全文HTML
--> --> -->微调带隙以及价带和导带的位置, 对于设计二维材料设备的性能是非常重要的, 而在众多的二维材料中, 二维过渡金属硫化物材料已经显示出优异的电子和光学特性[11?17], 如高功率/质量光收集[18]和极高灵敏度光子检测[19]. 在这些二维过渡金属硫化物材料中, 单层MoS2因在柔性电子器件[20]、光致发光[21,22]、谷电子[23,24]和场效应晶体管中[25]发挥了重要的作用而备受关注. 单层MoS2作为层状二维过渡金属硫化物中的典型代表, 可以通过机械或基于溶剂的剥离方法从块体中剥离出单层MoS2纳米片. MoS2单层结构类似于石墨烯, 三明治夹心的S-Mo-S层通过弱范德瓦耳斯力相互作用结合在一起. 块体MoS2是1.20 eV的间接带隙[26], 相比之下, 单层MoS2 1.80 eV的直接带隙使其具有更优异的催化性能[27?29]、光学性能[30]和润滑性能[31?33]等. 此外, 它还具有良好的电子特性, 包括高迁移率[25]和室温下大的开关比[34], 使其在许多科学领域中都有广泛的应用.
Elias等[35] 报道, 石墨烯的电子性质可以通过可逆氢化来进行调控. 受到该研究结果的启发, 人们对其他二维材料也进行了类似的研究[36], 其中包括对氢化单层MoS2的研究. 人们预计氢化单层MoS2将显示出许多与原始MoS2不同的独特性质. 氢化单层MoS2可以有效地提高电导率并且可能引起其从半导体到金属的转变. 此外, MoS2表面上的氢化团簇结构可以产生导电的纳米通道[37]. 如果使用具有导电属性的纳米图案结构作为导体区域, 电子或光学器件的直接制造可以通过使用传统的器件制造工艺就能实现[38]. Sundberg等[39]报道, 在不同的温度下, 块体MoS2可以实现可逆的氢化, 将表面吸附的H原子从边缘位置移动到MoS2表面的基底区域进行调控. 然而, 对于氢化程度而言, 氢化MoS2单层的稳定性和其电子性质变化的机理仍未解释清楚. 因此, 为了利用氢化的单层MoS2来制造新颖的电子和光学器件, 有必要深入研究氢化的单层MoS2的稳定性和电子特性.
本文基于第一性原理计算, 研究了氢化单层二硫族化合物(MX2 (M = Mo, W; X = S, Se, Te))的稳定性和电子特性. 研究发现, 在单层MX2的层间有一个比其表面更稳定的氢吸附位点, 其结合能的变化与阴阳离子序数存在线性关系. 氢原子从MX2的表面经层间穿越到另一表面具有一个相对较小的扩散势垒, 约为0.8 eV, 这和热质子穿过单层石墨烯的势垒值非常接近[40]. 氢化对单层MX2的电子特性也会产生较大的影响, 表面氢化诱导出自发的磁性, 使得带隙急剧减少, 但依然保持原单层MX2体系的半导体性质. 然而, 层间氢化使得单层MX2直接实现了从半导体到金属的过渡; 层间氢化单层MX2 (M = Mo, W; X = S, Se)使体系产生磁性, 而阴离子为Te2? 时, 磁性却几乎消失. 这些结果可以为理解MX2层的氢功能化提供理论指导.


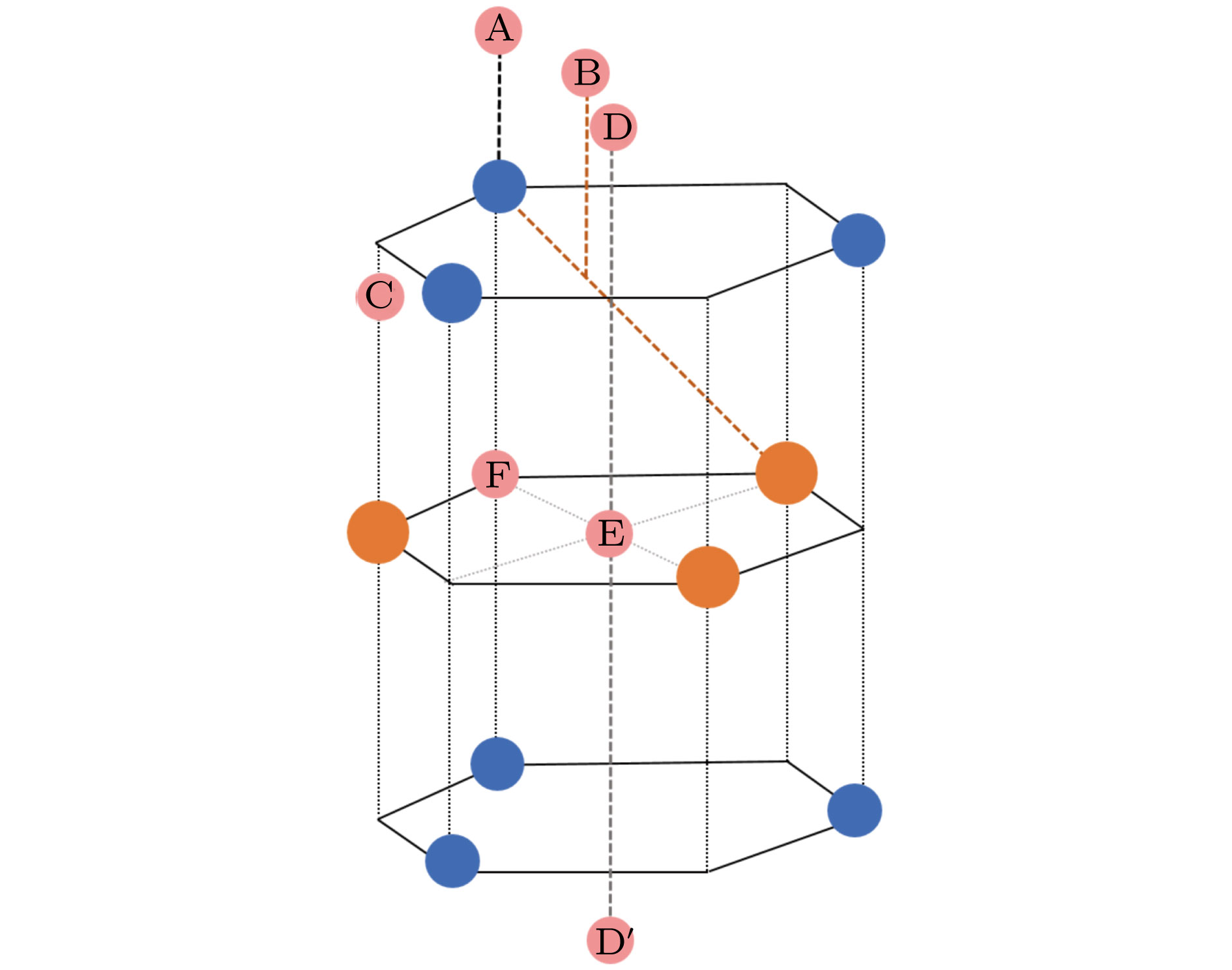 图 1 单层MX2的结构模型及可能的H吸附位点示意图, 其中蓝色, 橙色和粉色分别表示阴离子(X2?)、阳离子(M4+)和氢原子; A—F表示氢原子可能的吸附位点
图 1 单层MX2的结构模型及可能的H吸附位点示意图, 其中蓝色, 橙色和粉色分别表示阴离子(X2?)、阳离子(M4+)和氢原子; A—F表示氢原子可能的吸附位点Figure1. Structural model and possible H adsorption sites of single-layer MX2. Blue, orange and pink balls correspond to anions (X2?), cations (M4+) and hydrogen atoms, respectively. A?F shows a possible adsorption site for hydrogen atom.
| AS | Binding energy/eV | M—H键长/? | |||||
| A | B | C | D | E | F | ||
| MoS2 | ?0.38 | ?0.41 | 0.21 | ?0.27 | ?0.44 | 0.99 | 1.893 |
| MoSe2 | ?0.19 | ?0.16 | ?0.26 | ?0.30 | ?0.91 | 0.47 | 1.950 |
| MoTe2 | ?0.10 | ?0.35 | ?0.10 | ?0.52 | ?1.42 | ?0.19 | 1.996 |
| WS2 | ?0.09 | ?0.16 | 0.30 | ?0.06 | ?0.26 | 1.58 | 1.901 |
| WSe2 | ?0.01 | ?0.09 | 0.50 | ?0.18 | ?0.80 | 0.69 | 1.970 |
| WTe2 | 0.09 | ?0.05 | ?0.64 | ?0.45 | ?1.39 | ?0.18 | 2.005 |
表1氢原子吸附在单层MX2上的可能吸附位点(AS)的结合能, 及氢原子与阳离子(M4+ = Mo, W)的键长
Table1.Binding energy of possible adsorption sites (AS) of hydrogen atoms adsorbed on single-layer MX2, and M—H (M4+ = Mo, W) bond lengths.
从表1的计算结果可以发现, 氢原子吸附在单层MoS2和WS2表面上时最稳定的表面吸附位点为B点, 这和文献[27, 44]的计算结果一致. 有趣的是, 同作为二维过渡金属硫化物家族, 氢原子吸附在MoSe2, MoTe2和WSe2层表面上的最稳定的吸附位点为D点, 及WSe2层表面上的最稳定的吸附位点为C点. 氢原子在单层MX2表面的稳定吸附位点不同, 可能是由表面原子的电负性强弱造成的. 然而, 当氢原子吸附在单层MX2 (M = Mo, W; X = S, Se, Te)层间时, 所有的单层MX2体系都有相同的最稳定吸附位点E. 同时发现对于同一单层MX2体系, 层间吸附位点E比表面稳定吸附位点B (D/C)要相对更稳定一些, 因为层间吸附位点E具有相对更低的吸附能量. 例如, 当氢原子吸附在单层MoS2和WS2表面时, 最稳定的表面吸附位点B的结合能分别为?0.41 eV和?0.16 eV. 而当氢原子吸附在单层MoS2和WS2层间时, 吸附位点E的结合能分别为?0.44 eV和?0.26 eV. 这是因为H原子是呈电负性的, 而单层MX2中, 由于S的电负性相对于Se和Te的电负性更强, 使得MoS2和WS2与H的结合强度反而变弱. 因此, 对于单层MoS2和WS2来说, 层间稳定吸附位点和表面稳定吸附位点具有一个相对较小的能量差. 这个能量差在单层MoSe2, MoTe2, WSe2和WTe2中更容易发现. 如当氢原子吸附在单层MoSe2时, 可以很明显地看出, 最稳定的层间吸附位点E的结合能(?0.91 eV)低于最稳定的表面吸附位点D的结合能(?0.30 eV). 因此对于单层MX2来说, 其层间有一个比其表面更稳定的氢吸附位点(如图1中E点所示), 这是由于电负性的氢原子与表面被吸附原子形成共价键吸附, 而与层间被吸附原子形成配位键吸附, 并且由于H原子与金属之间的库仑相互作用力更强, 结合力更强, 使得H原子在层间吸附位点E时相对表面吸附位点更靠近阳离子, 从而更加稳定.
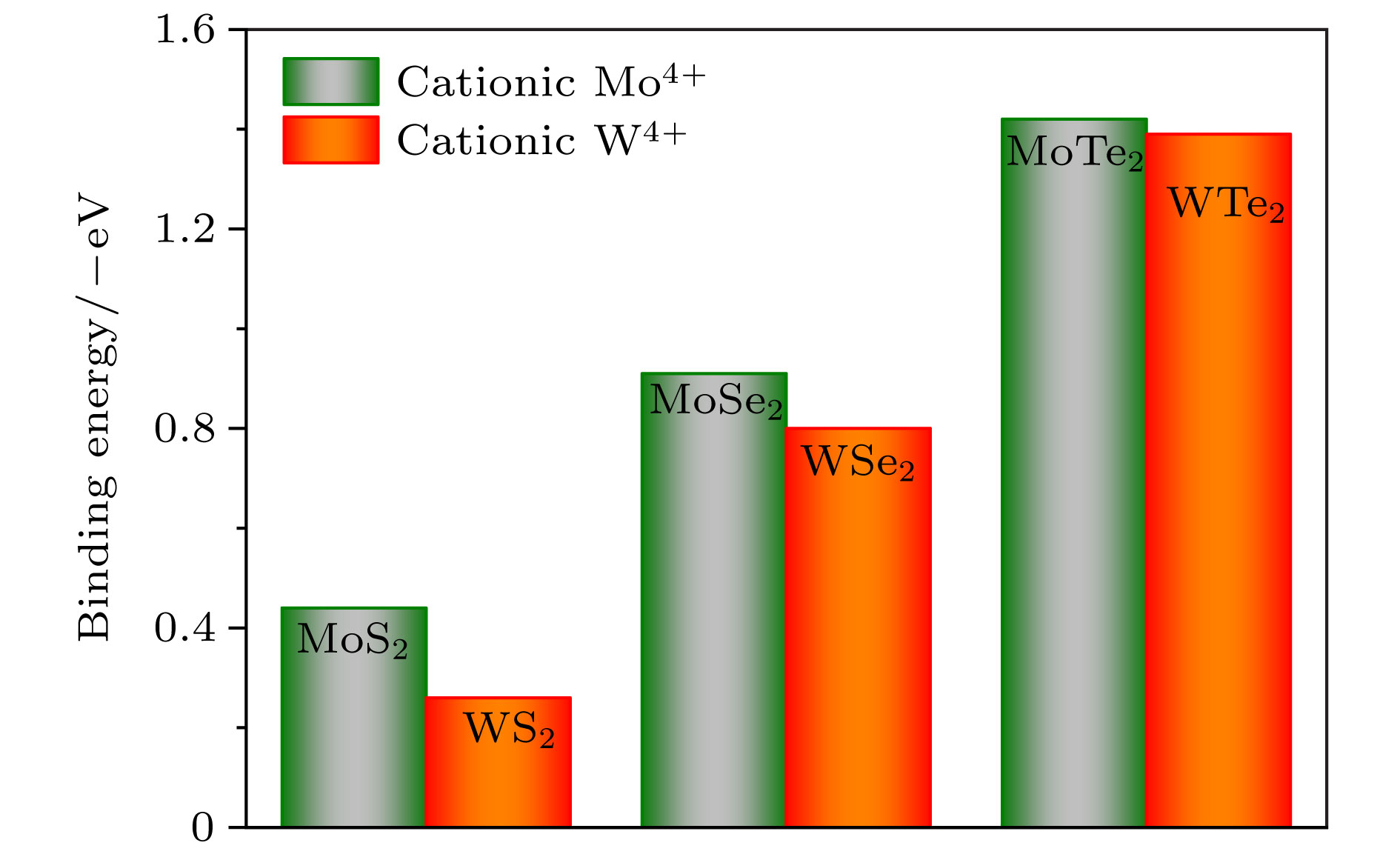 图 2 单层MX2的稳定吸附位点结合能统计图
图 2 单层MX2的稳定吸附位点结合能统计图Figure2. Binding energy of the stable adsorption sites of single-layer MX2.
在所考虑的吸附位点中, 选择出单层MX2(M = Mo, W; X = S, Se, Te)的最稳定的吸附位点, 这些最稳定吸附位点的Eb总结在图2中. 从图2中可以很直观地看出单层MX2稳定H吸附位点的变化规律. 由于氢原子显示出较大的电负性, 阴离子S, Se, Te的电负性(离子性)是依次减弱的, 共价键逐渐变强. 因此当同阳离子(

为了进一步解释氢原子稳定吸附在单层MX2层间的原因, 接下来计算了氢原子从MX2层一个表面(如图1中D点所示)穿过层间(图1中E点所示)扩散到另一个表面(图1中D'点所示)的势垒分布, 计算结果如图3所示. 从图3可以发现, 氢原子穿过MoS2层有一个相对较小的扩散势垒, 势垒大小约0.9 eV (< 1 eV), 穿过其他MX2层的扩散势垒都大于1.2 eV, 这和热质子在室温下穿过单层石墨烯的势垒0.8 eV是非常接近的. 我们知道, 当势垒超过1 eV时, 扩散就难以进行, 而1 eV以下的势垒扩散相对较容易发生. 因此, 氢原子穿过单层MS2仅有一个0.9 eV的扩散势垒, 只要在一定的条件下, 该扩散过程还是相对较容易进行的. 同时也观察到一个有趣的现象, 氢原子从表面D点扩散到D'点的过程中, 有一个比表面D(D')点更低的能量点, 该点正是图1中的E吸附位点. 此结果与结合能的计算结果是一致的.
 图 3 氢原子穿过单层MX2的扩散势垒(扩散路径为从单层MX2表面D点穿过层间E点扩散到另一个表面D'点)
图 3 氢原子穿过单层MX2的扩散势垒(扩散路径为从单层MX2表面D点穿过层间E点扩散到另一个表面D'点)Figure3. Diffusion barrier of hydrogen atoms through a single layer of MX2. The diffusion path is from the D point of surface through the interlayer E point to the other surface D' point.
电荷转移是衡量掺杂原子和被掺杂体系之间相互作用的一个标准. 掺杂原子与被掺杂体系之间电荷转移的越多, 电子得失程度越大, 说明掺杂原子与被掺杂体系之间的电子输送越大, 体系越稳定. 采用Bader电荷分析方法, 计算了氢原子稳定吸附在单层MX2的电荷转移情况(稳定吸附在图1中的E位点), 如图4所示. 氢原子吸附在单层MoS2, MoSe2和 MoTe2层间E位点时, 氢原子从单层MoS2, MoSe2和 MoTe2体系分别得到0.235, 0.267, 0.338个电子. 因此, 可以很容易知道, 同阳离子Mo4+时, 随着阴离子原子序数的增加, 由于阴离子电负性的逐渐减弱, 氢原子从该单层体系中得到的电子数也是逐渐增加. 类似的情况在单层WS2, WSe2和 WTe2中也可以见到, 如图4(b)所示. Bader电荷计算再次验证了同阳离子(M4+)的单层体系中, 随着阴离子(X2?)原子序数依次增加, 氢化单层MX2结构越稳定这一结论的正确性.
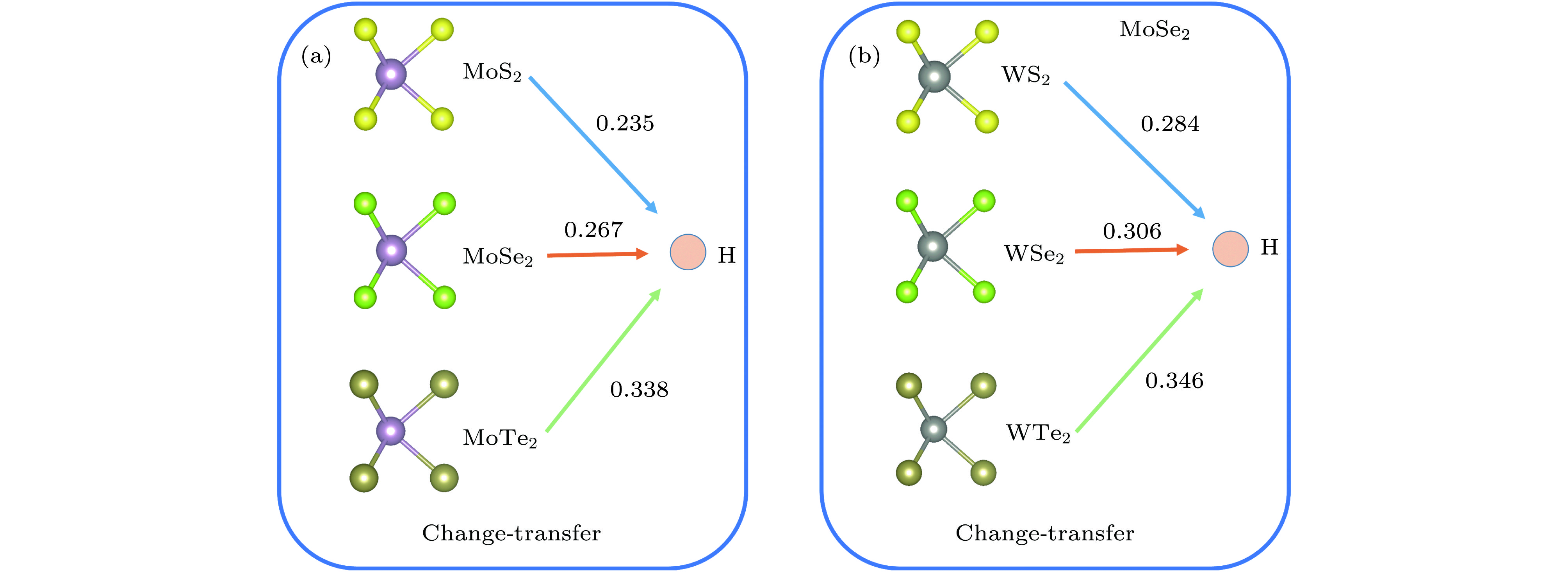 图 4 氢原子稳定吸附在单层 MX2的电荷转移分布图(计算的电荷转移为MX2层E位点的转移情况, 箭头指示电荷转移的方向)
图 4 氢原子稳定吸附在单层 MX2的电荷转移分布图(计算的电荷转移为MX2层E位点的转移情况, 箭头指示电荷转移的方向)Figure4. Charge transfer profile of hydrogen atoms adsorbed on a single-layer MX2. The calculated charge transfer is the transfer of the E site of the MX2 layer, and the arrows indicate the direction of charge transfer.
最后研究了氢化对单层MX2电子结构的影响. 为了更清晰地了解氢化时的能带结构变化, 首先计算了4 × 4超胞的单层纯MX2的电子能带结构以作为比较. 如图5所示, 单层纯MX2都是直接带隙的半导体, 其价带顶和导带底都位于K/K'高对称点, 带隙大小变化范围约为1.00—1.80 eV[45].
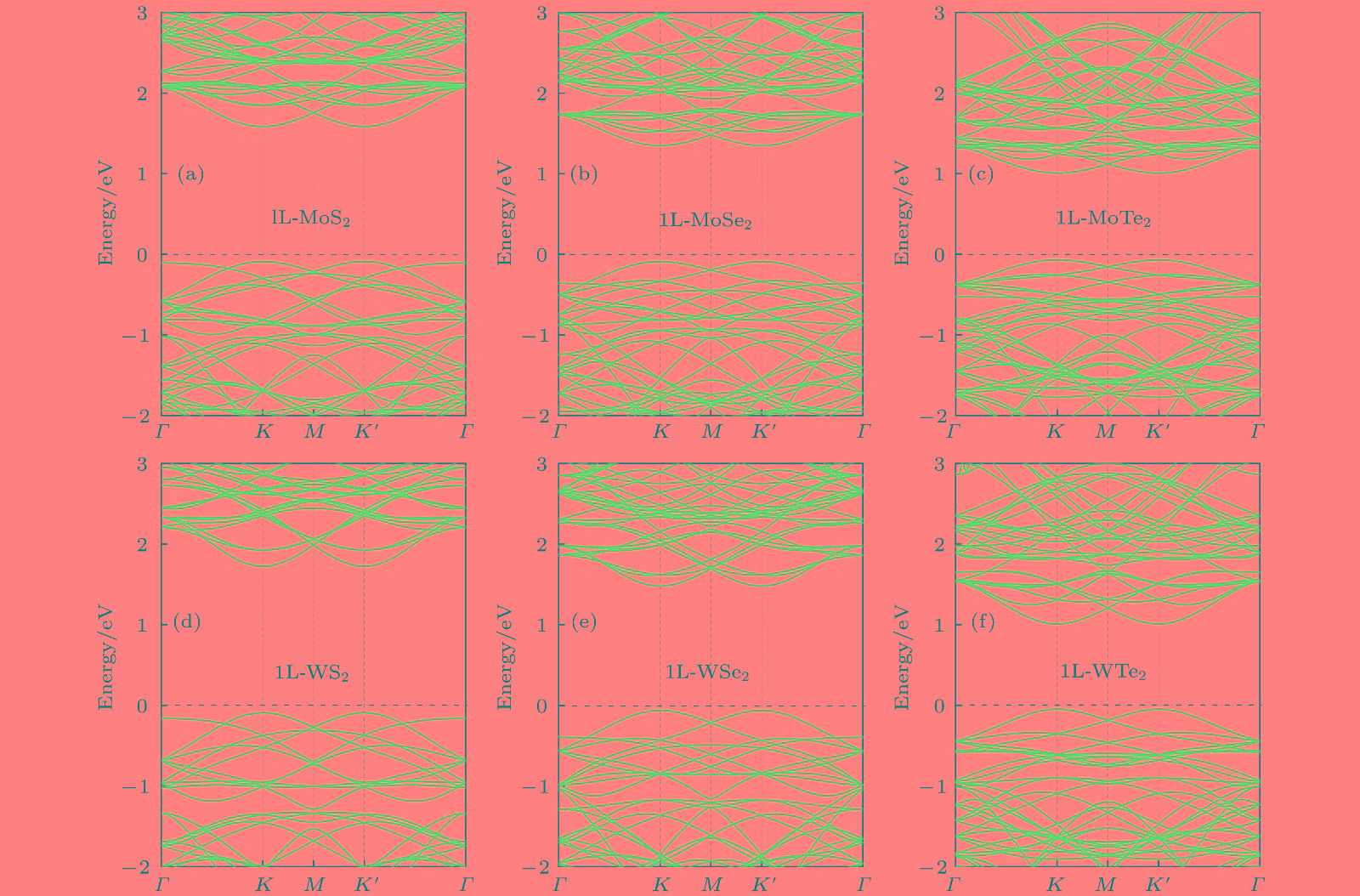 图 5 4 × 4单层MX2 (1L-MX2)超胞的能带结构图(费米能级用黑色虚线表述并设为0 eV)
图 5 4 × 4单层MX2 (1L-MX2)超胞的能带结构图(费米能级用黑色虚线表述并设为0 eV)Figure5. Band structures of 4 × 4 monolayer MX2 (1L-MX2) supercell. The fermi level is represented by dotted black line and set as 0 eV.
图6为单个H原子位于表面稳定吸附位点时的MX2能带结构, 通过对比纯单层MX2的能带结构图, 不难发现表面氢化时氢原子能级进入了纯的MX2的带隙, 从而导致体系带隙急剧减小, 其带隙也相应从1.0—1.8 eV减小到约0.3—0.6 eV, 但依然保持原单层MX2体系的半导体性质. 这其实是由于表面氢化时, 氢原子与MX2中的X原子形成共价键吸附, 此作用相当于吸附掺杂, 而能带急剧减小也正是由于杂质(氢)能级加入所导致. 此外, 从图5和图6中的能带图对比可以看出, 没氢化时, 纯MX2的能带是简并的, 即自旋向上和向下的能带具有相同的能量. 而表面氢化时, MX2的自旋向上和自旋向下的能带能量不再简并, 产生了自旋极化劈裂, 说明此时体系具有一定的磁性, 即发生了从无磁性体系到磁性体系的过渡. MX2中磁性产生的原因主要是由于H原子电负性较强于MX2中的X原子, 其有从X原子得到电子的趋势, 导致MX2体系的电荷分布将发生重新分配, 致使体系的电子云在H原子吸附位点产生小的畸变, 从而使体系表现出微弱且局域化的磁矩. 当然, 相较于直接用H原子替代掺杂X原子以使体系少电子从而产生较大的本征磁场而言(即p型替代掺杂), 这种通过吸附H原子使MX2体系诱导出的磁性是十分微弱且局域化的, 但这也不妨是一种使无磁性的MX2产生磁性的思路.
 图 6 表面氢化MX2能带结构图(蓝色和红色线分别表示自旋向上和自旋向下)
图 6 表面氢化MX2能带结构图(蓝色和红色线分别表示自旋向上和自旋向下)Figure6. Band structures of surface hydrogenation MX2. Blue and red correspond to spin-up and spin-down, respectively.
然而, 当一个H原子稳定吸附在4 × 4的单层MX2超胞中E吸附位点时, 如图7所示, 此时H原子与MX2形成更稳定的配体成键吸附, H原子的能级穿过了费米能级, 即层间氢化使得单层MX2直接实现了从半导体到金属的过渡. 有趣的是, 不同于表面氢化诱导的自发磁性, 层间氢化单层MX2 (M = Mo, W; X = S, Se)都会使体系产生磁性, 而阴离子为Te2? 时, 磁性却几乎消失. 这可能是由于Te元素的电负性弱于S和Se元素, 层间氢化时H原子与WTe2和MoTe2中形成了更稳定的配体成键, 因而导致磁性消失. 总之, 通过控制氢化的位置, 可以有效地调控单层MX2的电子性质.
 图 7 层间氢化MX2能带结构图(蓝色和红色线表示自旋向上和自旋向下)
图 7 层间氢化MX2能带结构图(蓝色和红色线表示自旋向上和自旋向下)Figure7. Band structures of interlayer hydrogenation MX2. Blue and red correspond to spin-up and spin-down, respectively.
感谢湖南大学物理与微电子科学学院陈克求教授的讨论; 感谢国家超级计算长沙中心提供的计算资源.
