全文HTML
--> --> -->太赫兹技术是目前光学领域最重要的新兴技术之一[5]. 在许多方面, 比如医学[6]、安全监测系统[7]、生命科学等领域都有应用[8,9]. 通常, 太赫兹波是指频率范围为0.1—10.0 THz之间的电磁波, 波长范围为0.03—3.00 mm, 介于微波与远红外波段之间. 由于太赫兹的频率较高, 所以具有良好的空间分辨率和时间分辨率[10,11].
在安全检查和医学检查方面, 由于X光的光子能量高, 对人体伤害大, 而低频率的红外光能量过低, 穿透力差, 不易被探测器接收, 同时, 红外探测器还易受环境的干扰[12]. 处于高频区域的太赫兹波克服了两者的缺点, 光子能量相比X光较低, 只有毫电子伏特, 不易破坏被检测物质[13]. 因此, 太赫兹波是安全检查和医学影像技术的理想波谱. 上述器件中, 液晶材料作为调制光波的必备材料, 其与太赫兹光的相互作用是关键因素[14,15]. 液晶分子的电光效应是建立在分子极性基础之上, 极性越强, 对THz波的响应与吸收都十分敏感. 如果对THz波吸收太强, 波能量的损失就越大. 另外, 液晶分子的振动和旋转也对波吸收有较大的贡献. 所以, 选择一种对THz波具有较小的吸收且敏感的电光响应材料是较为困难的事情. 红外光谱是材料分子吸收某些波长的红外光的谱线. 该波长的光能够引起分子振动能级和转动能级的跃迁, 根据分子内部原子间的相对振动和分子转动等信息可以确定物质的分子结构. 因此, 红外吸收光谱又称分子振动光谱或振转光谱[16].
通常将红外光谱按波长分为三个区域: 近红外区(0.75—2.50 μm)、中红外区(2.5—25.0 μm)和远红外区(25—300 μm). 中红外光谱属于分子的基频振动光谱; 远红外光谱则属于分子的转动光谱和某些基团的振动光谱. 太赫兹波谱是位于红外光谱上的中、远红外区的光波. 因此, 研究太赫兹波的问题, 事实上是研究中、远红外波的工作. 根据相关光谱文献[16], 关于物质对红外光谱吸收的原因基本上都有较为详细的结论. 例如, 有机化合物分子中的官能团特征吸收峰表现在中红外光的较高频段, 而在远红外波段, 产生吸收峰的主要原因是分子的振动与转动对波的吸收. 通常, 有机分子内低能电子跃迁、含氢原子团伸缩振动的倍频吸收位于近红外(大于120 THz)波段, 绝大多数有机化合物和无机离子的基频吸收带, 即分子内与单个化学键相关的伸缩和弯曲等振动模式出现在中红外区(6—120 THz), 而各种有机分子之间的氢键弱相互作用、偶极子的振动和转动跃迁以及晶体中声子的低频骨架振动所对应的吸收频率均位于远红外区(小于6 THz)的太赫兹波段[10].
由于THz波的产生对波源有较高的要求, 而实验研究有较多的不可控制因素, 因此, 关于液晶在THz波段的实验测量数据并不多, 关于THz吸收方面的物理和化学机理分析也较少.
由于理论模拟方法简单便利, 能够有效了解和研究物质分子的THz吸收特性, 工作效率高、成本低, 近些年在液晶THz吸收方面也有一些计算模拟工作[17,18]. 这些工作能够对液晶分子的性质进行设计与预测, 对材料的合成具有指导意义.
本研究参考了具有THz实验测量数据的液晶nCHBT(4-(trans-4-n-alkylcyclohexyl) isothiocy-anatobenzenes) (n = 0—15)系列分子[19], 通过计算得到分子的中远红外吸收光谱, 分析分子在0.1—5.0 THz范围内的波吸收状况, 讨论烷基链的碳原子数目与THz吸收的关联性.
图1(a)和图1(b)展示了液晶分子5CB[18]和MBBA(N-(4-methoxybenzylidene)-4-butylaniline)[17]在THz波段的实验以及计算结果. 由于实验测量中液晶分子的吸收是由于氢键弱相互作用、偶极子的振动和转动跃迁引起的, 有些振动频率简并或十分接近不易分辨, 还有一些简正振动无红外活性, 使分子不产生红外吸收, 所以吸收谱难以观察到吸收峰. 另一方面, 由于实验误差和噪音的影响, 低频率附近的测量结果总是难以表征. 而理论模拟采用的是单分子模型, 没有考虑分子之间的相互作用, 因此没有体现氢键的作用, 吸收峰值体现了偶极子振动和转动的吸收, 吸收峰值比较稀疏. 但是可以看出计算结果和实验测量的吸收趋势十分相似, 说明计算模拟得到偶极子振动和转动的吸收值与实验测量相比是准确的. 通常, 在较高频段, 液晶分子产生的吸收绝大部分为官能团产生的特征吸收峰, 因而计算结果与实验测量符合得较好. 因此, 使用Gaussian计算模拟结果是可靠的, 可以详细地展示分子对波吸收的复杂性. 这些计算结果为本工作的可靠性提供支撑[27-29].
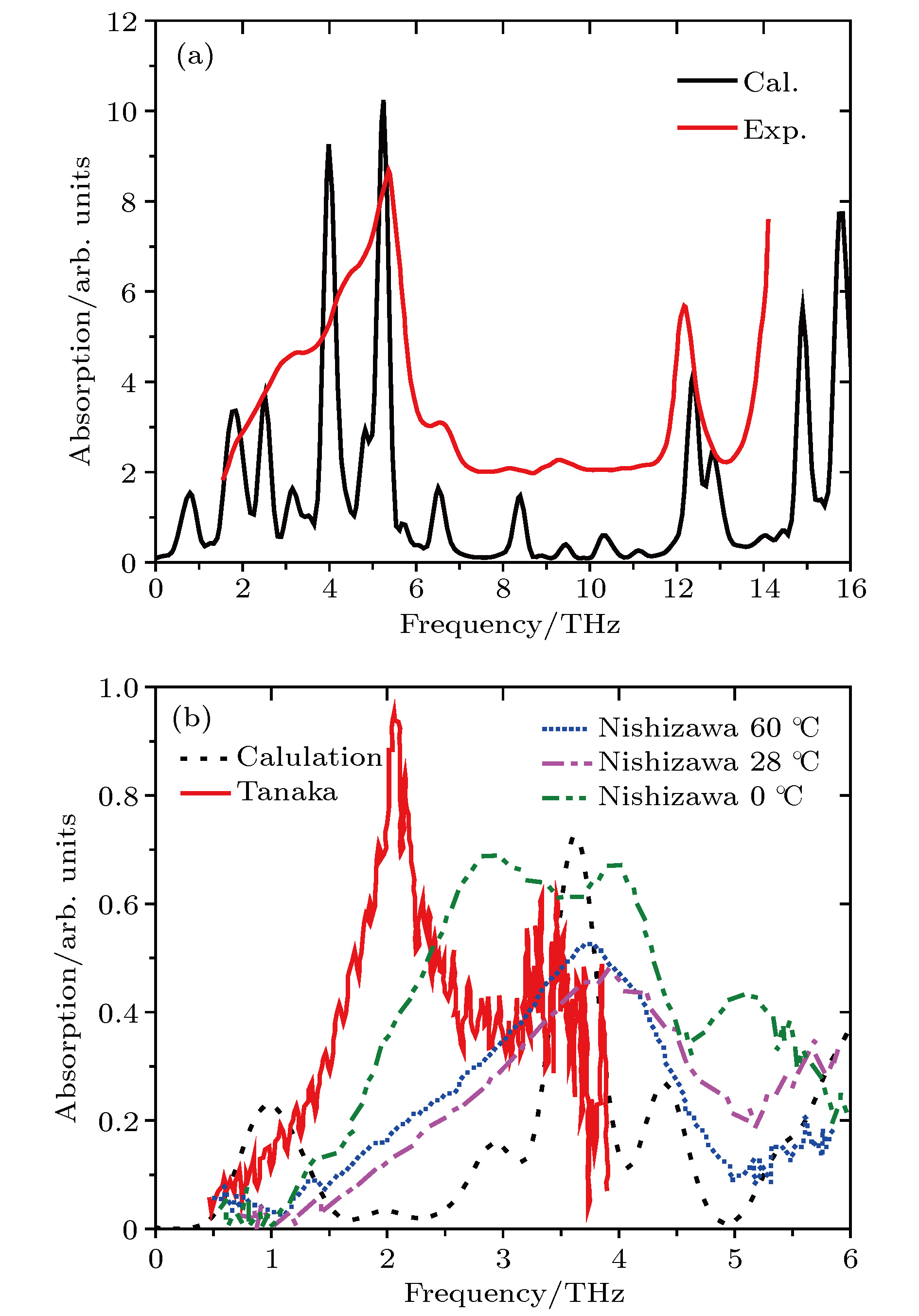 图 1 (a) 5CB计算值和实验值对比; (b) MBBA计算值和实验值对比
图 1 (a) 5CB计算值和实验值对比; (b) MBBA计算值和实验值对比Figure1. (a) Comparison of calculations and experimental results about 5CB; (b) comparison of calculations and experimental results about MBBA.
本文所研究的16种液晶分子, 均是在CHBT分子的基本结构基础上, 通过改变烷基链碳原子的数目n构成不同的液晶分子. 其结构如图2所示.
 图 2 16个棒状液晶分子的化学结构 (n = 0—15)
图 2 16个棒状液晶分子的化学结构 (n = 0—15)Figure2. Chemical structure of 16 liquid crystal molecules (n = 0?15)
在使用Gaussian09程序包过程中, 以Opt + Freq计算类型, 使用B3LYP/6G311g基组优化了16种液晶分子, 得到了红外吸收光谱等数值[20]. 通过这些数值, 对影响分子吸收的因素进行了讨论. 讨论总体分为两部分:第一部分对0.1—5.0 THz频率范围内分子的吸收进行了分析;第二部分针对Chodorow等[19]的实验数据, 比较了0.3—3.0 THz波段范围内3—12 CHBT液晶分子的吸收光谱.
2
3.1.0.1—5.0 THz波段的波吸收分析
图3展示了0.1—5.0 THz波段16个分子的吸收状况. 可以看出, 所有分子的吸收都有一个较高的峰值, 该吸收峰的分布十分相似, 大约分布在0.4—0.9 THz之间. 但分子峰值的大小有所有不同. 当n = 3, 4, 5, 6, 7时, 分子的吸收峰值较高. 0.1—5.0 THz属于远红外波段, 对于单个分子模型而言, 分子的振动与转动是对波吸收的主要因素. 因此, 这些吸收峰差异是由分子自身振动与转动差异带来的[10,16]. 图 3 0—5 THz波段分子吸收光谱
图 3 0—5 THz波段分子吸收光谱Figure3. Molecular absorption spectroscopy at 0?5 THz.
另一方面, 红外吸收谱的强度是分子振动跃迁概率的表征, 与分子振动时偶极矩的变化相关. 因此, 与基团固有偶极矩有关, 极性越强, 振动时偶极矩变化越大, 吸收谱峰值越高; 分子中的原子对称性好, 振动时偶极矩变化就小, 吸收谱峰值越弱. 液晶分子在低太赫兹波段的吸收同时又是分子转动的一个重要体现. 分子的转动取决于分子内原子质量分布和转动惯量. 因此, 为了分析0.1—5.0 THz波段吸收峰产生的原因, 我们计算了这些分子的偶极矩和转动惯量.
16个分子转动惯量和偶极矩的计算结果随烷基链长度的变化展示在图4和图5中. 从图4和图5可以看出, 分子的转动惯量随碳原子数目的增加而增大. 偶极矩只有n = 3, 5候有些异常, 可能是烷基链碳氢的增加对分子重心从苯环到环己烷的迁移产生的电荷中心的差异造成的. 其他分子的偶极矩整体差异不大, 但是微小差别显示出偶极矩的奇偶效应. 这说明分子的电荷分布随烷基链的增加变化较小, 显示烷基链对分子的极性贡献不大.
 图 4 16个分子在其长轴的转动惯量值的变化趋势
图 4 16个分子在其长轴的转动惯量值的变化趋势Figure4. Trend of the rotational inertia of 16 molecules on their long axis.
 图 5 16个分子在其长轴的总偶极矩值的变化趋势
图 5 16个分子在其长轴的总偶极矩值的变化趋势Figure5. Trend of the total dipole moment of 16 molecules on their long axis.
影响分子振动和转动的因素主要考虑分子的重心和转动惯量, 因此, 需要讨论分子内的原子质量分布(如表1)和转动惯量对吸收的影响. 我们把整个分子分为NCS-Benzene-Cyclohexane-CnH2n+1几个部分. 根据各部分的原子质量之和, 确定分子的重心位置. 表1和图6展示了分子质量分布和重心位置的标定. 以n = 1为例, 经过计算发现, 分子的质量中心在靠近环己烷一端的苯环位置.
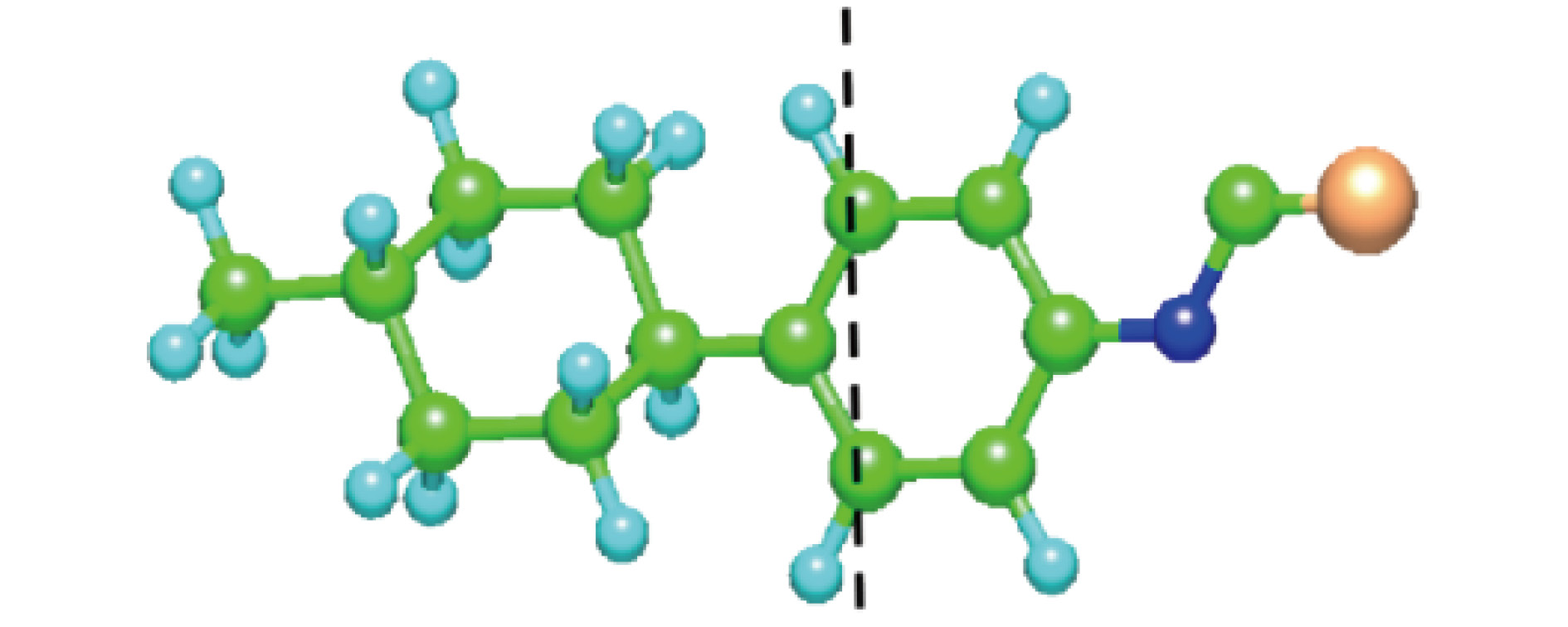 图 6 1CHBT的分子质量分布和重心位置标定
图 6 1CHBT的分子质量分布和重心位置标定Figure6. Calibration of mass distribution and center of gravity for 1CHBT.
| nCHBT | NCS/ g·mol–1 | Benzene/ g·mol–1 | NCS+ Benzene/ g·mol–1 | Cyclohexane/ g·mol–1 | NCS+ Benzene + Cyclohexane/g· mol–1 | CnH2n+1/ g·mol–1 | Cyclohexane + CnH2n+1/ g·mol–1 |
| 0CHBT | 58 | 76 | 134 | 82 | 216 | 1 | 83 |
| 1CHBT | 58 | 76 | 134 | 82 | 216 | 15 | 97 |
| 2CHBT | 58 | 76 | 134 | 82 | 216 | 29 | 111 |
| 3CHBT | 58 | 76 | 134 | 82 | 216 | 43 | 125 |
| 4CHBT | 58 | 76 | 134 | 82 | 216 | 57 | 139 |
| 5CHBT | 58 | 76 | 134 | 82 | 216 | 71 | 153 |
| 6CHBT | 58 | 76 | 134 | 82 | 216 | 85 | 167 |
| 7CHBT | 58 | 76 | 134 | 82 | 216 | 99 | 181 |
| 8CHBT | 58 | 76 | 134 | 82 | 216 | 113 | 195 |
| 9CHBT | 58 | 76 | 134 | 82 | 216 | 127 | 209 |
| 10CHBT | 58 | 76 | 134 | 82 | 216 | 141 | 223 |
| 11CHBT | 58 | 76 | 134 | 82 | 216 | 155 | 237 |
| 12CHBT | 58 | 76 | 134 | 82 | 216 | 169 | 251 |
| 13CHBT | 58 | 76 | 134 | 82 | 216 | 183 | 265 |
| 14CHBT | 58 | 76 | 134 | 82 | 216 | 197 | 279 |
| 15CHBT | 58 | 76 | 134 | 82 | 216 | 211 | 293 |
表116个分子各个组成部分的原子质量
Table1.Atom mass of each component of 16 molecules.
下面具体分析所有分子的质量分布和重心位置与吸收的关系. 从表1可以看出, 当n ≤ 3时, 异硫氰酸脂加苯环的质量大于环己烷加烷基链的质量, 分子的重心在苯环上. 因为苯环的结构坚硬, 所以分子短而且不容易发生弯曲, 振动吸收较弱. 但当n = 3时, 分子的重心十分接近苯环与环己烷相连的C—C键, 导致该C—C键容易弯曲, 分子的振动变得容易, 从而增加3CHBT对光的振动吸收, 所以3CHBT峰值较高, 而(0—2)CHBT峰值较低. 当4 ≤ n ≤ 7时, 异硫氰酸脂加苯环的质量小于环己烷加烷基链的质量, 分子的重心迁移在环己烷上. 因为环己烷的结构柔软, 导致环己烷的C—C—H与连接苯环的C—C—C形成的面角扭曲或者弯曲振动变得容易, 从而增加4CHBT, 5CHBT, 6CHBT和7CHBT对THz的吸收, 所以它们的峰值较高. 当n ≥ 8时, 虽然分子的重心继续迁移, 结构稳定性较差. 但是, 随着烷基链上碳原子的数目不断增加, 分子逐渐变长, 质量逐渐增大, 转动惯量增大变得显著, 相对于同样的温度和分子内能, 分子的转动速度变低, 振动与转动状态变得稳定, 对THz波的吸收变得较弱, 所以吸收峰值较低.
2
3.2.计算与实验结果对比
Chodorow等[19]通过实验测量了0.3—3.0 THz波段(n = 3—12)CHBT分子的吸收光谱. 图7对比了10组实验数据与计算结果. 由于实验测量分为o光和e光, 根据相关理论, 我们将o光与e光吸收数据加和绘制为一条曲线. 由于实验测量和计算结果吸收的单位不同, 实验测量的单位是吸收系数, 而计算结果只是吸收的相对数值大小, 实验结果和计算结果的吸收峰值高低的绝对值比较没有意义, 只需要观察不同分子相对峰值的高低. 因此, 为了方便对比和观察, 我们将计算结果的数值进行倍数放大.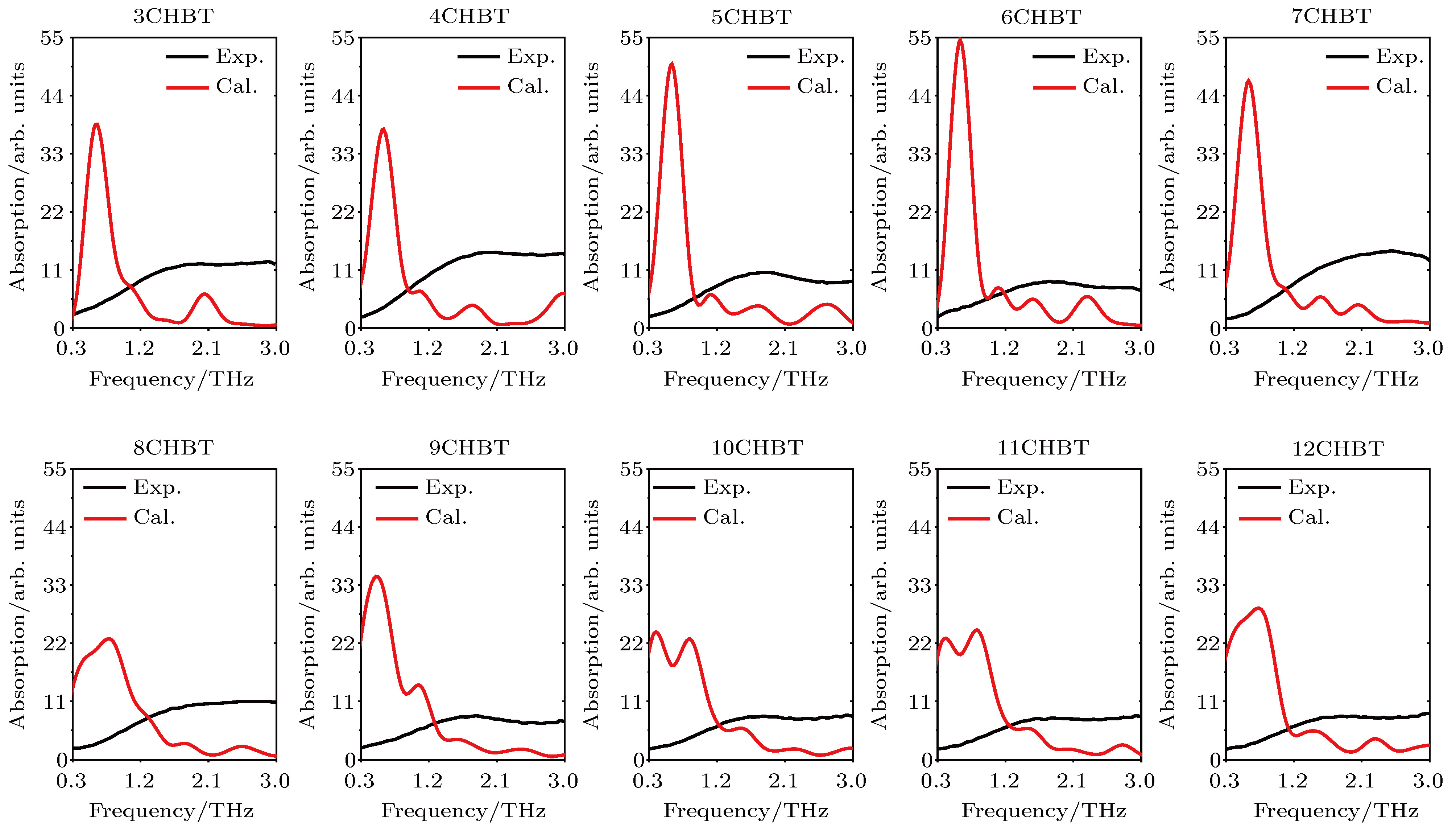 图 7 0.3—3.0 THz波段(3—12)CHBT吸收光谱的实验值与计算值对比
图 7 0.3—3.0 THz波段(3—12)CHBT吸收光谱的实验值与计算值对比Figure7. Comparison of calculations and experimental results about (3?12)CHBT in 0.3?3.0 THz.
通过图7的数据对比, 可以看出, 就像其他计算得到的结果一样[17,18], 在0.3—0.8 THz之间计算得到的强吸收峰, 实验测量都没有得到. 实验测量没有发现明显的吸收峰, 吸收只是随着频率的增加而增加, 但是在1.7 THz之后, 液晶开始出现吸收, 大部分材料都有平坦的吸收峰. 而计算值在0.5 THz处有特别明显的吸收峰, 在之后又有一些较低的吸收峰.
实验测量在低频段峰值很少, 在这个频段, 有机分子之间的氢键(N—H)弱相互作用在吸收中占据主要成分, 而不同液晶材料的分子结构与分子间的氢键是相似的. 偶极子的振动-转动跃迁以及晶体中声子的低频骨架振动所对应的吸收频率占据次要成分, 因此吸收的峰值位置差异难以体现, 而吸收的强度差别体现在偶极子跃迁. 换句话说, 实验测量更取决于氢键和声子的贡献, 由于液晶分子有些振动频率简并或十分接近不易分辨, 还有一些简正振动无红外活性, 使分子不产生红外吸收, 所以从吸收谱难以观察到吸收峰. 另一方面, 理论模拟采用的是在300 K设置温度下的单分子模型, 没有考虑分子之间的相互作用, 因此氢键的作用没有体现. 计算得到的峰值取决于偶极子的振动跃迁, 这个跃迁吸收只对吸收强度有贡献, 而吸收峰值比较稀疏.
为了说明上述现象和便于观察, 图8展示了计算结果在0.5 THz之后的较低峰值与实验值的对比. 可以发现, 对于实验测量中吸收强度较大的((n = 3—7)CHBT)的分子, 计算结果在相应的位置也表现出强的吸收, 表明该强度来源于偶极子振动吸收的贡献(图8(b)中标出). 相反, 其他几个分子具有较低吸收峰. 也就是说, 计算结果的峰值体现的是偶极子振动和转动吸收, 对吸收强度的贡献结果与实验测量体现出来的规律是一致的.
 图 8 计算结果与实验测量的吸收峰值分析 (a)实验测量; (b) 计算在1.2—3.0 THz的吸收峰值
图 8 计算结果与实验测量的吸收峰值分析 (a)实验测量; (b) 计算在1.2—3.0 THz的吸收峰值Figure8. Analysis of the absorption peaks between the calculation and the experimental data: (a) The experimental measurements; (b) the calculation of absorption peaks in 1.2?3.0 THz.
遗憾的是, 文献中的实验数据仅仅只有0.3—3.0 THz的结果, 不像5CB分子那样具有较宽的波段[18], 因此, 难以对计算值的吸收峰和实验值的趋势进行更宽域的对应. 就像前边讨论的那样, 液晶材料5CB的理论计算也是在较高的频率才能够与实验结果在吸收峰位置有重合, 在低频段, 实验测量也只是得到了线性增加的类似结果, 也说明了实验测量可能是来源于氢键的贡献. MBBA的测量中, TANAKA的数据让人难以置信, 我们已在前边的工作中做过陈述[17]. NISHIZAWA的测量数据也像5CB一样显示出了上述特性, 特别是温度对波吸收的影响也不是很大, 但是零度的测量明显具有较多的吸收峰[17]. 在本研究中, 我们尝试了使用其他计算方法和关键词, 但是得到的结果与本工作使用的参数相差不大, 规律相同. 因此, 实验测量的起始阶段, 是否是仪器设备无法精确探测, 我们不得而知, 只能寄希望于未来有更多的实验数据进行比对.
