1.School of Materials Science and Engineering, Xi’an Shiyou University, Xi’an 710065, China 2.State Key Laboratery of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, China
Fund Project:Project supported by the Young Scientists Fund of the National Natural Science Foundation of China (Grant No. 51702257), the Natural Science Basic Research Plan in Shaanxi Province of China (Grant No. 2018JQ5123), and the Provincial Superiority Discipline of Materials Science and Engineering of Xi’an Shiyou University, China (Grant No. ys37020203).
Received Date:31 August 2018
Accepted Date:15 January 2019
Available Online:01 March 2019
Published Online:20 March 2019
Abstract:L-arginine phosphate monohydrate (LAP) crystal is an excellent nonlinear optical material, its effective nonlinear optical coefficient is about 2?3.5 times that of potassium dideuterium phosphate (KDP) crystal, and its conversion efficiency can achieve up to 90%. The deuterated crystal of LAP has a very high laser damage threshold. Thus, once it was considered as a preferred material to replace KDP crystal for laser inertial confinement fusion and other fields. In addition, the LAP crystal has a much higher stimulated Brillouin scattering (SBS) reflectivity than quartz crystal and also has a lower SBS threshold. Moreover, it exhibits a special reversible phase-change in the variable temperature process, and shows an ultra-long spin-lattice relaxation time at solid-state NMR. In a word, the LAP crystal has shown its uniqueness under the action of energy such as light, heat and magnetic field. However, for these special phenomena, there is no reasonable explanation. Phosphate arginine is responsible for the biological energy storage and transfer in invertebrates as an important phosphorus source, which has a similar chemical composition to that of LAP crystal. The special electrostatic or hydrogen bonding interaction between guanidine and phosphate plays an important role in protein molecule interaction and their biochemical functions. Moreover, the conformational transitions of L-arginine molecule in phosphoric acid solution at different energies have been reported, and the fluorescence emission of L-arginine molecule aggregates can be changed by the interaction between phosphoate and guanidine group. The interaction between phosphoate and guanidine group in crystal structure is also studied as a model of biomolecular interaction. In order to further study the mechanism of interaction between phosphoate and guanidine group and the crystal macroscopic properties, phosphate bis-guanidinoacetate (PBGA) crystal containing the similar phosphoate and guanidine groups has been synthesized and reported. In this paper, the geometry parameters, band structure, electronic density of states, and optical properties of PBGA crystal are investigated by first-principles based the density functional theory. The energy gap of PBGA crystal is 4.77 eV, much smaller than 5.96 eV of KDP crystal. Therefore, the photon transition becomes easier and the corresponding photon absorption is relatively large in PBGA crystal. The top states of crystal valence band are mainly composed of the N-2p of guanidine and the O-2p of carboxyl and phosphate groups. There exists the electron interaction among guanidine, carboxyl and phosphate groups. The optical properties of PBGA crystal are similar in the [100] and [010] orientation, whose linear optical properties are better than those of [001] when the incident photon energy is less than 10 eV. The strong energy loss peak at 9.46 eV in the [001] orientation is due to the electronic transition of N-2p on guanidine group in the valence band, and its distribution is narrow. Thus the optical properties of [001] orientation are limited. The present research establishes a good foundation for further understanding and studying the intergroup interactions and optical properties in PBGA crystal. Keywords:phosphate bis-guanidinoacetate/ first principles/ electronic structure/ optical properties
全文HTML
--> --> --> 1.引 言在无脊椎动物体内, 精氨酸磷酸(phosphate arginine, PA)作为主要的磷源和能量存储单元而广泛存在. PA分子实现其生化功能的主要机制在于其分子中磷酸与精氨酸胍基间特殊的静电作用[1-4], 除此之外, 磷酸与胍基间特殊的非共价键作用在很多其他生物分子功能中也扮演着重要角色[5,6]. 在晶体中也存在类似的磷酸胍基. 20世纪70年代, Cotton等[7]就以磷酸甲基胍、磷酸二甲基胍晶体结构中磷酸与胍基间氢键作用作为生物体中该类非共价键作用的模型, 其研究中提到L-精氨酸磷酸盐(LAP)晶体结构中磷酸与胍基的关系更类似于生物分子的化学状态. LAP晶体是一种性能优异的非线性光学材料, 其有效非线性光学系数约为磷酸二氢钾(KDP)晶体的2—3.5倍, 可以实现高达90%以上的转化效率, 且具有超高的激光损伤阈值, 其氘化晶体(DLAP)曾被认为是可以取代KDP晶体用于激光惯性约束核聚变等领域的首选材料[8-10], 因而有关该晶体的结构与性质, 以及L-精氨酸衍生物晶体已有大量研究与报道[11,12]. 在探索LAP晶体特异性的过程中, 非生物环境下, 磷酸溶液中L-精氨酸分子在不同能量下的构象转变被发现[13]; 磷酸与胍基间的作用会影响L-精氨酸分子聚集体产生的荧光发射现象也有报道[14]. 为了进一步研究磷酸胍基间的作用与晶体宏观性质机制的重要关联, 已设计制备了含有类似磷酸与胍基的磷酸双乙酸胍(phosphate bis-guanidinoacetate, PBGA)晶体[15]. PBGA晶体是属于三斜晶系, P-1空间群, 晶胞参数为: a = 7.776(2) ?, b = 8.113(2) ?, c = 12.459(3) ?, $\alpha $ = 89.591(2)°, $\beta $ = 89.146(3)°, $\gamma $ = 61.37°, V = 689.8(3) ?3, D = 1.599 g /cm3, Z = 1. 图1为PBGA晶体的分子构型及沿a轴的晶胞投影图, 两个胍基乙酸与一个磷酸根阴离子组成了PBGA分子, 其中两个乙酸胍解离形式不同, 一个的羧基完全失去质子形成COO-基团, 另一个羧基则没有失去质子. 从投影图可以看出, 沿晶体a轴方向的氢键连接形成了明显的层状结构. 图 1 PBGA晶体的(a)分子构型和(b)沿a轴方向的投影图 Figure1. (a) Molecular configuration and (b) projection viewed along a-axis of PBGA crystal
图3(a)是PBGA晶体的总态密度与轨道分态密度, 根据轨道态密度对总态密度的不同贡献, 价带可以分为如下三个区域: –15 eV以下的低能级, –15 eV至–9 eV的中能级以及–9 eV至0 eV的高能级. 可以看出价带低能级主要是s轨道的贡献, 价带中能级是s与p轨道的综合贡献, 价带高能级与导带中的底部主要由p轨道组成, 导带顶部主要来自s轨道. PBGA晶体的能带间隙为4.77 eV, 比KDP晶体的5.96 eV小得多[23]. 根据电子跃迁理论, 价带和导带之间的光子跃迁至少需要克服与带隙大小相同的能量窗. 因此, 当355 nm激光辐照晶体时, 它可以吸收两个光子引起电子跃迁. PBGA的窄带隙使得价带和导带之间的光子跃迁变得容易, 对应光子的吸收相对较大. 图 3 PBGA晶体的分态密度 Figure3. Partial density of states of PBGA crystal
图3(b)给出了总态密度与晶体中各原子分态密度, 结合图3(a)可以看出, 价带中低能级主要是由O-2s和N-2s贡献, –15 eV至–5 eV间能态由多个原子s与p轨道综合贡献, –5 eV至0 eV间能态主要来自于C-2p, O-2p和N-2p, 其中价带顶部主要由O-2p和N-2p组成, 导带的底部主要来自C-2p. 图4(a)为O-2p总态密度与不同基团上O-2p态密度分布, 图4(b)为N-2p总态密度与不同基团上N-2p态密度分布, 可以看出价带顶部电子能态主要来自于乙酸胍分子胍基上N-2p, 磷酸根与羧基上O-2p的贡献, 其中带电乙酸胍分子相对不带电乙酸胍分子, 其O-2p占据比例更高. 因此, 当光子入射进入PBGA晶体时, 胍基、磷酸根与羧基上最容易发生电子跃迁. 图 4 PBGA晶体中(a)氧和(b)氮的p轨道分态密度 Figure4. The p-orbital partial density of states of (a) oxygen and (b) nitrogen in PBGA crystal
23.2.光学性质 -->
3.2.光学性质
PBGA晶体在[100], [010]以及[001]三个方向介电函数虚部与能量的关系曲线如图5所示. 从图5可以看出, PBGA晶体在三个方向的首个介电峰分别位于6.22, 5.78和6.07 eV, 且在[001]方向强度最强, [100]方向最弱. 结合态密度图3可知, 首个特征峰主要是由PBGA中N-2p与O-2p态由价带顶部跃迁至导带引起. 图 5 PBGA晶体的介电函数虚部与能量关系 Figure5. Relationship between energy and imaginary part of dielectric function of PBGA crystal



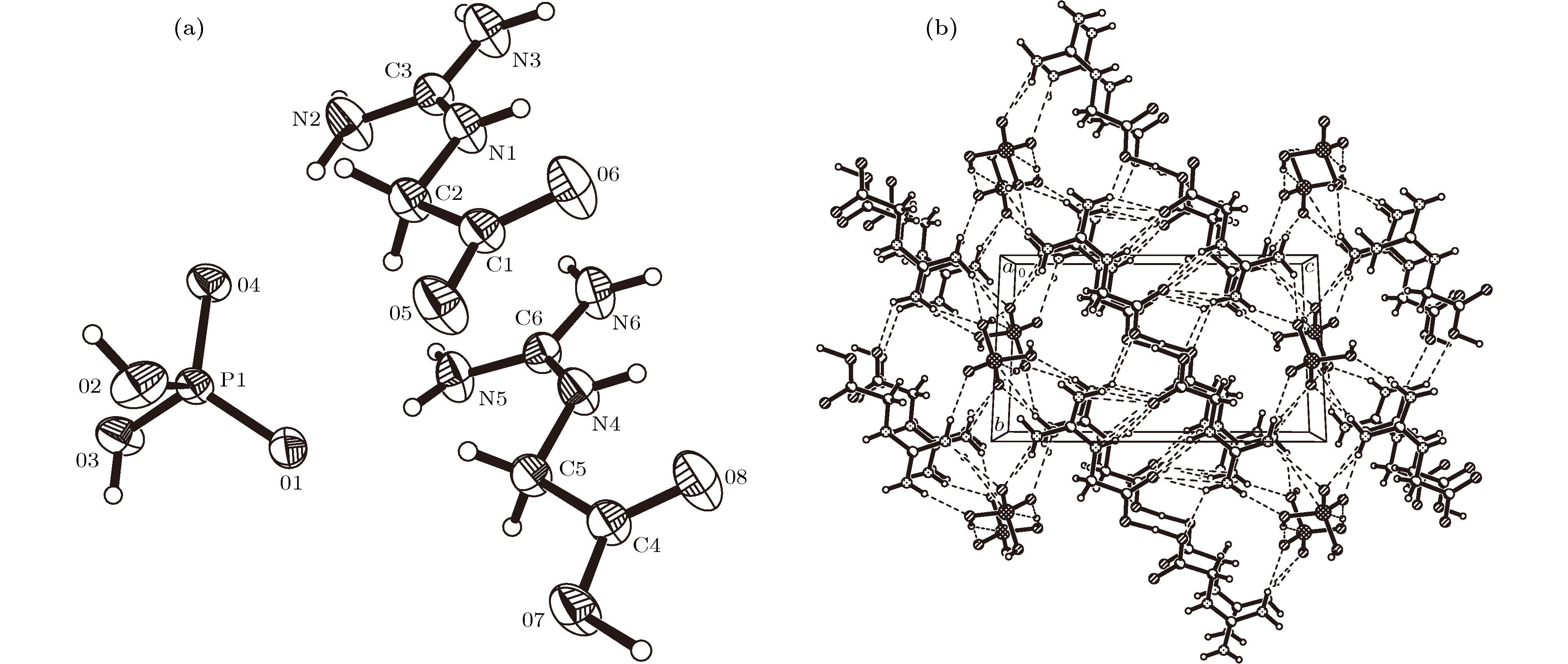 图 1 PBGA晶体的(a)分子构型和(b)沿a轴方向的投影图
图 1 PBGA晶体的(a)分子构型和(b)沿a轴方向的投影图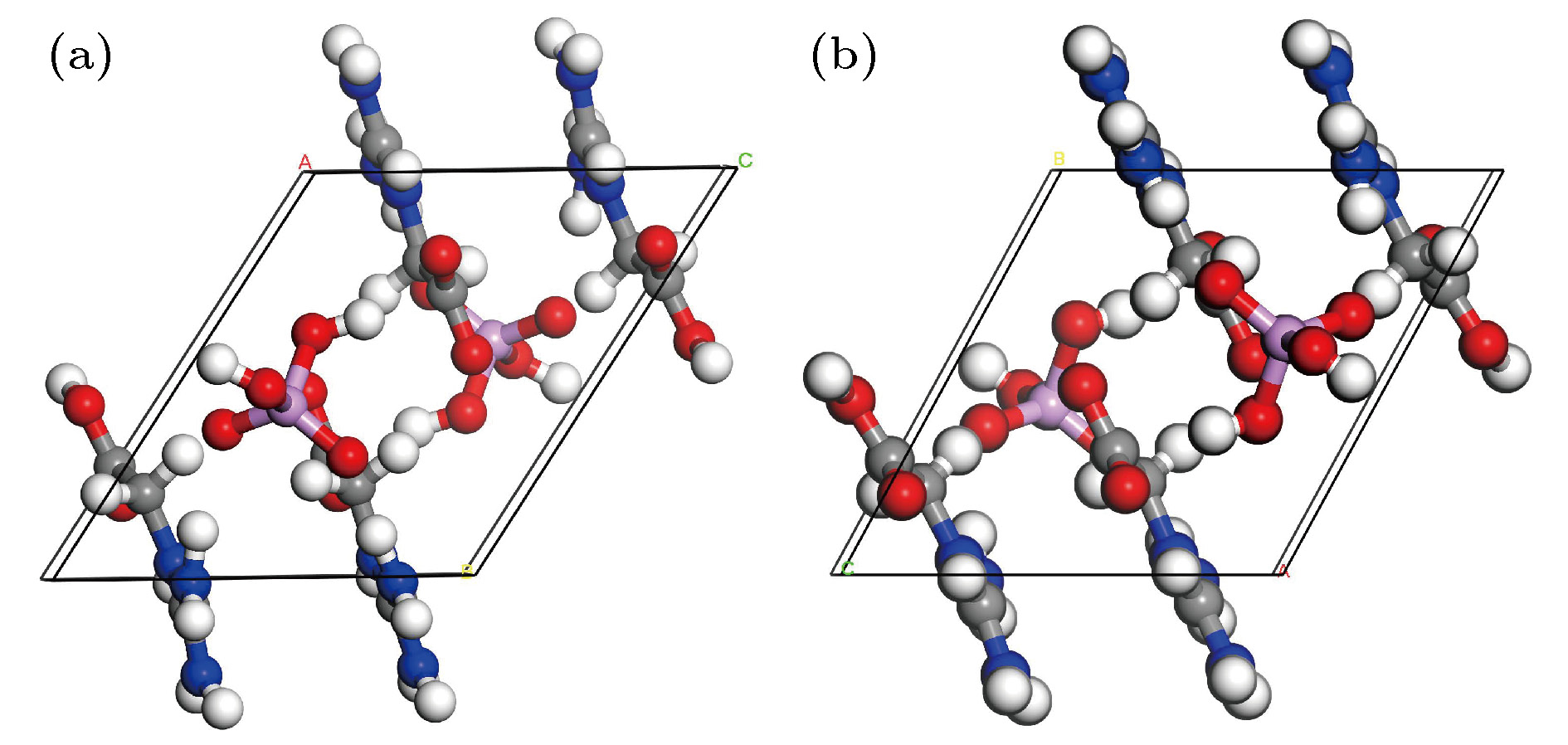 图 2 PBGA晶体(a)原始模型与(b)优化后模型
图 2 PBGA晶体(a)原始模型与(b)优化后模型









 图 3 PBGA晶体的分态密度
图 3 PBGA晶体的分态密度 图 4 PBGA晶体中(a)氧和(b)氮的p轨道分态密度
图 4 PBGA晶体中(a)氧和(b)氮的p轨道分态密度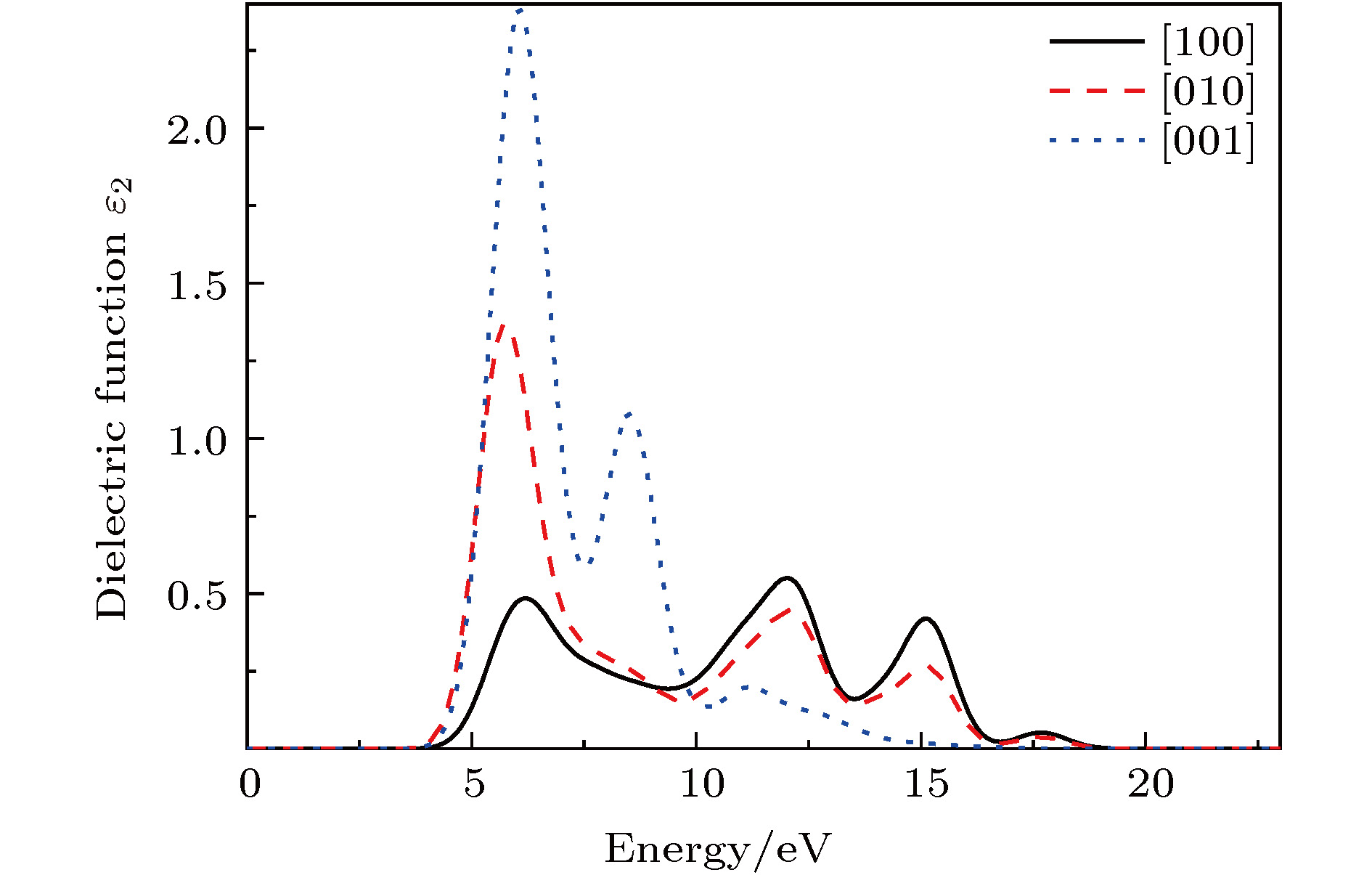 图 5 PBGA晶体的介电函数虚部与能量关系
图 5 PBGA晶体的介电函数虚部与能量关系 图 6 PBGA晶体的(a)折射率和(b)吸收系数
图 6 PBGA晶体的(a)折射率和(b)吸收系数
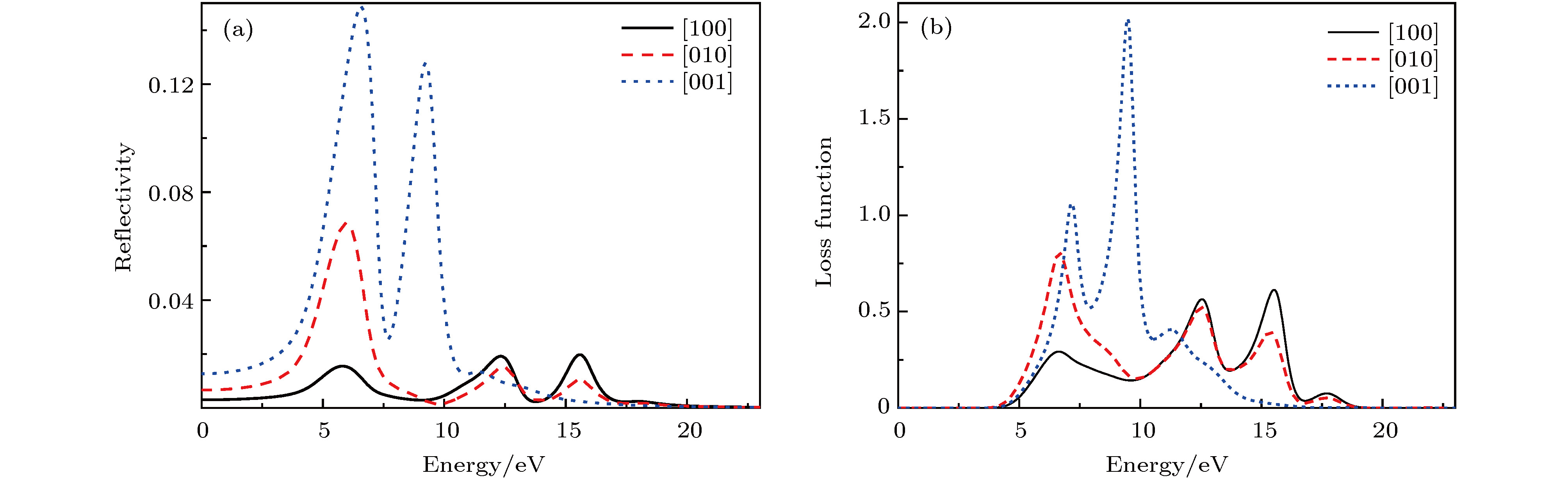 图 7 PBGA晶体的(a)反射率与(b)能量损失函数
图 7 PBGA晶体的(a)反射率与(b)能量损失函数