全文HTML
--> --> -->实验上, 1987年Burley等[9]采用导向离子束质谱测量了基态氧离子与H2分子及其同位素反应的速率常数、分子比以及积分截面等动力学信息. 采用电荷转移探测方法、八极离子囚禁技术和2—4倍的八极装置, Li等[12]在0.02—12.0 eV的碰撞能范围内测量了O+(4S, 2D, 2P)+ H2(D2)反应的积分截面.
理论上, 2004年Martínez等[16]采用599个从头算能量点构建了OH2+体系的解析势能面. Martínez等[16-19]基于该势能面进行了一系列的动力学研究. 2005年, 采用准经典轨线方法(QCT), Martínez等[17]计算了O+与H2分子及其D2, HD同位素的反应. 紧接着采用非含时的量子机制的紧密耦合方法结合超球坐标, Martínez等[18]在0.04—1.24 eV的碰撞能范围内进一步研究了O+ + H2反应. 2006年, 采用含时量子机制的实波包方法, Martínez等[19]重新计算了O+离子与H2分子及其同位素的反应. 文中把反应概率和积分截面与之前应用非含时量子方法计算的结果进行了比较, 以此来检测科里奥利耦合效应在反应过程中的作用. 之后基于Martínez等[16]的势能面, Xu等[21], Gómez-Carrasco等[22], Bulut等[23]使用含时量子波包方法(TDWP)进行了O+ + H2反应体系的动力学计算, 报道了该反应的反应概率、积分截面和速率常数等动力学性质. 由于Martínez等[16]的势能面仅包含599个从头算能量点, 不足以精确描述具有两个势阱一个势垒的复杂反应体系, 2018年, Li等[24]采用置换不变多项式神经网络方法对63000个从头算能量点进行了拟合, 构建了新的势能面. 并采用TDWP方法在新的势能面基础上进行了动力学计算. 文中对反应概率、积分截面和微分截面等动力学信息进行了报道, 并与之前的实验与理论结果进行了比较.
之前的研究主要集中于介绍势能面的性质, 并且对于长程相互作用势的描述还不充分. 本文对该体系在核间距较远的地方增加了一些从头算点并重新拟合构建了新势能面. 图1给出了当氢分子固定在平衡位置, O+离子线性接近氢分子的图像. 由图1不难看出, 新势能面在长程相互作用势上对于老势能面有了很大的改善. 在新的势能面的基础上, 进行了O+ + H2反应的动力学计算. 本文的结构如下: 第2小节简略介绍TDWP方法; 第3小节对动力学结果进行详细的分析和讨论; 第4小节给出结论.
 图 1 新旧两个势能面的长程相互作用势比较
图 1 新旧两个势能面的长程相互作用势比较Figure1. Comparison of the long range interaction potential for the old and new potential energy surfaces



在采用TDWP方法计算过程中, 二阶分裂算符[31]用来传播初始波包. 如果





最终, 通过传播初始波包和抽取态-态散射矩阵, 可以得到体系的反应概率、积分截面、微分截面和速率常数:


 图 2 90°, 120°, 150°和180°的最小能量路径
图 2 90°, 120°, 150°和180°的最小能量路径Figure2. The minimum energy paths at 90°, 120°, 150°, and 180° angles
为了获得收敛的动力学结果, 在进行动力学计算之前需要对诸多动力学参数进行调试. 本文计算了总角动量J = 0的反应概率, 得到了最优的动力学参数, 并将这些参数用于J大于0的计算中. 具体的参数见表1
| 格点范围和大小 | $R \in \left[ {0.1,16} \right],{N_R} = 279,N_R^{{\rm{int}}} = 159$ |
| $r \in \left[ {0.1,15} \right],{N_r} = 279,N_r^{{\rm{asy}}} = 159$ | |
| 初始波包 | Rc = 11.0, ${k_0} = \sqrt {2{E_0}{\mu_R}} $ |
| ${\varDelta _R} = 0.2 {\rm{exp}}\left[ { - \dfrac{{{{\left( {R - {R_{\rm{c}}}} \right)}^2}}}{{2\varDelta _R^2}}} \right]{\rm{cos}}\left( {{k_0}R} \right)$; 其中E0 = 0.5 eV | |
| 总传播时间 | 30000 |
| 最大总角 动量J | 70 |
表1计算中使用的参数(除了特殊声明, 均采用原子单位a.u.)
Table1.Parameters used in the calculation (The atomic unit is used in the calculation unless otherwise stated)
2
3.1.反应概率
图3给出了若干总角动量J的反应概率. 由图可知, 在反应概率上有很多共振峰, 并且这些共振峰随着能量和总角动量J的增加而变宽, 振幅变小. 这是由于在最小能量路径上有一个支持很多束缚态和准束缚态的深势阱. 随着碰撞能和J的增加, 中间络合物O+-H-H的寿命变短, 因此反应概率上的共振峰变得不那么明显了. 此外, 还可以发现, 对于J < 40, 随着碰撞能的增加其反应概率逐渐降低. 这是一个典型的放热反应特征, 这是因为随着碰撞能以及离心势的增加O+离子的速率越来越快, H2分子来不及与O+离子形成OH键, O+离子就跑开了, 因此反应概率会越来越低. 而对于J > 60, 其反应概率随着碰撞能的增加逐渐升高. 结合图1, 本文认为这是因为随着碰撞能和离心势的增加, O+ + H2反应已经不局限在最小反应路径上, 反应可以在其他角度的能量路径上进行, 在反应路径上将会有势垒的存在, 这导致反应概率随着碰撞能的增加而增加.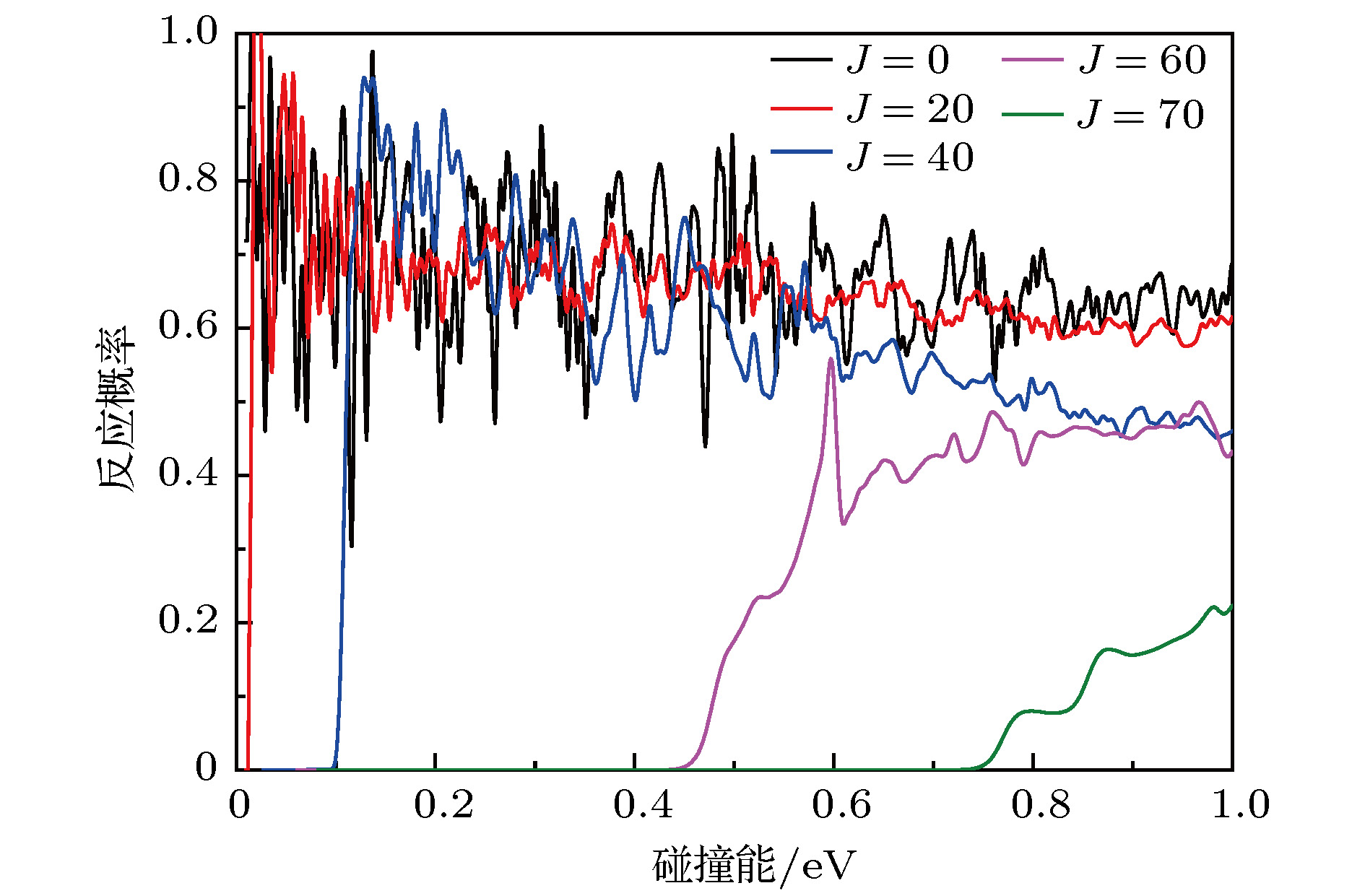 图 3 对于几个选定的总角动量J在能量范围0—1.0 eV的反应概率
图 3 对于几个选定的总角动量J在能量范围0—1.0 eV的反应概率Figure3. The total reaction probabilities for several selected total angular momentum J in the collision energy from 0 to 1.0 eV
图4给出了J = 0反应概率的振动分辨. 由图可知, 产物OH+ (v′ = 0, 1)的振动态在低能时就是打开的, 随着碰撞能的增加, 其他振动激发态陆续打开, 这表明随着碰撞能的增加有更多的能量转化为体系的内能. 从图4中不难看出, v′ = 0和v′ = 1所占据的反应概率的比重一直是最高的. 随着产物振动态的增加, 其所占的比例越低. 这表明在O+ + H2→ OH+ + H反应过程中振动激发的效率不高, 这可能是因为OH+产物振动态之间的能隙间隔过宽所致.
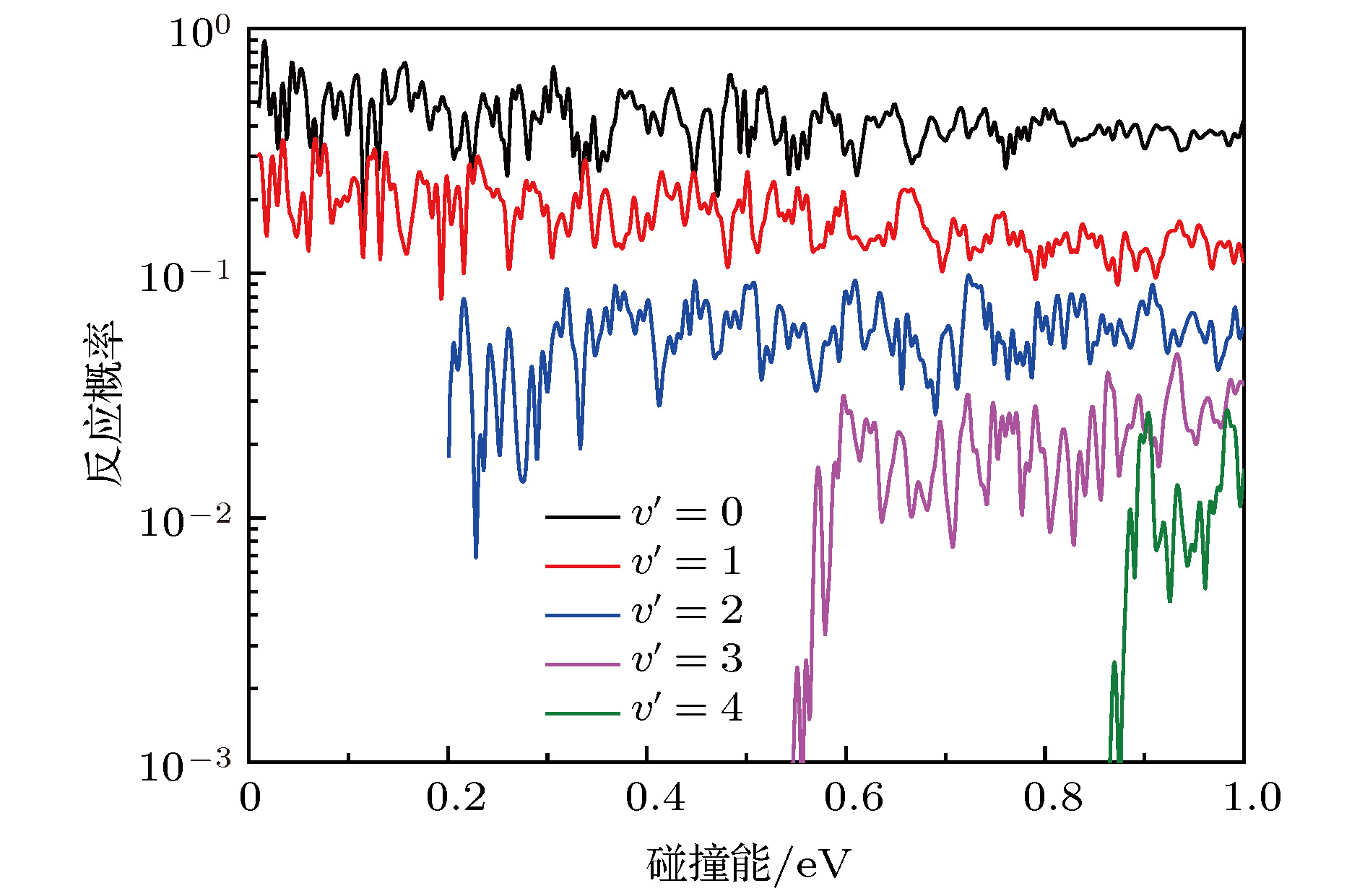 图 4 总角动量J = 0反应概率的振动分辨
图 4 总角动量J = 0反应概率的振动分辨Figure4. The vibrational resolved reaction probabilities of the total angular momentum J = 0
图5给出了J = 0反应概率的转动分辨. 图5展示了所有共振结构的特征, 如上所述, 这是由反应路径上的深势阱所致. 产物OH+ (v′= 0)的转动通道在j′= 11之前都是打开的. 在图5中仅列出j′从0到5的反应概率. 对比J = 0反应概率的振动分辨, 发现OH+的转动激发更容易实现. 图5有一个奇怪的现象, 产物OH+(v′= 0, j′ = 1)的激发效率远低于其他转动态. 这可能是过渡态产物的特性导致的.
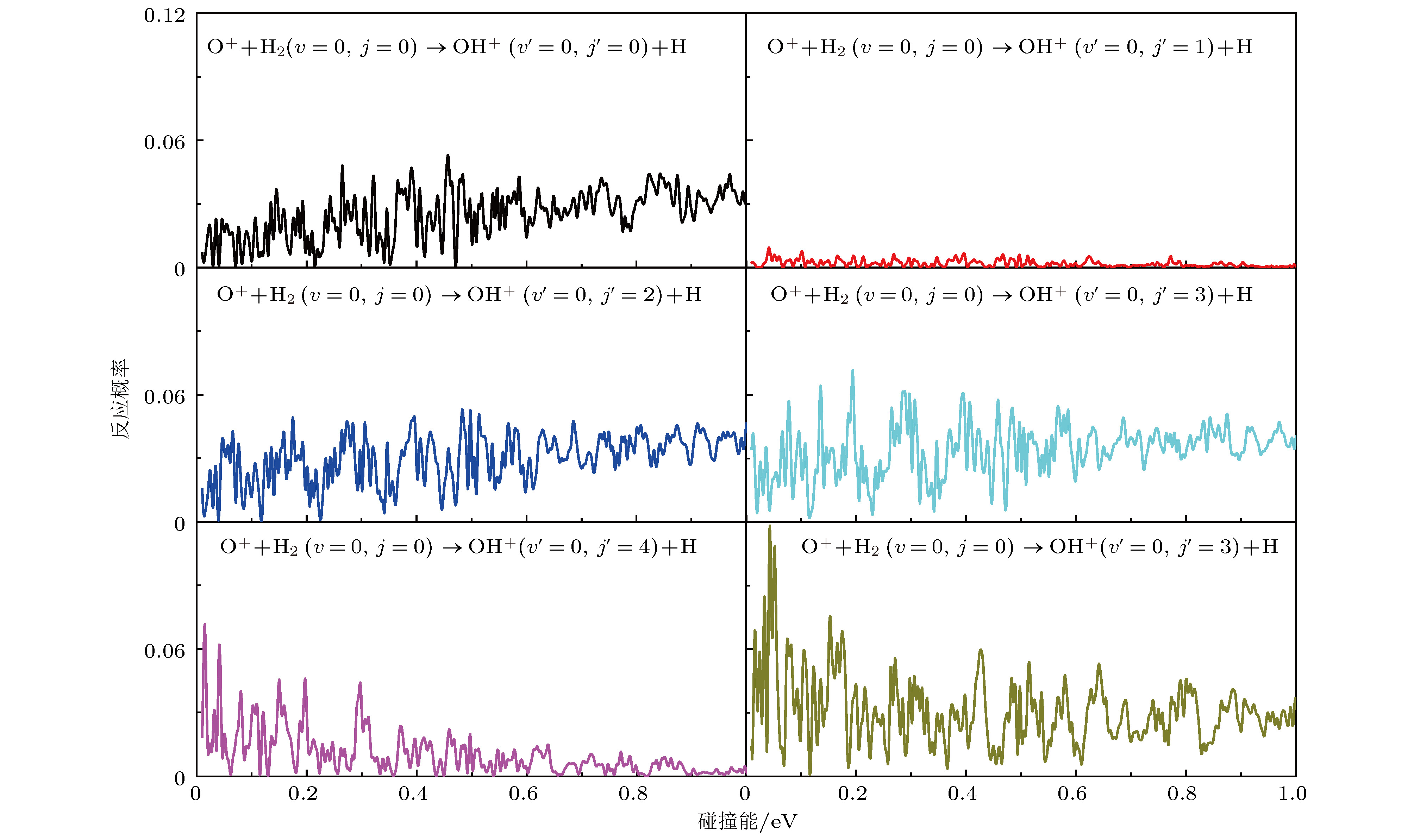 图 5 O+ + H2(v = 0, j = 0) → OH+ (v' = 0, j' ) + H反应在总角动量J = 0时转动分辨的反应概率
图 5 O+ + H2(v = 0, j = 0) → OH+ (v' = 0, j' ) + H反应在总角动量J = 0时转动分辨的反应概率Figure5. Rotationally resolved reaction probabilities for the O+ + H2 (v = 0, j = 0) → OH+(v' = 0, j' ) + H reaction at total angular momentum J = 0
2
3.2.积分截面
图6给出了O+ + H2反应体系在0.01—1.0 eV能量范围内的总积分截面. 为了与理论和实验文件进行比较, 由文献[22]获得理论结果和文献[8]得到的实验结果也在图6中给出. 由图6可知, 总积分截面随着碰撞能的增加而降低. 比较文献[22]的理论结果与本文的结果, 当碰撞能小于0.1 eV时, 文献[22]的结果高于本文结果, 当碰撞能大于0.4 eV时, 本文的结果略高于文献[22]的结果. 通过与实验结果的比较可知, 本文结果与实验结果符合得更好, 而文献[22]的结果在低碰撞能范围内略高于实验结果.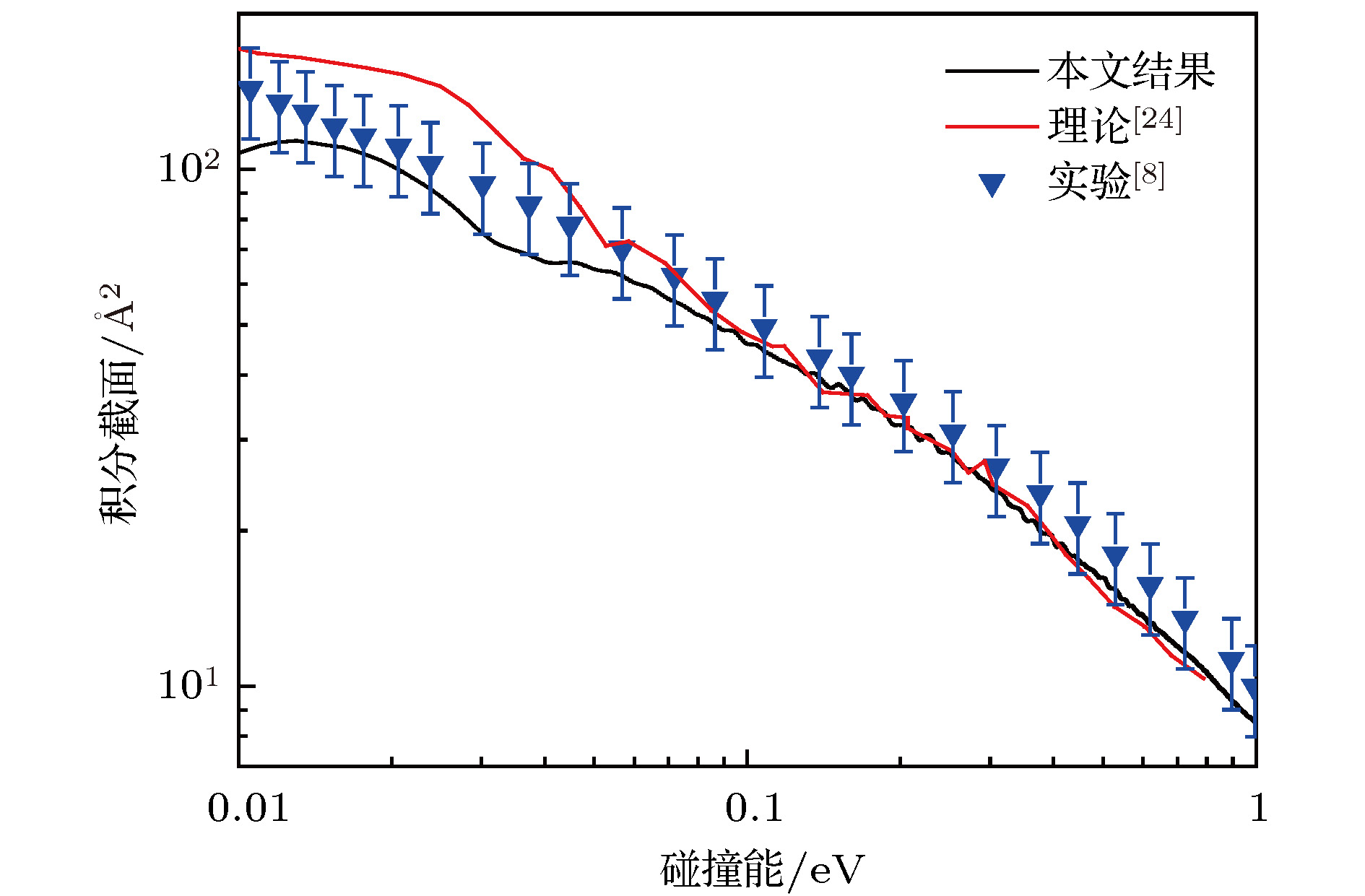 图 6 O+ + H2反应的积分截面以及文献[22]的理论结果和文献[8]的实验结果
图 6 O+ + H2反应的积分截面以及文献[22]的理论结果和文献[8]的实验结果Figure6. The integral cross section of the O+ + H2 reaction as well as the theoretical and experimental results obtained from Ref. [22] and Ref. [8], respectively
2
3.3.微分截面
图7给出了O+ + H2反应若干能量点的微分截面. 由图7可知, 微分散射的信号主要集中在前向(0°)和后向(180°)的方向上. 边缘散射的信号分布很少, 并且随着碰撞能的升高逐渐降低. 从图7左图相应的低能部分可以看出, 前向散射的信号和后向散射的信号都非常明显, 但是偏向于前向散射. 在低碰撞能时, 反应主要是在最低能量路径上进行, 反应路径上的深势阱所产生的中间络合物具有很长的寿命. 因此, 产物既可以向前又可以向后散射, complex-forming反应机制占据主导地位. 从图7右图可以看出, 随着碰撞能的增加, 反应已经不局限于最小能量路径上, 反应路径上出现了一个势垒两个势阱, 前向散射明显高于后向散射. 这说明O+离子碰撞H2分子直接形成OH键并带走一个H原子, 没有经过过多的振荡, 表明反应机制逐渐由间接的插入机制转变为直接的抽取机制.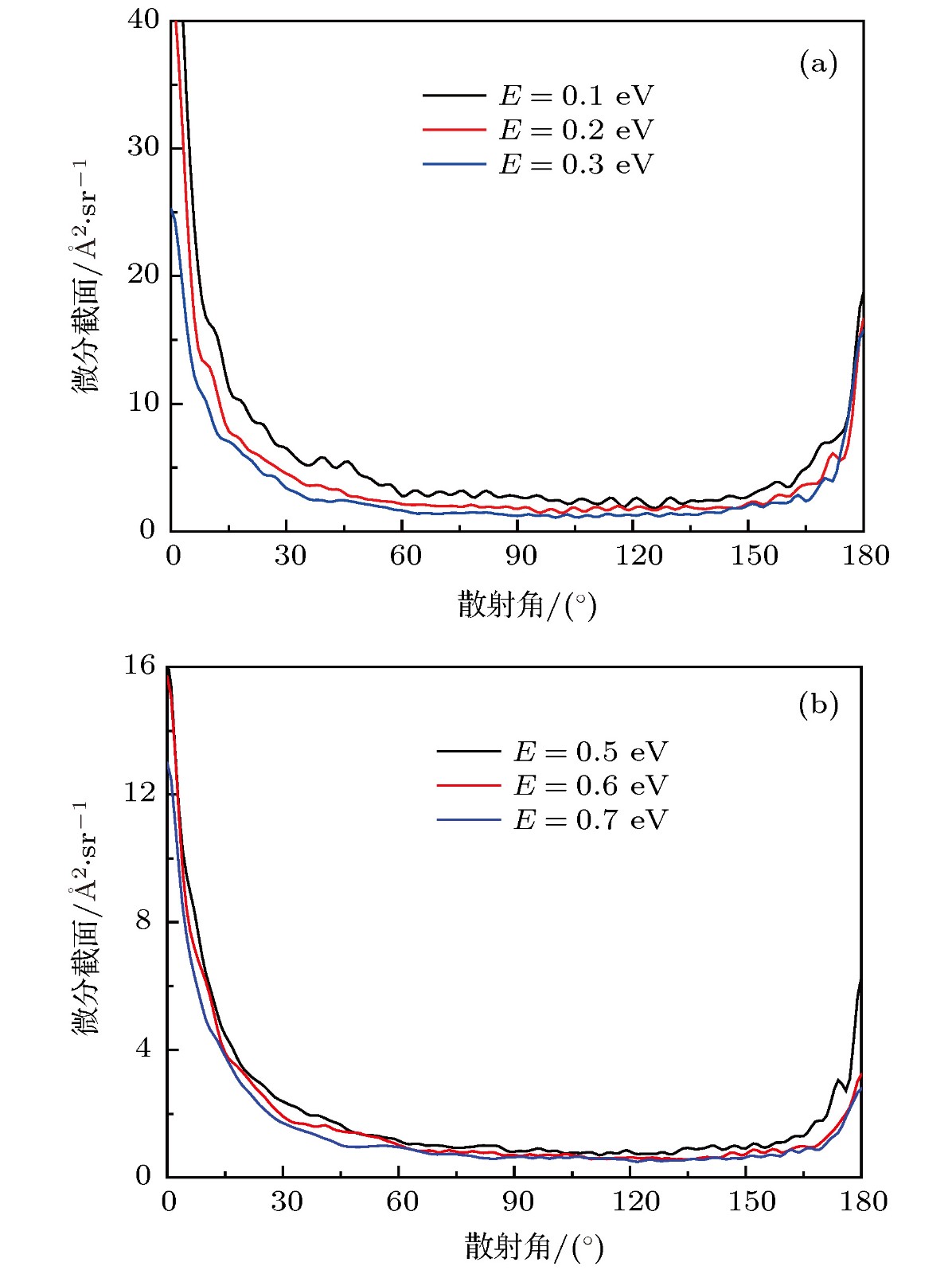 图 7 O+ + H2反应若干能量点的微分截面 (a)低能部分; (b) 高能部分
图 7 O+ + H2反应若干能量点的微分截面 (a)低能部分; (b) 高能部分Figure7. The differential cross sections of O+ + H2 reaction for several collision energies: (a) The low energy; (b) the high energy part
2
3.4.速率常数
图8给出了O+ + H2反应在200—1000 K温度范围内的速率常数. 从图8可以看出, 随着温度的增加, 速率常数以约1.25的因子增加. 通过与实验比较发现, 本文的理论结果明显偏低. 速率常数可以通过积分截面获得, 由图6可以看出本文的积分截面与实验结果十分吻合, 所以预期本文的速率常数结果将会与实验结果有比较好的符合. 然而通过图8可以看出, 两者有较大的差别. 本文认为导致这个差距的原因主要源于和实验数据进行对比需要包含反应物H2分子的转动激发态的影响, 而本文的计算为初始态为(v = 0, j = 0)的动力学计算, 没有包含转动激发的影响. 图 8 O+ + H2反应的速率常数和文献[5]的实验结果
图 8 O+ + H2反应的速率常数和文献[5]的实验结果Figure8. The rate constant of the O+ + H2 reaction and the experimental values obtained from Ref. [5] in the temperature range of 200 to 1000 K
