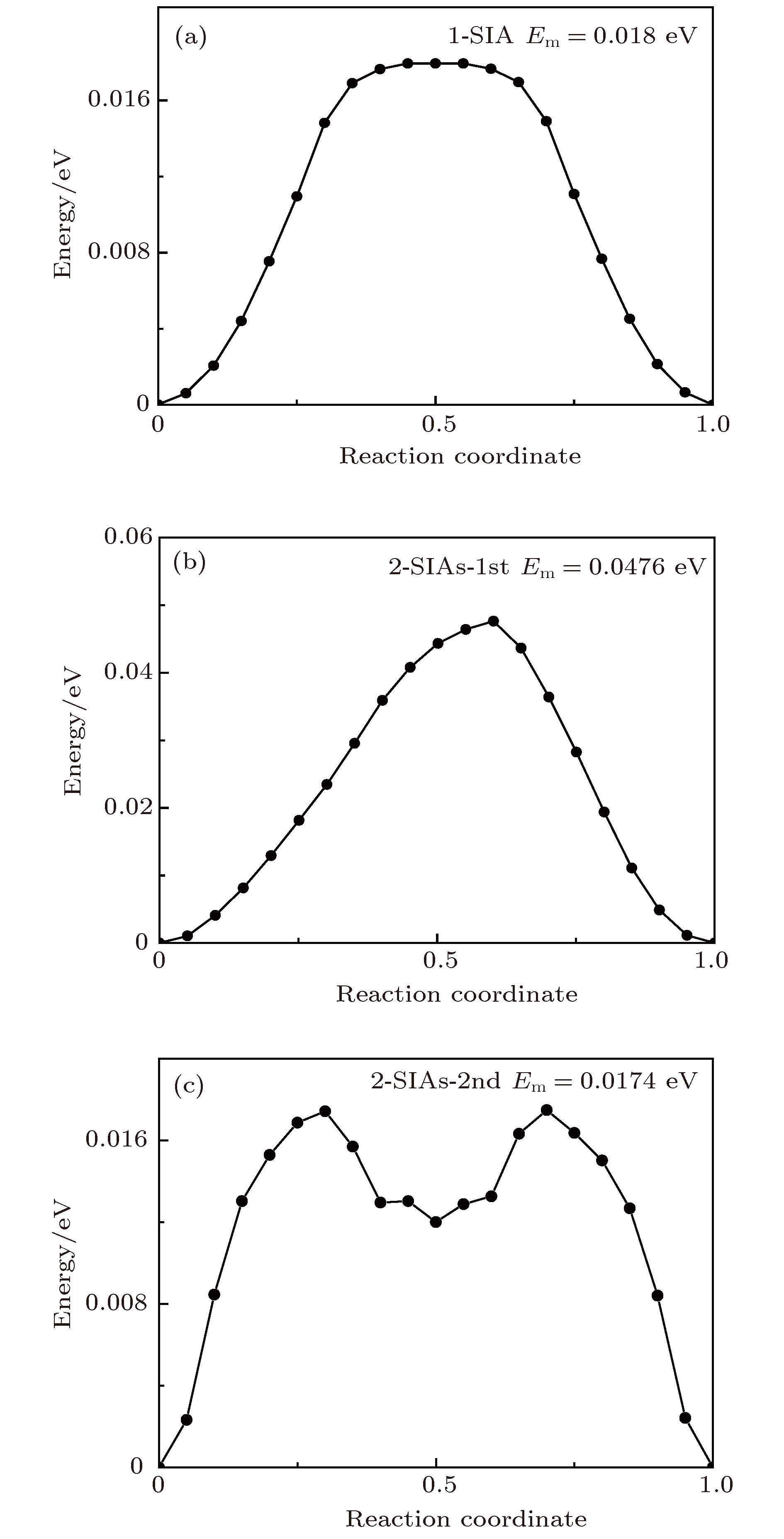 图 7 通过NEB方法所得不同结构的迁移能垒 (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd
图 7 通过NEB方法所得不同结构的迁移能垒 (a) 1-SIA; (b) 2-SIAs-1st; (c) 2-SIAs-2nd
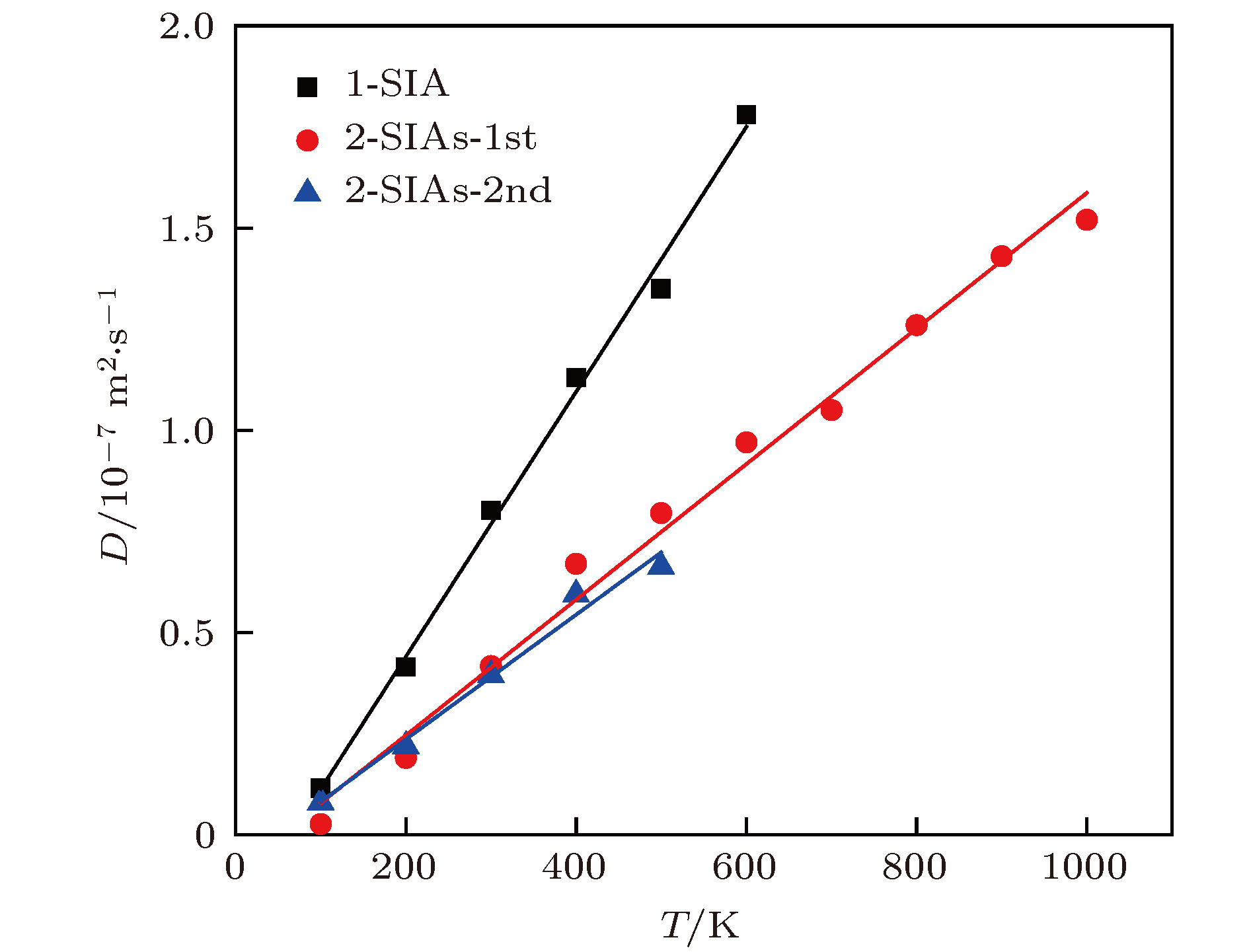 图 8 不同缺陷的扩散系数
图 8 不同缺陷的扩散系数

