全文HTML
--> --> -->
与此同时, NiTi合金还具有良好的生物相容性. 在NiTi合金表面与生物基体相接触的过程中, 应避免合金中有毒的Ni离子溶入体液. 换言之, NiTi合金表面原子结构、表面稳定性对其在生物医用领域具有重要意义. 因此, 现阶段关于NiTi合金表面的研究越来越多. Hua等 [8]研究了O2在B2-NiTi(110)和(100)表面的吸附特性, 发现O2仅与距离最近的Ti原子发生作用, 且利于在(100)面上吸附. 朱建新等[9]计算了 B2-NiTi (110)和(100)表面的原子结构和电子结构, 发现(100)表面化学活性较高, 而(110)表面稳定性更优. 此外, 研究人员还大量研究了NiTi合金的表面改性技术: 表面涂覆[10,11]、阳极氧化[12,13]和表面化学沉积[14,15]等, 提高了合金表面的抗腐蚀性能.
此外, NiTi合金可以作为陶瓷增强金属基复合材料的界面过渡物相, 以形成柔性界面, 提高界面结合强度. 2017年, Li等[16]研究了氧化锆增韧氧化铝(ZTA)陶瓷增强铁基复合材料, 发现在复合材料界面处添加NiTi合金可以显著提高陶瓷颗粒与铁基材料之间的润湿性, 提高界面的结合强度. 该界面性质的改善与NiTi合金表面的原子结构、电子结构以及化学性质密切相关.
Nigussa和St?vneng [17]研究了B2-NiTi (111), (110), (001)表面与O2结合特性, 发现相比于(110)和(001)表面, (111)表面最难与O2发生相互作用; Vishnu和Strachan[18]研究了B2-, B19-, B19'- 和BCO-NiTi (110)表面的原子结构和表面能, 发现B2-NiTi (110)的表面能最低; Sandoval和Haskins[19]研究了B19'-NiTi (100)和(001)表面结构和稳定性, 发现(001)表面在有限温度下的稳定性相较于0 K时较高. 以上文献均对B2-和B19'-NiTi合金的部分低指数表面稳定性、O2吸附特性等进行了深入研究. 然而, 综合B2-和B19'-NiTi合金的所有低指数表面, 进行更全面、系统的稳定性评价、电子结构表征和原子弛豫分析等却未见报道. 因此, 本文采用第一性原理计算方法, 系统地研究了B2-和B19'-NiTi所有低指数表面, 尤其是密排面的原子结构、电子结构、表面能及表面稳定性等.
本文在构建NiTi合金表面原子构型过程中, 考虑到B2和B19'晶格对称性的原因, 共构建出表面构型有:
1) B2-NiTi的(100) = (010) = (001), (101) = (110) = (011), (111)表面, 同时考虑到表面原子终止类型不同, (100)和(111)表面原子层既可以由Ni终止, 也可以由Ti终止, 而 (101)密排面表面原子层则由Ni和Ti共同终止, 分别标注为“_Ni”“_Ti”和“_NiTi”, 由此共构建出5种不同的B2- NiTi表面构型, 图1(a)—(c)分别给出了密排面(101), (100)和(111)以Ni原子终止的表面构型.
 图 1 NiTi合金的低指数表面原子构型 (a) B2-NiTi(101)_NiTi; (b) B2-NiTi_Ni; (100); (c) B2-NiTi(111)_Ni; (d) B19’-NiTi(010)_NiTi; (e) B19'-NiTi(001)_Ni; (f) B19'-NiTi(110)_Ni; 绿色球和黑色球分别代表Ni, Ti原子
图 1 NiTi合金的低指数表面原子构型 (a) B2-NiTi(101)_NiTi; (b) B2-NiTi_Ni; (100); (c) B2-NiTi(111)_Ni; (d) B19’-NiTi(010)_NiTi; (e) B19'-NiTi(001)_Ni; (f) B19'-NiTi(110)_Ni; 绿色球和黑色球分别代表Ni, Ti原子Figure1. Atomic low-index surface configurations of NiTi alloy: (a) B2-NiTi(101)_NiTi; (b) B2-NiTi(100)_Ni; (c) B2-NiTi(111)_Ni; (d) B19’-NiTi(010)_NiTi; (e) B19'-NiTi(001)_Ni; (f) B19'-NiTi(110)_Ni.
2) B19'-NiTi的(100), (010), (001), (101), (110), (011)和(111)表面, 其中(100), (001), (101), (110)和(111)五种低指数表面既可以由Ni终止, 也可以由Ti终止, 而(010)密排面和(011)面则由Ni和Ti共同终止, 共得到12种不同的B19'-NiTi表面构型, 图1(d)—(f)分别给出了密排面(010), (001)和(110)以Ni原子终止的表面构型.
表面模型的厚度通过对表面层数测试基础上获得: 首先, 通过几何优化列出优化后各层原子的相对位移(弛豫)量; 然后, 得到每个低指数表面构型中间层原子相对位移可忽略(


3.1.NiTi合金体相特性
表1为本文计算所得晶格常数(a, b, c, V)、体模量(B)和剪切模量(G), 与其他参考文献值[3,21-28]对比, 误差在2%以内, 证实了本文计算的可靠性.


| Compounds | a/? | b/? | c/? | V/?3 | G/MPa | B/MPa | ${\Delta _{\rm{r}}}H$/eV·atom–1 |
| B2-NiTi | 3.015 (3.033a, 3.016b, 3.01c) | — | — | 27.402 (27.901a, 27.434b, 27.27c) | 69.0 (73d) | 155.5 (142.3a, 150.0b, 142e) | –0.374 (–0.35f) |
| B19'-NiTi | 4.646 (4.685g, 4.813h, 4.631i) | 4.108 (4.035g, 4.121h, 4.10i) | 2.898 (2.941g, 3.007h, 2.885i) | 55.705 (55.080g, 58.610h, 54.84i) | 26.2 (23j) | 148.9 (147k, 158f) | –0.328 |
| 注: a, b, d, g, h, j, k为理论参考值, Ref.[21-22,23,3,26,27-28]; c, 实验参考值, Inorganic Crystal Structure Database (ICSD) #105413; e, 实验参考值Ref.[24]; f, 实验参考值, Ref.[25]; i, 实验参考值, ICSD #240195. | |||||||
表1NiTi合金的晶格常数、密度、剪切模量、体模量及生成焓
Table1.Calculated cell parameters, density, shear modulus, bulk modulus and formation enthalpy.
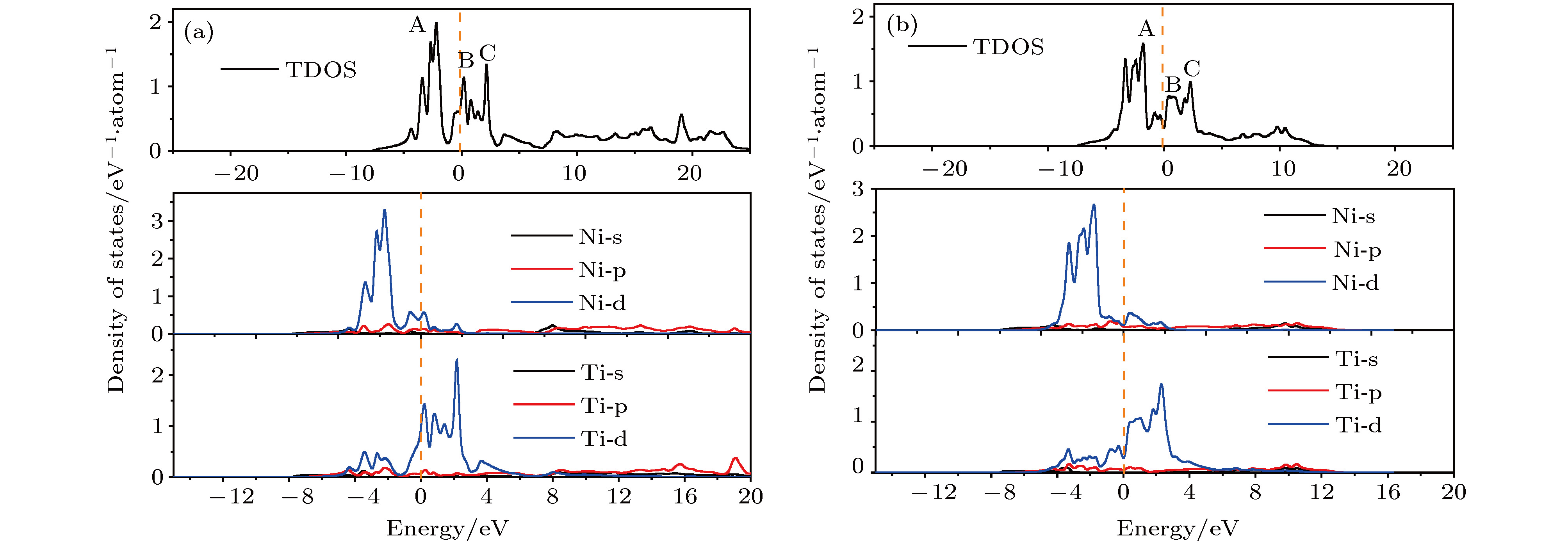 图 2 NiTi合金的DOS曲线 (a)B2-NiTi; (b)B19'-NiTi
图 2 NiTi合金的DOS曲线 (a)B2-NiTi; (b)B19'-NiTiFigure2. DOS curves for NiTi alloys: (a) B2-NiTi; (b) B19'-NiTi.
2
3.2.NiTi合金的低指数表面
33.2.1.表面结构弛豫
本文研究的表面构型在结构弛豫过程中并未发现表面结构坍塌、重构等物理现象. 为了进一步考察以上B2-和B19'-NiTi共17种表面原子构型在弛豫过程中原子位移情况, 本文以

| Surface | Termination | Interlayer | Slab thickness | ||||
| 3 | 5 | 7 | 9 | 11 | |||
| (101) | Ni, Ti | ${\varDelta _{12}}$ | –9.67 | –10.42 | –9.99 | –9.88 | –9.91 |
| ${\varDelta _{23}}$ | –0.19 | 1.18 | 1.99 | 1.78 | |||
| ${\varDelta _{34}}$ | –0.83 | –0.65 | –0.47 | ||||
| ${\varDelta _{45}}$ | 0.43 | 1.15 | |||||
| ${\varDelta _{56}}$ | 0.57 | ||||||
| (100) | Ni | ${\varDelta _{12}}$ | –1.77 | –8.13 | –8.89 | –8.78 | –8.93 |
| ${\varDelta _{23}}$ | 3.85 | 3.48 | 2.79 | 2.76 | |||
| ${\varDelta _{34}}$ | –0.72 | –0.41 | 0.17 | ||||
| ${\varDelta _{45}}$ | 1.11 | –0.37 | |||||
| ${\varDelta _{56}}$ | 2.18 | ||||||
| Ti | ${\varDelta _{12}}$ | –2.24 | –21.68 | –17.08 | –15.16 | –16.68 | |
| ${\varDelta _{23}}$ | 15.42 | 12.14 | 7.11 | 10.78 | |||
| ${\varDelta _{34}}$ | 0.01 | 4.03 | 2.82 | ||||
| ${\varDelta _{45}}$ | –5.72 | –1.93 | |||||
| ${\varDelta _{56}}$ | 1.53 | ||||||
表2B2-NiTi表面原子层位移相对体相间距的变化率随切片厚度的变化 (%)
Table2.Relaxations of B2-NiTi surfaces with different terminations and slab thickness given in terms of the change of interlayer spacing in percent of the bulk spacing (%).
由表2可知, 原子位移变化率主要集中在表面的3个原子层内, 第1, 2原子层的原子弛豫最为剧烈, 并且最外层和次外层间的原子振荡以Ti终止表面最为显著, Ni终止表面次之, Ni和Ti共同终止的(101)密排面的原子振荡最小. 当B2-NiTi合金表面层数大于7时,


3
3.2.2.表面稳定性
表面能描述了将体相材料建成表面构型所需的能量, 可用来衡量表面构型的稳定性, 表面能越小, 表面越稳定. 对于密排面和其他符合化学计量比表面构型的表面能按照(1)式计算:而非化学计量比表面的表面能按照(2)式计算:




本文首先研究了表面原子层厚度对表面能的影响. 图3为富Ti条件下B2-和B19'-NiTi合金密排面及其他非密排面的表面能随表面构型原子层数的变化曲线. 由图可知, 当B2-NiTi合金的密排面原子层厚度大于7层时, 表面能开始迅速收敛. 因此为了获得合理的热力学能量数据, 本文B2-NiTi的表面构型需选用7层及以上原子切片, 这与前述表面构型的原子层结构驰豫结果推论一致. 针对B19'-NiTi表面, 以富Ti侧B19'-NiTi (001)表面构型为例, 在保证构型中心对称的基础上, 分别研究了10, 14, 18, 22和26层表面原子层厚度时的表面能. 由图3可见, 当原子层数在12层以上时, 表面能均得到了完美的收敛. 本文B19'-NiTi(001)表面构型即选用了14层的表面原子层厚度.
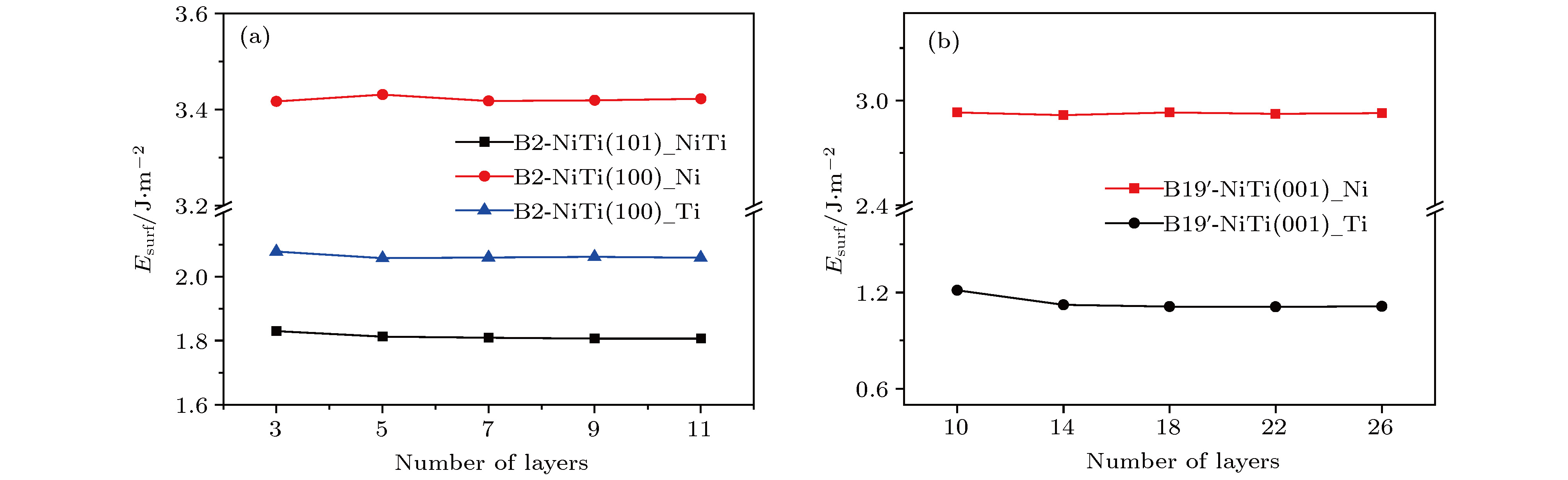 图 3 富Ti条件下表面能随表面构型原子层数目变化曲线 (a) B2-NiTi; (b) B19'-NiTi
图 3 富Ti条件下表面能随表面构型原子层数目变化曲线 (a) B2-NiTi; (b) B19'-NiTiFigure3. Under the condition of Ti-rich, the surface energy varies with the number of atomic layers of the surface configuration: (a) B2-NiTi; (b) B19'-NiTi.
在进一步确定表面构型原子层厚度的基础上, 本文首先根据(1)式计算得到B2-NiTi (101) 密排面的表面能为1.81 J/m2, B19'-NiTi (010) 密排面的表面能为1.93 J/m2, 计算结果与文献[18]计算所得B2-NiTi(101)表面能1.62 J/m2相差较小, 并且B2-NiTi密排面的表面能低于B19'-NiTi密排面. 这与两种异构体的电子密度分布密切相关. 图4为B2-和B19'-NiTi合金密排面的电荷密度分布. 图中任何区域的电子密度均大于0, 离域性的价电子云证明了NiTi合金的金属性特征. NiTi合金中化学键主要表征为Ni—Ti—Ni—Ti链式金属键, 这些链式金属键弥散分布于自由电子气中, 结合图2所示的DOS曲线可知NiTi化合物体系金属性主要归因于Ni和Ti原子d轨道的贡献. B2-NiTi密排面上的电荷密度分布相对于B19'-NiTi密排面更均匀, 同时测得B2-NiTi (101)表面Ni和Ti原子平均间距为2.632 ?, 而B19'-NiTi (010)表面原子平均间距为2.662 ?. 可见B2-NiTi (101)表面较小的原子间距为Ni和Ti原子间的金属键结合强度提升提供了良好环境, 这种较均匀分布的高结合强度Ni—Ti键会提高(101)密排面的稳定性.
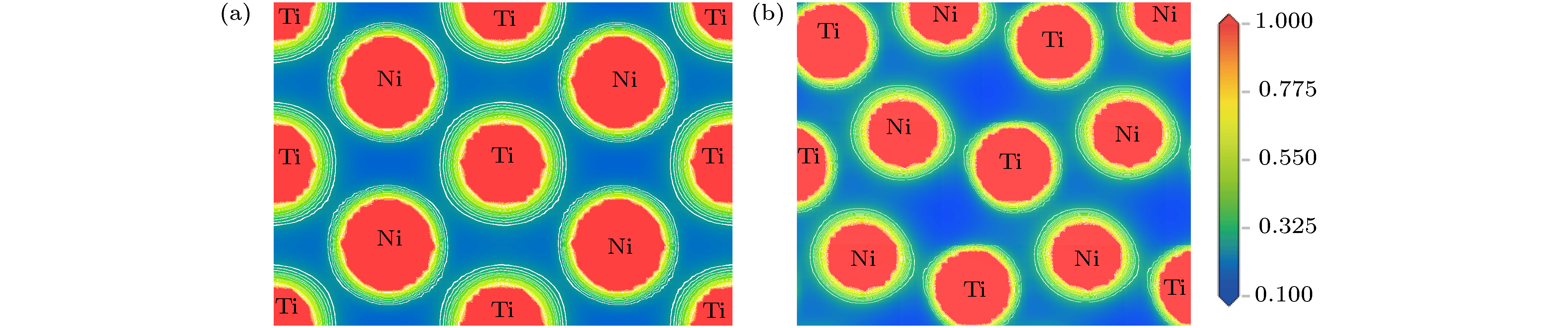 图 4 B2-NiTi和B19'-NiTi体相密排面的电荷密度分布 (a) B2-NiTi (101); (b) B19'-NiTi (010)
图 4 B2-NiTi和B19'-NiTi体相密排面的电荷密度分布 (a) B2-NiTi (101); (b) B19'-NiTi (010)Figure4. Charge density distribution of dense plane of bulk B2-NiTi and B19'-NiTi: (a) B2-NiTi (101); (b) B19'-NiTi (010).
非化学计量比表面的表面能计算结果如图5所示. 对于B2-NiTi而言, 其密排面的表面能显著低于其他各低指数表面的表面能, 显示出较高的稳定性. B19'-NiTi非化学计量比表面的表面能曲线见图5(b), 可知在大部分Ti的化学势范围内, B19'-NiTi (001)_Ti的表面能最低, 其表面稳定性最高, 而(100)_Ni的表面能最高, 稳定性最差. 在大部分Ti化学势范围内, (010)密排面的表面能低于其余各表面的表面能. 并且从图5可以看出, 所有非化学计量比表面的表面能与Ti化学势密切相关. 随着Ti化学势改变, 非化学计量比表面的稳定性会发生轻微变化. 因此非化学计量比表面的稳定性取决于Ti的化学势, 在实验过程中可通过改变Ti的性质来控制这类表面的稳定性.
 图 5 非化学计量比表面的表面能随Ti化学势的变化 (a) B2-NiTi; (b) B19'-NiTi
图 5 非化学计量比表面的表面能随Ti化学势的变化 (a) B2-NiTi; (b) B19'-NiTiFigure5. Surface energies of non-stoichiometric surface versus Ti chemical potentials: (a) B2-NiTi; (b) B19'-NiTi.
以B2-NiTi (100), (111)表面和B19'-NiTi (110), (001)表面为例, 考察其表面能与表面原子结构的关系, 发现无论Ni或者Ti终止, B2 (100)面的表面能均大于(111)面; B19' (110)面的表面能大于(001)面. 对于B2-NiTi合金, 比较图1(b)和(c), 其(100)面上第一层Ni原子成键为双重键(two-fold bonding); 而(111)面上Ni原子成键为四重键, (111)表面原子的配位数高. 因此, 表面稳定性通常与构型表层原子配位数有关. 配位数越高, 表面原子密度越高, 原子之间的结合强度越强, 表面的稳定性提高. 密排面的原子配位数最大, 且在大多数Ti化学势范围内, 密排面的稳定性也最高. 这进一步说明了表面能与表面原子的配位数有关, 表面稳定性随着配位数的增加而提升[31], 密排面的稳定性较高.
3
3.2.3.表面电子性质
1) (100), (010), (001)表面以B2-NiTi (100)_Ni和B19'-NiTi (010)_NiTi表面为例, 体相总DOS和表面层投影DOS曲线如图6(a)和图6(b)所示. 从图中可以看出, 费米面处的DOS值存在较大差异. 对于B2(100)_Ni表面, 第1, 3, 5层由Ni原子构成, 第2, 4层由Ti原子构成, 但第1层Ni原子的DOS和第5层Ni原子的DOS曲线显示出较大差异, 这是因为第5层Ni原子的性质已经接近于体相NiTi的性质, 而第1层Ni原子仅形成了表面态. 并且对于B2-NiTi表面, 第3层Ni原子的DOS分布曲线已经近似于体相总DOS曲线, 这也为上文中表面层数的选择提供了依据. 对于B19'-NiTi (010)_NiTi密排面, 层投影DOS曲线与体相DOS曲线相似, 这是因为由Ni和Ti原子共同终止的密排面上, 原子密度和配位数较高, 表面上Ni和Ti原子之间的相互作用可补偿表面悬键的存在, 因此, 密排表面稳定性较高, 表面性质更接近于体相. 相反, 非密排面上配位数低, 存在相对较多的悬键, 表面稳定性变差.
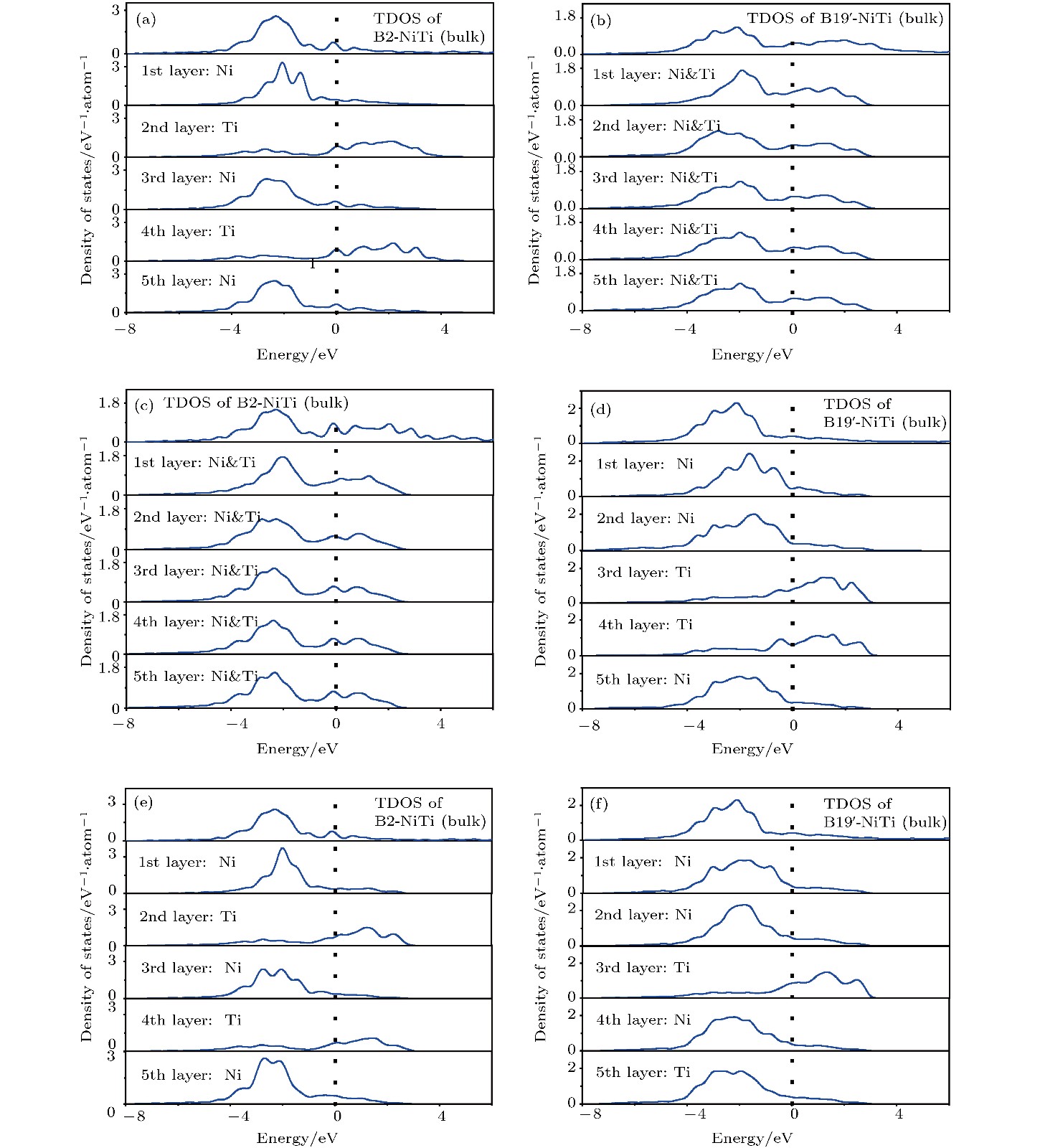 图 6 B2-和B19'-NiTi体相总DOS和表面构型层投影DOS曲线 (a) B2-NiTi (100)_Ni; (b) B19'-NiTi (010)_NiTi; (c) B2-NiTi (101)_NiTi; (d) B19'-NiTi (101)_Ni; (e) B2-NiTi (111)_Ni; (f) B19'-NiTi (111)_Ni
图 6 B2-和B19'-NiTi体相总DOS和表面构型层投影DOS曲线 (a) B2-NiTi (100)_Ni; (b) B19'-NiTi (010)_NiTi; (c) B2-NiTi (101)_NiTi; (d) B19'-NiTi (101)_Ni; (e) B2-NiTi (111)_Ni; (f) B19'-NiTi (111)_NiFigure6. Total DOS of B2 and B19'-NiTi body phase and surface configurations layer projected DOS curves: (a) B2-NiTi (100)_Ni; (b) B19'-NiTi (010)_NiTi; (c) B2-NiTi (101)_NiTi; (d) B19'-NiTi (101)_Ni; (e) B2-NiTi (111)_Ni; (f) B19'-NiTi (111)_Ni.
2) (101), (110), (011)表面
图6(c)和图6(d)为B2-NiTi (101)_NiTi和B19'-NiTi (101)_Ni表面层投影DOS和体相总DOS分布. 如图所示, 相比于B2-NiTi (101)密排面, B19'-NiTi (101)非密排面的最外层附近Ni原子的分DOS逐渐向高能区转移, 这可能是因为Ni原子周围配位数的减少导致表面存在高能化倾向, 进而表面稳定性降低.
为了深入分析表面电子结构, 本文绘制了B2-和B19'-NiTi部分低指数晶面弛豫后的总电子密度分布. 如图7所示, 表面弛豫仅发生在表面3个原子层, 且电荷密度沿真空层方向快速递减. 对于图7(a)和(d)所示的B2-NiTi (101)_NiTi和B19'-NiTi (101)_Ni而言, 可以看出强Ni-Ti金属键存在于B2-NiTi (101)_NiTi密排面的相邻原子层, B19'-NiTi (101)_Ni非密排面相邻原子层仅存在较弱的Ni—Ti金属键, 表面与亚表面层间电子密度值较低. 另外, B2-NiTi (101)_NiTi密排面的第1和第2层原子之间存在Ni—Ti—Ni链式金属键, 这一原子链能使表面的表面能降低, 有助于提高表面构型的稳定性, 而B19' (101)_Ni表面原子层之间Ni和Ti原子距离较远, 表面链式金属键不明显, 稳定性较差.
 图 7 B2-和B19'-NiTi表面构型总电子密度分布 (a) B2-NiTi (101)_NiTi; (b) B2-NiTi (111)_Ni; (c) B19'-NiTi (010)_NiTi; (d) B19'-NiTi (101)_Ni
图 7 B2-和B19'-NiTi表面构型总电子密度分布 (a) B2-NiTi (101)_NiTi; (b) B2-NiTi (111)_Ni; (c) B19'-NiTi (010)_NiTi; (d) B19'-NiTi (101)_NiFigure7. Total electron density distribution of B2- and B19'-NiTi surface configurations: (a) B2-NiTi (101)_NiTi; (b) B2-NiTi (111)_Ni; (c) B19'-NiTi (010)_NiTi; (d) B19'-NiTi (101)_Ni.
3) (111)表面
对于B2-和B19'-NiTi, 其Ni终止类型(111)表面的总DOS和层投影DOS如图6(e)和图6(f) 所示, 可以看出从第1层到第5层, 层投影DOS曲线越来越接近于体相总DOS曲线的形状. B2-NiTi(111)_Ni表面的电子密度分布如图7(b)所示, 从图中可知(111)表面原子间距较大, 表面层原子倾向于与亚表面的原子成键, 导致表面原子分布呈现波浪状, 粗糙度大; 结合计算所得B2-NiTi (111)_Ni表面Ni—Ti键的mulliken重叠布居数为–0.38, 原子间显示出较强的排斥力, 因此表面稳定性变差.
2) B2-NiTi (101)密排面上的原子间距相对于B19'-NiTi (010)密排面小, 原子间更容易成键且电子密度分布更为均匀, 保证了B2-NiTi (101)密排面较高的稳定性.
3) NiTi合金中非化学计量比表面的表面能与Ti的化学势密切相关, 因此在实验过程中可通过控制Ti的偏压来改变这类表面的相对稳定性.
4) NiTi合金表面稳定性与表面原子的配位数有关. 表面原子的配位数越高, 表面稳定性越高. (101)密排面的表面原子成键表现为Ni—Ti—Ni链式金属键, 在大多数Ti的化学势范围内, 稳定性较高.
