关键词:热化学非平衡;直接蒙特卡罗方法;反应模型;微观分析 Abstract The non-equilibrium phenomenon of thermochemical coupling has been a difficult problem in high temperature aerothermal dynamics, and hinders to analyze phenomena such as cell structure of detonation wave and ignition speed of low temperature combustion. In this paper, typical chemical reaction models (TCE, VFD, QK models) employed in the direct simulation Monte Carlo (DSMC) simulation are analyzed using two examples (namely, N dissociation at high temperature, and chain displacement reaction in H? O combustion) from microscopic reaction probability, vibrational state specific reaction rates, total reaction rate under thermal nonequilibrium condition, and post-collision redistribution of internal energy. It is found that the probability distribution of vibrational energy of reacted molecules deviates from the equilibrium Boltzmann distribution for both the high temperature dissociation reaction having high activation energy and the chain displacement reaction having low activation energy. The VFD model with strong vibrational favored contribution can predict well the high temperature dissociation reaction, whereas the TCE model (a special case of VFD model) and QK model are better for the chain displacement reaction. Besides, the post-collision redistribution of internal energy should follow the principle of detailed balance, as small deviations may cause inequality between the translational and vibrational energy under final equilibrium state. The DSMC simulation results also show that the vibrational favor of chemical reactions has an obvious effect on the thermochemical coupling process. Particularly, because molecules having high vibrational energy are more easily to have chemical reactions, the decrease of the average vibrational energy of the gas will affect the subsequent chemical reactions.
在高温时(>15 000 K)由于氮气的离解和振动松弛的特征时间比较接近, 会导致再入激波附近的非平衡区域存在所谓的振动与离解耦合现象(CVD). 这一现象对飞行再入中的高温流场有重要影响, 但由于存在复杂的微观物理和苛刻的高温环境, 实际流动问题的数值模拟结果和实验结果都存在一定误差, 二者在定量上的准确对比目前还有困难[26]. 本节的主要目的是通过空间均匀的热化学耦合问题对反应模型的动力学性质开展应用评估. DSMC模拟的初始条件为数密度为 的纯净氮气, 其平动、转动、振动温度分别为30 000 K, 20 000 K, 5 000 K, 初始模拟粒子总数为 . DSMC模拟所需的转动能和振动能的松弛碰撞数 和 由特征松弛时间 转化得到, 转换中都引入了相应的DSMC 微观修正[35]. 此外振动能的特征松弛时间 还考虑了高温修正[7]. 为了突出离解前期的振动与离解耦合过程, 只考虑了单向的离解反应, 忽略了对前期影响很小的复合反应. 另外由于QK模型和TCE模型的动力学性质比较接近, DSMC模拟只选择了TCE 和VFD模型进行评估比较. 计算结果如图7所示, 其中图7(a)给出了反应过程的 浓度变化, 图7(b)为平动温度和振动温度变化, 而转动温度很快和平动温度达到平衡并未在图中给出. 在反应前期, 振动温度明显低于平动温度, VFD模型预测的离解反应速率明显低于TCE模型结果, 反应吸热相对少, 因此系统整体温度高. 比较结果显示在同一时刻VFD和TCE模型预测的温度相差可达1000 K以上, 表明化学模型细节差异对于实际应用的重要性. 显示原图|下载原图ZIP|生成PPT 图7离解过程中的浓度和温度变化 ||||(a) 浓度随时间的变化;(b) 温度随时间的变化 -->Fig.7Evolution of concentration and temperature during dissociation |||| (a)Evolution of concentration;(b) Temperature history -->
通过微观模拟还可以分析化学反应对系统热力学状态的影响. 图8为采用VFD模型的算例在8.3955 ns 时分子的振动能概率分布. 与相同振动温度(12 698 K)的平衡态Boltzmann分布相比, 模拟结果在低振动能级与平衡态基本一致, 但高振动能级的概率明显降低. 原因是离解反应在短时间内消耗了超出平衡的高振动能级的分子, 而热松弛过程(非弹性碰撞)尚未补充恢复. 这说明即使采用双温反应速率的宏观化学反应模型, 在计算热化学耦合问题时仍会带来一定的误差, 而微观粒子模拟或态指定的反应速率求解方法则能包含更多反应细节、模拟结果更为可信. 显示原图|下载原图ZIP|生成PPT 图8VFD模型计算的离解过程中振动能级的瞬时概率分布 -->Fig.8Vibrational energy distribution of during dissociation with VFD model -->
(ChenSong, SunQuanhua.Analysis of maximum dissociation degree of oxygen during hypersonic flight .Chinese Journal of Theoretical and Applied Mechanics, 2014, 46(1): 20-27 (in Chinese))
(PengAoping, LiZhihui, WuJunlin, et al.Validation and analysis of gas-kinetic unified algorithm for solving Boltzmann model equation with vibrational energy excitation .Acta Physica Sinica, 2017, 66(20): 204703 (in Chinese))
(ZhangZijian, LiuYunfeng, JiangZonglin.Effect of vibration excitation on hypersonic aerodynamic and aerothermodynamic .Chinese Journal of Theoretical and Applied Mechanics, 2017, 49(3): 616-626 (in Chinese))
[4]
FiévetR, VoelkelS, KooH, et al.Effect of thermal nonequilibrium on ignition in scramjet combustors .Proceedings of the Combustion Institute, 2017, 36(2): 2901-2910 [本文引用: 1]
[5]
ShiL, ShenH, ZhangP, et al.Assessment of vibrational non-equilibrium effect on detonation cell size .Combustion Science and Technology, 2017, 189(5): 841-85 [本文引用: 1]
(FangYishen, HuZongmin, TengHonghui, et al.Numerical study of the oblique detonation initiation induced by spheres .Chinese Journal of Theoretical and Applied Mechanics, 2017, 49(2): 268-273 (in Chinese))
[7]
ParkC.Assessment of two-temperature kinetic model for ionizing air .Journal of Thermophysics and Heat Transfer, 1989, 3(3): 233-244 [本文引用: 2]
[8]
Park C. The limits of two-temperature model. AIAA Paper, 2010- 911, 2010 [本文引用: 1]
[9]
VoelkelS, RamanV, VarghesePL.Effect of thermal nonequilibrium on reactions in hydrogen combustion .Shock Waves, 2016, 26(5): 539-549 [本文引用: 1]
[10]
BirdGA.Molecular Gas Dynamics and the Direct Simulation Monte Carlo of Gas Flows. Oxford: Clarendon Press, 1994 [本文引用: 4]
(FanJing.Rarefied gas dynamics: Advances and applications .Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese)) [本文引用: 2]
[12]
HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation .Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489 [本文引用: 4]
[13]
BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations .Physics of Fluids, 1997, 9(4): 1162-1170 [本文引用: 2]
[14]
BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects .AIP Conference Proceedings, 2005, 762(1): 607-613 [本文引用: 2]
[15]
BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant . AIAA Paper, 2007-614, 2007 [本文引用: 2]
[16]
WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen .AIP Conference Proceedings, 2016, 1786(1): 050021 [本文引用: 5]
[17]
BirdGA.The QK model for gas-phase chemical reaction rates .Physics of Fluids, 2011, 23(10): 106101 [本文引用: 3]
[18]
BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions .AIP Conference Proceedings, 2016, 1786(1): 090005
[19]
LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations .The Journal of Chemical Physics, 2017, 146(7): 074303
[20]
SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models .Physics of Fluids, 2017, 29(1): 017102
[21]
RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method .Computers & Fluids, 2016(140): 111-121
[22]
SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55th AIAA Aerospace Sciences Meeting, 2017
[23]
BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen .Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185 [本文引用: 4]
[24]
KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections .Physics of Fluids, 2014, 26(1): 012006 [本文引用: 2]
[25]
WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis .Physics of Fluids, 2012, 24(4): 042002 [本文引用: 2]
[26]
WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements .Physics of Fluids, 2014, 26(4): 043101 [本文引用: 2]
[27]
ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface .Physics of Fluids, 2015, 27(8): 086102 [本文引用: 2]
[28]
Bird GA. The DSMC Method .Create Space Independent Publishing Platform, 2013 [本文引用: 5]
BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method .AIP Conference Proceedings, 2011, 1333(1): 1209-1214 [本文引用: 1]
[31]
YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation .AIP Conference Proceedings, 2014, 1628(1): 1261-1267 [本文引用: 1]
[32]
GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method .Physics of Fluids, 2004, 16(7): 2442-2451 [本文引用: 2]
[33]
SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide .Combustion and Flame, 2006, 145(1): 316-323 [本文引用: 1]
[34]
BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions .The Journal of Chemical Physics, 2015, 143(5): 054304
[35]
GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method .Physics of Fluids, 2002, 14(12): 4452-4455 [本文引用: 1]
[36]
MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures .Combustion and Flame, 1988, 74(1): 53-69 [本文引用: 1]
[37]
DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr .Chemical Physics, 1974, 6(3): 431-444 [本文引用: 1]



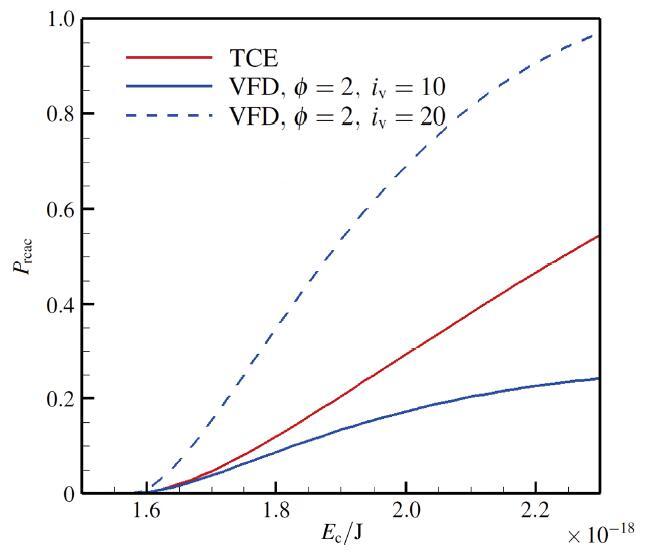 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT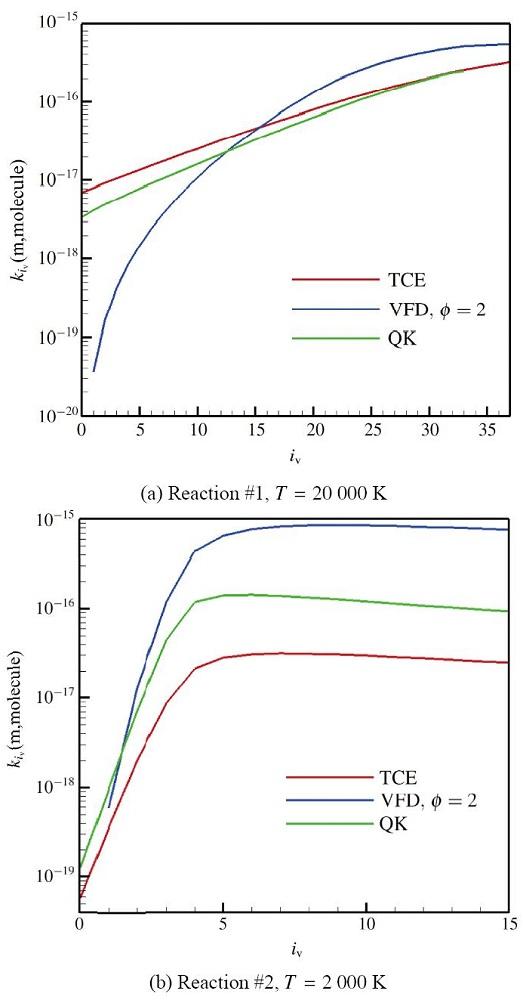 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT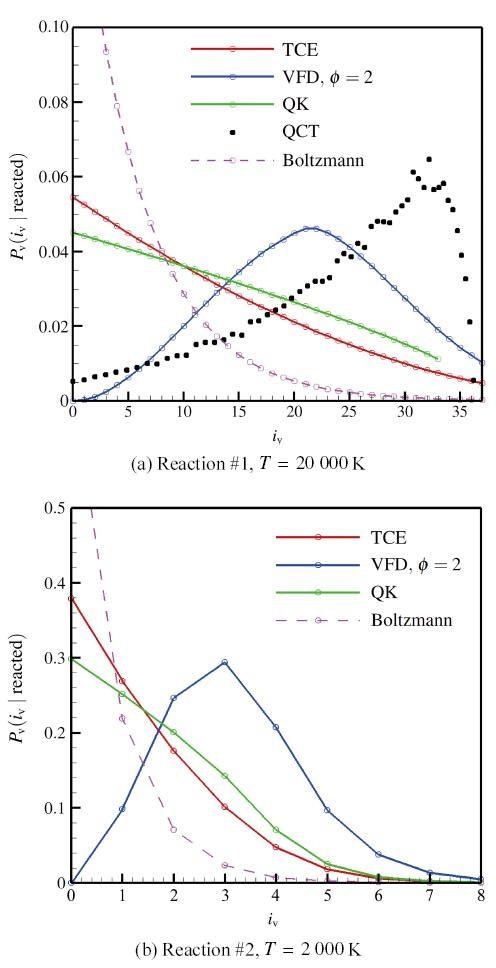 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT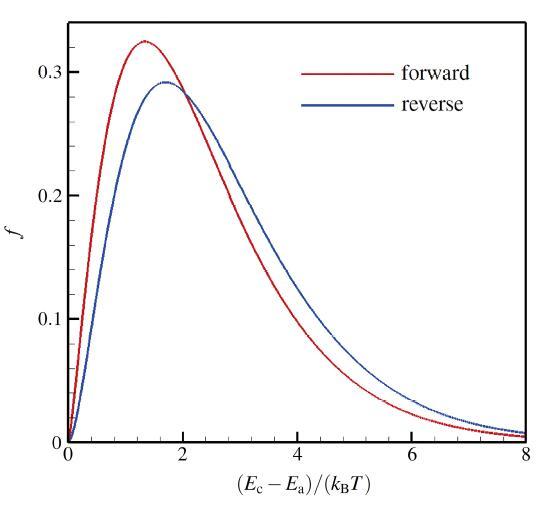 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT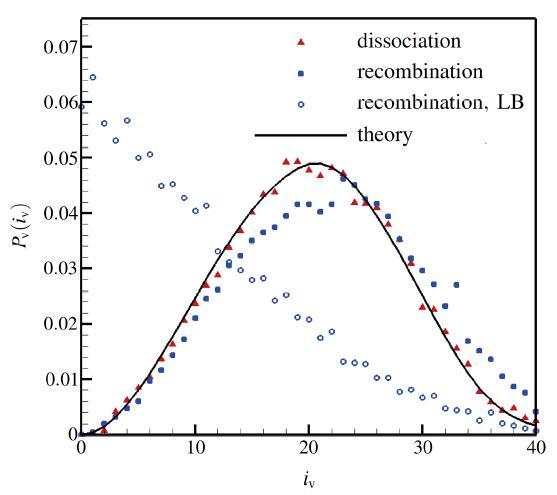 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT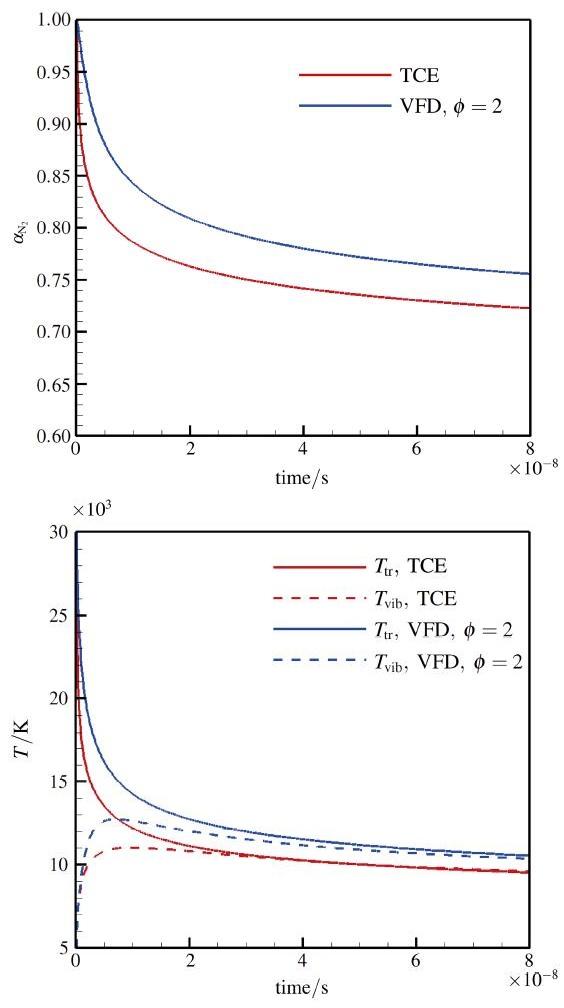 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT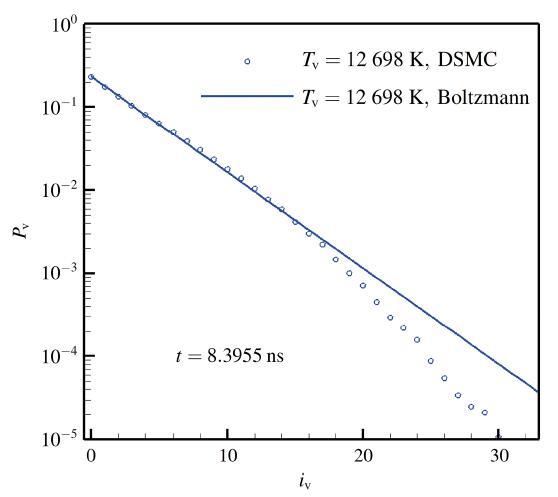 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}