全文HTML
--> --> -->2.1.HFR方法概述
本文计算采用Cowan程序包中的HFR理论方法[11]. 在HFR理论中, N电子原子体系的单电子径向波函数通过自洽场方法求解HFR方程获得. 体系波函数在所有可能的组态基矢空间展开, 表示为





原子体系的哈密顿算符(单位: Ry, 1 Ry = 13.606 eV)为










下面描述具体的计算方法[12-14]. 首先在HFR理论计算时考虑了质量速度项和达尔文项修正, 同时还包括了近似Breit修正. 自洽场计算时, 所有离子径向积分参数统一标度为: 组态平均能为95%、自旋-轨道参数为100%、库仑相互作用(含组态相互作用)参数为85%. 计算过程中, 奇宇称和偶宇称组态能级值均以基态能级能量值作为参考点. 然后, 通过已有可靠实验能级值采用最小二乘拟合(least-square fitting, LSF)方法计算出离子的实验径向积分参数. 在此基础上, 再运用自行设计的FORTRAN程序, 分析径向参数的HFR和LSF值之比值随着Zc变化的规律, 采用广义最小二乘拟合(generalized least-square fitting, GLSF)外推(内插)计算方法, 得到径向积分参数的最佳优化计算结果. 最后, 利用这些积分参数计算出各能级值和有关的跃迁数据.
2
2.2.MCDHF方法概述
本文MCDHF方法[15]计算采用的是全相对论grasp2K-DEV程序包[9,16?18]. 在MCDHF理论方法中, 对于N电子原子体系, 相对论单电子波函数采用狄拉克四分量旋量表示










ASF展开中, 采用价电子与价电子之间的关联模型(VV)[21,22]. 对于类砷离子, 原子实记为C0≡1s22s22p63s23p63d10, 基组态为C04s24p3 (宇称量子数




C0 {4s24p3}(


→VV4 = C0{4s, 4p, 4d, 4f} →VV5 = VV4 + {5s, 5p, 5d, 5f, 5g}
→VV6 = VV5 + {6s, 6p, 6d, 6f, 6g, 6h}
→VV7 = VV6 + {7s, 7p, 7d, 7f, 7g, 7h, 7i}.











| 离子能级 | Rb V | Sr VI | Y VII | Zr VIII | Nb IX | Mo X | |||||||||||
| ${\varDelta _{\rm{s}}}$ | ${\varDelta _{\rm{m}}}$ | ${\varDelta _{\rm{s}}}$ | ${\varDelta _{\rm{m}}}$ | ${\varDelta _{\rm{s}}}$ | ${\varDelta _{\rm{m}}}$ | ${\varDelta _{\rm{s}}}$ | ${\varDelta _{\rm{m}}}$ | ${\varDelta _{\rm{s}}}$ | ${\varDelta _{\rm{m}}}$ | ${\varDelta _{\rm{s}}}$ | ${\varDelta _{\rm{m}}}$ | ||||||
| 2D5/2 | 218 | 7 | 260 | –14 | 293 | 3 | 332 | –5 | 373 | –15 | 435 | –29 | |||||
| 4P5/2 | –327 | –8 | –359 | –50 | –329 | –33 | –316 | –7 | –320 | 18 | –334 | 36 | |||||
| 2P3/2 | –214 | 4 | –25 | –15 | 60 | –7 | 100 | –2 | 126 | –1 | 151 | –8 | |||||
| 2D3/2 | –212 | 12 | –258 | –71 | –294 | –47 | –336 | 5 | –382 | 66 | –452 | 122 | |||||
| 4P3/2 | 121 | 4 | 150 | 36 | 163 | 39 | 183 | 43 | 215 | 53 | 259 | 64 | |||||
| 2S1/2 | –259 | 8 | –275 | –25 | –265 | 69 | –271 | 1 | –289 | –75 | –319 | –140 | |||||
| 2P1/2 | 468 | 17 | 300 | 102 | 210 | –12 | 181 | 1 | 180 | 17 | 196 | 32 | |||||
| 4P1/2 | 45 | 9 | 206 | –4 | 162 | –21 | 127 | –36 | 97 | –50 | 66 | –59 | |||||
表1Rb V—Mo X离子4s4p4组态能级实验值与理论计算值之差(单位cm–1)的比较
Table1.Comparasion of differences between observed and calculated levels values for the 4s4p4 configuration in Rb V?Mo X
| 离子 | 能级 | Eexp/cm–1 | ELSF/cm–1 | EGLSF/cm–1 | EMCDHF/cm–1 | 本征矢纯度 |
| Rh XIII | 4s24p3 | |||||
| 4S3/2 | 0 | 0 | 0 | 0 | 71% + 21%2P3/2 + 9%2D3/2 | |
| 2D3/2 | 35762 | 35820(58) | 35863(101) | 35992(230) | 66% + 22%4S3/2 + 12%2P3/2 | |
| 2D5/2 | 49151 | 49072(–79) | 49110(–41) | 49284(133) | 100% | |
| 2P1/2 | 72267p | 72237(–30)p | 72279(12)p | 72915(648) | 100% | |
| 2P3/2 | 100673 | 100642(–31) | 100722(49) | 100668(–5) | 68% + 25%2D3/2 + 7%4S3/2 | |
| 4s4p4 | ||||||
| 4P5/2 | 284236 | 284230(–6) | 284111(–125) | 283365(–871) | 87% + 6%2D5/2 + 5%4s24p24d 4P5/2 | |
| 4P3/2 | 308739 | 308504(–235) | 308770(31) | 307548(–1191) | 86% + 6%4s24p24d 4P3/2 + 5%2D3/2 | |
| 4P1/2 | 313989 | 314205(36) | 314507(518) | 313218(–771) | 82% + 11%2S1/2 + 6%4s24p24d 4P1/2 | |
| 2D3/2 | 349977 | 350012(35) | 349934(–43) | 352413(2436) | 69% + 14%4s24p24d 2D3/2 + 7%4P3/2 | |
| 2D5/2 | 359733 | 359722(–11) | 359614(–119) | 361457(1724) | 76% + 15%4s24p24d 2D5/2 + 7%4P5/2 | |
| 2S1/2 | 395765 | 395592(–173) | 395707(–58) | 399771(4006) | 47% + 26%2P1/2 + 9%4s24p24d 2S1/2 | |
| 2P3/2 | 401975 | 401960(–15) | 401883(–92) | 408136(6161) | 55% + 28%4s24p24d 2P3/2 + 8%2D3/2 | |
| 2P1/2 | 439005 | 439001(–4) | 439405(400) | 444364(5359) | 33% + 25%2S1/2 + 23%4s24p24d 2P1/2 | |
| Pd XIV | 4s24p3 | |||||
| 4S3/2 | 0 | 0 | 0 | 0 | 66% + 23%2P3/2 + 10%2D3/2 | |
| 2D3/2 | 40021p | 40092(71)p | 40175(154)p | 40147(126) | 64% + 26%4S3/2 + 11%2P3/2 | |
| 2D5/2 | 54958p | 54852(–106)p | 54935(–23)p | 54996(38) | 100% | |
| 2P1/2 | 79147p | 79112(–35)p | 79189(42)p | 79766(619) | 100% | |
| 2P3/2 | 113280p | 113224(–56)p | 113295(15)p | 113113(–167) | 66% + 26%2D3/2 + 8%4S3/2 | |
| 4s4p4 | ||||||
| 4P5/2 | 305545 | 305506(–39) | 305289(–256) | 304542(–1003) | 87% + 7%2D5/2 + 5%4s24p24d 4P5/2 | |
| 4P3/2 | 333774 | 333809(35) | 333389(–385) | 332461(–1313) | 86% + 6%2D3/2 + 6%4s24p24d 4P3/2 | |
| 4P1/2 | 339032 | 339356(324) | 339321(289) | 338267(–765) | 80% + 13%2S1/2 + 6%4s24p24d 4P1/2 | |
| 2D3/2 | 376537p | 377555(1019)p | 376256(–281)p | 378921(2384) | 67% + 13%4s24p24d 2D3/2 + 9%4P3/2 | |
| 2D5/2 | 388385p | 387860(–525)p | 387887(–498)p | 389905(1520) | 76% + 14%4s24p24d 2D5/2 + 8%4P5/2 | |
| 2S1/2 | 425277p | 424079(–1189)p | 424752(–525)p | 429012(3735) | 45% + 28%2P1/2 + 9%4P1/2 | |
| 2P3/2 | 432003p | 431953(–50)p | 431264(–739)p | 438183(6180) | 53% + 27%4s24p24d 2P3/2 + 10%2D3/2 | |
| 2P1/2 | 468322p | 468558(236)p | 474603(6281)p | 480060(11738) | 30% + 25%2S1/2 + 21%4p24d 2P1/2 | |
| Ag XV | 4s24p3 | |||||
| 4S3/2 | 0 | 0 | 62% + 26%2P3/2 + 12%2D3/2 | |||
| 2D3/2 | 45260 | 45098 | 62% + 29%4S3/2 + 9%2P3/2 | |||
| 2D5/2 | 61485 | 61457 | 100% | |||
| 2P1/2 | 86808 | 87360 | 100% | |||
| 2P3/2 | 127267 | 127015 | 65% + 26%2D3/2 + 9%4S3/2 | |||
| 4s4p4 | ||||||
| 4P5/2 | 327358 | 326566 | 86% + 8%2D5/2 + 5%4s24p24d 4P5/2 | |||
| 4P3/2 | 358887 | 358483 | 81% + 8%4s24p24d 4P3/2 + 6%2D3/2 | |||
| 4P1/2 | 364986 | 364313 | 78% + 15%2S1/2 + 6%4s24p24d 4P1/2 | |||
| 2D3/2 | 403533 | 406661 | 64% + 12%4p24d 2D3/2 + 12%4P3/2 | |||
| 2D5/2 | 417320 | 419813 | 76% + 14%4s24p24d 2D5/2 + 9%4P5/2 | |||
| 2S1/2 | 454883 | 459653 | 43% + 29%2P1/2 + 10%4s24p24d 2S1/2 | |||
| 2P3/2 | 461039 | 469489 | 47% + 26%4s24p24d 2P3/2 + 12%2D3/2 | |||
| 2P1/2 | 510793 | 517520 | 26% + 24%2S1/2 + 20%4s24p24d 2P1/2 | |||
| Cd XVI | 4s24p3 | |||||
| 4S3/2 | 0 | 0 | 58% + 28%2P3/2 + 14%2D3/2 | |||
| 2D3/2 | 51168 | 50914 | 59% + 33%4S3/2 + 8%2P3/2 | |||
| 2D5/2 | 68806 | 68729 | 100% | |||
| 2P1/2 | 95174 | 95759 | 100% | |||
| 2P3/2 | 142703 | 142486 | 64% + 27%2D3/2 + 9%4S3/2 | |||
| 4s4p4 | ||||||
| 4P5/2 | 350419 | 349499 | 85% + 7%2D5/2 + 5%4s24p24d 4P5/2 | |||
| 4P3/2 | 385245 | 385637 | 78% + 9%4s24p24d 4P3/2 + 5%2D3/2 | |||
| 4P1/2 | 391550 | 391382 | 76% + 17%2S1/2 + 5%4s24p24d 4P1/2 | |||
| 2D3/2 | 431691 | 435771 | 60% + 15%4s24p24d 2D3/2 + 10%4P3/2 | |||
| 2D5/2 | 447887 | 451301 | 76% + 13%4s24p24d 2D5/2 + 9%4P5/2 | |||
| 2S1/2 | 486090 | 491836 | 41% + 30%2P1/2 + 12%4s24p24d 2S1/2 | |||
| 2P3/2 | 489808 | 502144 | 33% + 23%4s24p24d 2P3/2 + 15%2D3/2 | |||
| 2P1/2 | 547585 | 556756 | 21% + 29%2S1/2 + 23%4s24p24d 2P1/2 |
表3Rh XIII—Cd XVI离子4s24p3, 4s4p4组态能级和本征矢纯度
Table3.Energy levels and percentage compositions of the 4s24p3 and 4s4p4 configurations for ions from Rh XIII to Cd XVI.
为了得到未知离子组态能级就需要运用GLSF外推(或内插)的方法. 首先计算这些离子的所有能级结构参数, 由这些参数, 进一步计算各离子4s24p3和4s4p4 组态能级值. 具体方法是: 对于已有4s24p3和4s4p4组态各能级实验值的离子, 本文以这些实验能级数据为基础, 用LSF方法获得所有这些离子组态能级结构参数. 对于目前还没有能级实验值的离子, 则通过分析这些参数随着Zc变化的特性, 运用GLSF外推(或内插)方法首先得到这些离子的所有能级结构参数.
图1给出了在GLSF外推计算过程中, 得到的各离子4s24p3, 4s4p4 和4s24p24d组态平均能量Eav随着Zc的变化情况. 根据本文计算, 对于Rb V离子4s4p4 和4s24p24d组态平均能分别为183473和238442 cm–1, 组态平均能之差为54969 cm–1. 而对于Cd XVI离子4s4p4 和4s24p24d组态平均能分别为457425 cm–1和631363 cm–1, 组态平均能之差为173938 cm–1 . 这一结果表明, 尽管这两个组态的平均能之差随着Zc 的增大在扩大, 但是这一差值远小于弱相互作用组态平均能差值的半经验判定标准106 cm–1 (详细分析说明请参阅文献[11]), 表明对于Rb V—Cd XVI离子, 4s24p24d与4s4p4 组态之间存在较强的相互作用. 从图1可以看到各离子组态平均能Eav随Zc变化十分光滑, 因此, 在本文的计算中各未知离子的Eav是通过比值外推计算得到的.
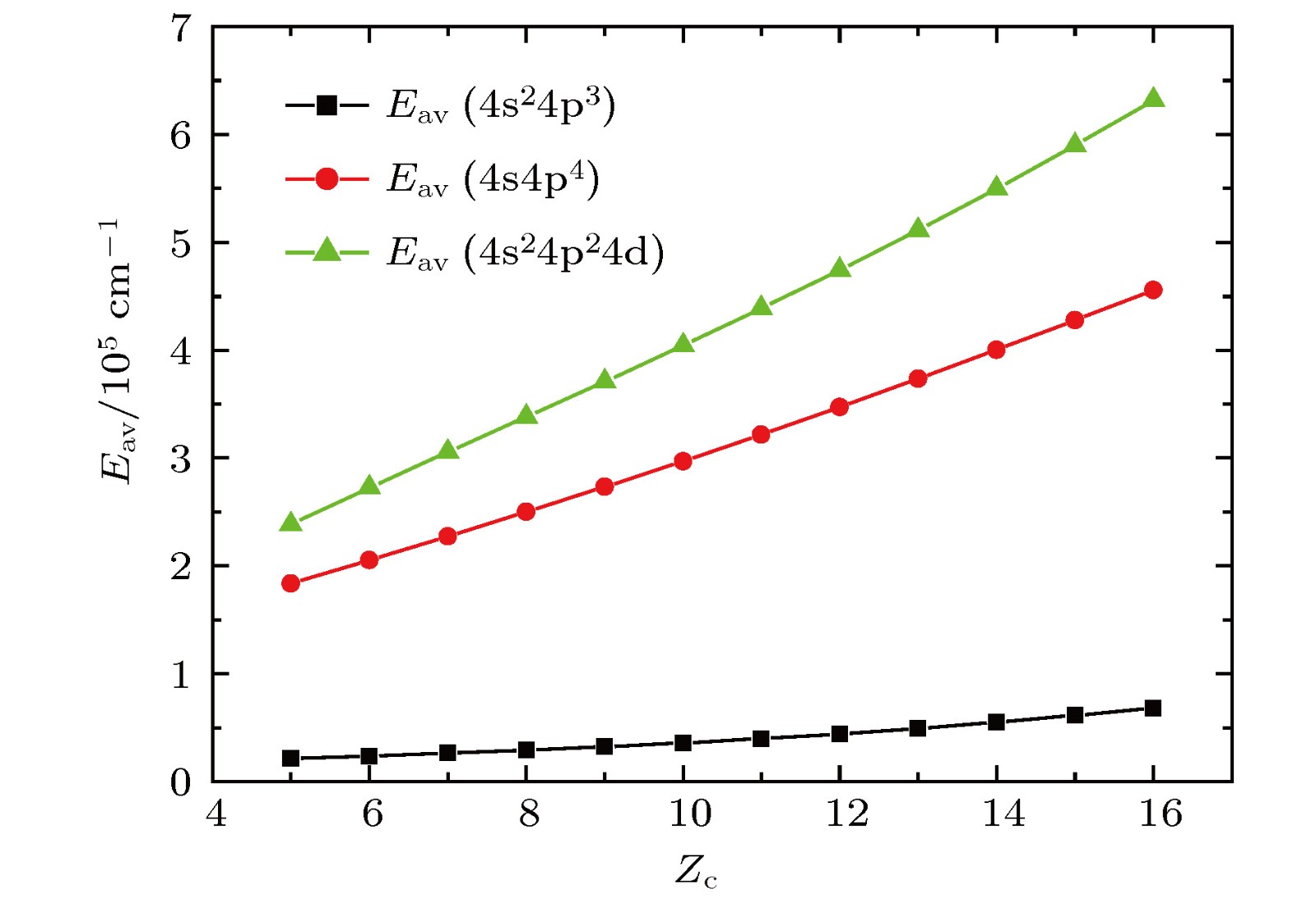 图 1 4p3, 4s4p4和4p24d组态GLSF平均能Eav随Zc的变化
图 1 4p3, 4s4p4和4p24d组态GLSF平均能Eav随Zc的变化Figure1. Variations of GLSF average energy of 4p3, 4s4p4 and 4p24d configurations with Zc.
图2为4s24p3组态能级结构参数中, 库仑相互作用直接积分F2(4p, 4p), 自旋与轨道相互作用积分








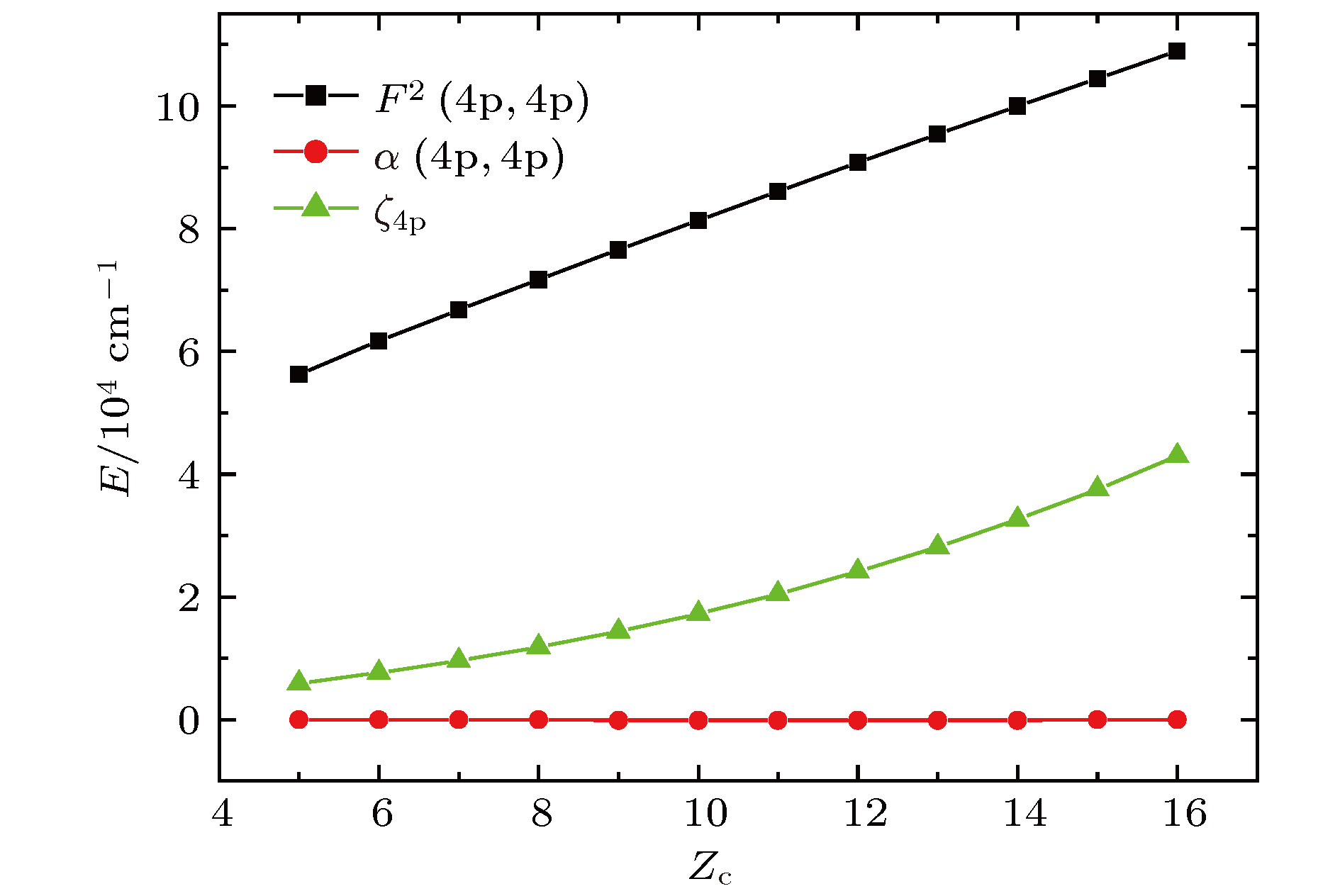 图 2 4p3组态GLSF参数随Zc的变化
图 2 4p3组态GLSF参数随Zc的变化Figure2. Variations of 4p3 configuration GLSF parameters with Zc.
图3表示了在GLSF外推计算过程中各离子4s4p4组态能级结构参数中, 库仑相互作用直接积分F2(4p, 4p)、交换积分G1(4p, 5s)、自旋与轨道相互作用积分




 图 3 4s4p4组态GLSF参数随Zc的变化
图 3 4s4p4组态GLSF参数随Zc的变化Figure3. Variations of 4s4p4 configuration parameters with Zc
表2列出了本文计算得到的Rh XIII, Pd XIV和Ag XV, Cd XVI离子4s24p3, 4s4p4和4s24p44d组态能级结构径向积分参数值(单位为1000 cm–1), 其中HFR, LSF和GLSF分别表示能级结构参数的HFR, LSF和GLSF计算值. 因为Ag XV和Cd XVI离子截至目前没有任何4s24p3和4s4p4组态能级实验值, 所以表2就没有列出LSF结果, 因而Ag XV和Cd XVI离子4s24p3和4s4p4组态能级结构参数值仅为本文GLSF外推计算结果, 所有离子能级与跃迁谱线波长均由这些参数计算得到. 这些参数可以为进一步研究这些离子结构提供重要的参考.
| 离子参数 | Rh XIII | Pd XIV | Ag XV | Cd XVI | |||||||||
| HFR | LSF | GLSF | HFR | LSF | GLSF | HFR | GLSF | HFR | GLSF | ||||
| Eav(4s24p3) | 84293 | 49254 | 49278 | 93187 | 55055 | 54835 | 103082 | 61632 | 114034 | 67464 | |||
| F2(4p, 4p) | 135018 | 95436 | 95372 | 139395 | 99964 | 100188 | 143769 | 104494 | 148147 | 109733 | |||
| $\alpha $(4p, 4p) | 50 | –65 | –70 | 50 | –67 | –90 | 50 | –61 | 50 | –127 | |||
| ${\zeta _{4{\rm{p}}}}$ | 52094 | 28120 | 28143 | 58686 | 32620 | 32403 | 65899 | 37541 | 73770 | 42137 | |||
| Eav(4s4p4) | 465733 | 373048 | 373412 | 494416 | 399046 | 401829 | 524621 | 428489 | 556932 | 460700 | |||
| F2(4p, 4p) | 134929 | 108077 | 108197 | 139305 | 112434 | 113134 | 143679 | 117141 | 148056 | 122299 | |||
| $\alpha $(4p, 4p) | 50 | 25 | 27 | 50 | 68 | 28 | 50 | 34 | 50 | 32 | |||
| ${\zeta _{4{\rm{p}}}}$ | 52046 | 28688 | 28919 | 58636 | 43974 | 34588 | 65846 | 37300 | 73714 | 47415 | |||
| G1(4s, 4p) | 157248 | 125955 | 126094 | 162275 | 130973 | 131787 | 167303 | 136397 | 172339 | 142349 | |||
| Eav(4s24p44d) | 607730 | 512551 | 511233 | 639800 | 1064552 | 547714 | 673345 | 589678 | 708331 | 624580 | |||
| F2(4p, 4p) | 134980 | 113841 | 113545 | 139352 | 231866 | 119342 | 143723 | 125896 | 148098 | 130840 | |||
| $\alpha $(4p, 4p) | 50 | –8170 | –8516 | 50 | –31872 | –12109 | 50 | –9682 | 50 | –19122 | |||
| ${\zeta _{4{\rm{p}}}}$ | 52183 | 26219 | 28282 | 58781 | 37685 | 32566 | 66000 | 44423 | 73875 | 44653 | |||
| ${\zeta _{4{\rm{d}}}}$ | 6735 | 7573 | 6146 | 7648 | 361714 | 9397 | 8649 | 7711 | 9744 | 17964 | |||
| F2(4p, 4d) | 118694 | 100105 | 99845 | 122677 | 204120 | 105062 | 126654 | 110945 | 130628 | 115408 | |||
| G1(4p, 4d) | 146907 | 123899 | 123578 | 151575 | 252202 | 129812 | 156229 | 136856 | 160878 | 142141 | |||
| G3(4p, 4d) | 94580 | 79768 | 79561 | 97657 | 162489 | 83636 | 100724 | 88236 | 103787 | 91702 | |||
| R(4p4p, 4s4d) | 151283 | 121177 | 121309 | 156089 | 125980 | 126756 | 160886 | 131151 | 165684 | 136827 | |||
表2Rh XIII—Cd XVI离子4s24p3, 4s4p4和4s24p44d组态能级结构参数(单位: cm–1)
Table2.Energy parameters of configurations 4s24p3, 4s4p4 and 4s24p44d for ions from Rh XIII to Cd XVI.
表3 列出了Rh XIII, Pd XIV, Ag XV和Cd XVI离子4s24p3和4s4p4组态能级(单位为cm–1)和本征矢纯度(本征矢分量平方)的百分比构成情况, 本征矢纯度小于1%的分量没有列出. Eexp为文献[8]报道的实验值, 数据后面p表示该能级值为该文作者通过分析MCDF理论中的EAL模型计算的能级值与相应实验值之差(EDF – EOBS)随着Zc变化规律得到的预言值. ELSF, EGLSF和EMCDHF 分别为本文LSF, GLSF和MCDHF理论方法(VV7)的计算值, 圆括号“( )”内的数据为本文理论计算值与实验值之差, 圆括号后面有p表示该能级值为本文理论计算值与文献[8]的预言值之差. 对于Rh XIII和Pd XIV离子的4s24p3和4s4p4组态能级, 与已有实验值比较, 本文计算值与实验值之差一般不超过150 cm–1, 而Pd XIV离子4s24p3和4s4p4截止目前还没有足够的实验跃迁数据来建立完整的实验能级结构, 只有4s4p4(4P1/2, 3/2, 5/2)组态三个能级有实验数据, 本文LSF和GLSF计算的结果与这三个实验值十分一致. 基于MCDHF理论的全相对论grasp2K-DEV程序包是当前原子结构计算研究中重要的从头算程序之一, 为了给今后相关理论和实验研究提供更多一些的参考信息, 第六列为采用本文2.2节描述的MCDHF方法计算的结果, 圆括号里的数据为本文计算值与文献[8]的实验值或预言值之差, 显然, MCDHF计算结果(VV7)与实验值的绝对偏差普遍大于1000 cm–1. 例如, 对于Rh XIII离子4s4p4 2P3/2 能级, MCDHF理论值与实验值的绝对偏差最大为6161 cm–1, 最小的4s4p4 4P1/2能级绝对偏差也达到了771 cm–1. 从表3的本征矢纯度构成的百分比就可以看出, 偶宇称组态4s4p4和4s24p24d组态本征矢之间具有非常强的混合. 例如, 对Rh XIII, Pd XIV, Ag XV和Cd XVI离子, 在4s4p4 2P3/2能级本征纯度构成中, 4s24p24d 2P3/2能级本征纯度依次为28%, 27%, 26%和23%, 而在4s4p4 2P1/2的本征矢纯度构成中, 4s24p24d 2P1/2本征矢纯度均超过了20%. 这些情况表明, 在MCHF理论高精度的计算研究中, 对于类砷离子4s4p4组态而言, 4s24p24d对其组态能级结构的组态相互作用效应是不能被忽略的.
表4列出了本文GLSF能级结构参数计算得到的Rh XIII, Pd XIV, Ag XV和Cd XVI离子4s24p3—4s4p4组态能级电偶极跃迁谱线波长(单位: nm)和相应跃迁的加权振子强度(gf × 10,g为下能态的统计权重). 其中,




| 离子跃迁 | $\lambda $/nm | ${\lambda _{\exp }}$/nm | ${\Delta _\lambda }$/nm | gf × 10 |
| Rh XIII | ||||
| 2P3/2—2D3/2 | 40.126 | 40.079b | –0.047 | 0.01 |
| 4S3/2—4P5/2 | 35.197 | 35.186 | –0.011 | 2.58 |
| —4P3/2 | 32.386 | 32.394 | 0.008 | 2.42 |
| 2D5/2—2D5/2 | 32.205 | 32.197 | –0.008 | 3.77 |
| 4S3/2—4P1/2 | 31.795 | 31.852 | 0.057 | 1.17 |
| 2D3/2—2D3/2 | 31.839 | 31.829 | –0.01 | 3.74 |
| 2P1/2—2S1/2 | 30.918 | 1.87 | ||
| 2P3/2—2P1/2 | 29.526 | 29.56 | 0.034 | 3 |
| 4S3/2—2D3/2 | 28.576 | 0.27 | ||
| 2D5/2—2P3/2 | 28.346 | 28.345 | –0.001 | 6.79 |
| 2D3/2—2S1/2 | 27.789 | 27.781 | –0.008 | 2.08 |
| —2P3/2 | 27.32 | 27.31 | –0.01 | 0.59 |
| —2P1/2 | 24.78 | 24.802 | 0.022 | 0.37 |
| 4S3/2—2P3/2 | 24.882 | 24.88 | –0.002 | 0.34 |
| —2P1/2 | 22.758 | 0.01 | ||
| Pd XIV | ||||
| 2P3/2—2D3/2 | 38.008 | 0.01 | ||
| 4S3/2—4P5/2 | 32.749 | 32.732 | –0.017 | 2.55 |
| —4P3/2 | 29.92 | 29.964 | 0.044 | 2.59 |
| 2D5/2—2D5/2 | 29.993 | 29.974 | –0.019 | 3.48 |
| 4S3/2—4P1/2 | 29.411 | 29.499b | 0.088 | 1.2 |
| 2D3/2—2D3/2 | 29.766 | 3.6 | ||
| 2P1/2—2S1/2 | 28.908 | 1.84 | ||
| 2P3/2—2P1/2 | 27.534 | 2.54 | ||
| 4S3/2—2D3/2 | 26.603 | 0.23 | ||
| 2D5/2—2P3/2 | 26.499 | 26.525 | 0.026 | 6.36 |
| 2D3/2—2S1/2 | 25.973 | 2.02 | ||
| —2P3/2 | 25.507 | 25.513 | 0.006 | 0.62 |
| —2P1/2 | 17.106 | 0.82 | ||
| 4S3/2—2P3/2 | 23.148 | 0.37 | ||
| —2P1/2 | 21.009 | 0.01 | ||
| Ag XV | ||||
| 2P3/2—2D3/2 | 36.196 | 0.01 | ||
| 4S3/2—4P5/2 | 30.547 | 2.38 | ||
| —4P3/2 | 27.863 | 2.73 | ||
| 2D5/2—2D5/2 | 28.102 | 3.96 | ||
| 4S3/2—4P1/2 | 27.398 | 1.27 | ||
| 2D3/2—2D3/2 | 27.911 | 3.94 | ||
| 2P1/2—2S1/2 | 27.168 | 1.94 | ||
| 2P3/2—2P1/2 | 26.739 | 0.01 | ||
| 4S3/2—2D3/2 | 24.781 | 0.36 | ||
| 2D5/2—2P3/2 | 25.027 | 6.14 | ||
| 2D3/2—2S1/2 | 24.412 | 2.27 | ||
| —2P3/2 | 24.051 | 0.21 | ||
| —2P1/2 | 21.93 | 0.07 | ||
| 4S3/2—2P3/2 | 21.69 | 0.19 | ||
| —2P1/2 | 19.95 | 0.77 | ||
| Cd XVI | ||||
| 2P3/2—2D3/2 | 34.603 | 0.01 | ||
| 4S3/2—4P5/2 | 28.537 | 2.26 | ||
| —4P3/2 | 25.957 | 2.87 | ||
| 2D5/2—2D5/2 | 26.379 | 4.05 | ||
| 4S3/2—4P1/2 | 25.539 | 1.32 | ||
| 2D3/2—2D3/2 | 26.279 | 3.97 | ||
| 2P1/2—2S1/2 | 25.58 | 1.96 | ||
| 2P3/2—2P1/2 | 26.066 | 0.01 | ||
| 4S3/2—2D3/2 | 23.164 | 0.35 | ||
| 2D5/2—2P3/2 | 23.752 | 4.35 | ||
| 2D3/2—2S1/2 | 22.992 | 2.37 | ||
| —2P3/2 | 22.797 | 0.01 | ||
| —2P1/2 | 21.045 | 0.03 | ||
| 4S3/2—2P3/2 | 20.416 | 0.01 | ||
| —2P1/2 | 18.999 | 1.54 | ||
| 注: ${\lambda _{\exp }}$表示实验值取自文献[8], b表示该谱线为混合谱线 | ||||
表4Rh XIII—Cd XVI离子4s24p3—4s24p4跃迁波长和振子强度(gf × 10)
Table4.Wavelengths and oscillator strengths of transitions 4s24p3?4s24p4 for ions from Rh XIII to Cd XVI.
