 ,复旦大学生命科学学院,植物科学研究所,上海 200433
,复旦大学生命科学学院,植物科学研究所,上海 200433Structural and functional characteristics of plant PHD domain-containing proteins
Tianyi Wang, Yingxiang Wang, Chenjiang You,Institute of Plant Science, College of Life Science, Fudan University, Shanghai 200433, China通讯作者: 尤辰江,博士,青年研究员,研究方向:植物表观遗传。E-mail:cjyou@fudan.edu.cn
编委: 宋任涛
收稿日期:2020-12-1修回日期:2021-02-9网络出版日期:2021-04-16
| 基金资助: |
Received:2020-12-1Revised:2021-02-9Online:2021-04-16
| Fund supported: |
作者简介 About authors
王天一,在读博士研究生,研究方向:植物减数分裂。E-mail:

摘要
植物同源结构域(plant homeodomain, PHD)是锌指结构域家族的一类转录调控因子,其最主要的功能是可以识别各种组蛋白修饰密码,包括组蛋白甲基化和乙酰化等;此外PHD结构域还可以与DNA结合。含有PHD结构域的蛋白,或者本身具有组蛋白修饰酶活性,或者可以与各类组蛋白修饰酶相互作用,还有部分与DNA甲基化相关,具有E3泛素连接酶活性,或者还可以作为染色质重塑因子,以各种不同的作用方式,在植物的生长发育过程中发挥了重要的作用。本文主要综述了结合各种类型组蛋白(包括H3K4me3/0、H3K9me3、H3R2和H3K14ac)以及DNA的PHD结构域的结构特点及其结合特异性、PHD结构域在植物中的进化保守性以及植物中已经发现的含有PHD结构域蛋白的功能及作用机制,为进一步了解该类蛋白在植物生长发育过程中如何发挥作用提供了参考。
关键词:
Abstract
Plant homeodomain (PHD) is a class of transcription factor in the Zinc finger domain family. The most important function of which is to recognize various histone modifications, including histone methylation and acetylation, etc. They can also bind to DNA. Proteins with PHD domains, some of which possess histone modification enzyme activity, or can interact with histone modification enzymes, and some are associated with DNA methylation, with E3 ubiquitin ligase activity, or even can be chromatin remodeling factors. As transcriptional regulators, they play an important role in plant growth and development. In this review, we summarize the structural features and substrate binding specificity of PHD domains (including H3K4me3/0, H3K9me3, H3R2, H3K14ac) and DNA, the conservation of plant PHD domain in evolution, the molecular mechanism of known PHD domain-containing proteins in plants, providing a reference for further understanding of the involvement of these proteins during plant growth and development.
Keywords:
PDF (1489KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
王天一, 王应祥, 尤辰江. 植物PHD结构域蛋白的结构与功能特性. 遗传[J], 2021, 43(4): 323-339 doi:10.16288/j.yczz.20-412
Tianyi Wang.
植物同源结构域(plant homeodomain, PHD)是真核生物进化过程中一种保守的锌指结构域。PHD结构域发挥功能最主要的方式是识别各类组蛋白修饰,它们对于各类组蛋白修饰的识别也具有一定的特异性,除此之外还可以识别一些DNA序列。含有PHD结构域的蛋白作为转录调控因子,参与生物体的各项生命过程。如在植物中,含有PHD结构域的蛋白参与了包括胚胎分生组织萌发、根系发育、发芽、开花、减数分裂,以及减数分裂后的花粉发育等重要过程,对植物的生长发育具有十分重要的作用[1]。
关于植物中含有PHD结构域的蛋白,Mouriz等[1]按照其功能分类进行了综述,本文则是从PHD结构域与各类组蛋白翻译后修饰的结合特异性、PHD结构域在植物中的进化保守性,以及各类PHD结构域蛋白的作用机制方面进行论述,主要针对拟南芥(Arabidopsis thaliana)中PHD蛋白的作用机制进行解析,为进一步了解其如何发挥调控功能提供参考。
1 PHD的结合特异性
组蛋白翻译后修饰主要包括甲基化、乙酰化等多种形式,这些翻译后修饰通过招募与组蛋白结合的效应蛋白来调控下游基因的表达。其中,识别组蛋白甲基化修饰的结构域主要为PHD结构域和Royal家族的Chromo、Tudor、PWWP (Pro-Trp-Trp- Pro)和MBT (malignant brain tumour)结构域等;而识别组蛋白乙酰化的主要为 Bromo、DPF (double PHD finger)和YEATS结构域[2]。除此之外,对于DNA的识别主要有螺旋-转角-螺旋(Helix-Turn-Helix, HTH)、锌指结构、亮氨酸拉链(basic Leucine zipper, bZIP)和碱性-螺旋-环-螺旋(basic Helix- Loop-Helix, bHLH)结构域等,而PHD结构域就属于锌指结构域家族。PHD结构域是一类比较小的蛋白结构域,由50~80个氨基酸残基组成,其不同家族成员序列表现出较低的氨基酸相似性,但该结构域可以折叠成高度保守的球状结构[3]。保守的PHD折叠,包含了一对反向平行的双链β-sheet和一个C端的α-helix (不是所有的PHD都有),2个锌原子被固定在Cys4-His-Cys3基序上,形成一个“cross-brace”的拓扑结构[4]。
PHD结构域最主要功能是与各种组蛋白修饰特异结合,作为组蛋白密码识别器来调控下游基因的表达。研究发现,PHD结构域可以识别的组蛋白密码包括H3K4me3/2/0[5,6]、H3K9me3[7,8,9]、H3K36[10]、H3R2me2/0[11,12]、H3K14ac以及H4乙酰化等[13,14]。研究发现,PHD结构域通常和其他识别模块(reader modules)成对出现,例如与Bromo、Chromo、Tudor、PWWP、MBD、SRA或者PHD finger本身,一起作为一个组合来发挥作用[15]。模块配对可以显著地提升PHD的识别能力,提高与组蛋白之间结合的亲和力和特异性,从而提供新的调控能力。例如BPTF通过PHD-Bromo结构域识别“H3K4me3+H4K16ac”[16]。
本文对已经研究出晶体结构以及结合特性的PHD蛋白进行了总结。
1.1 识别H3K4me3
PHD与组蛋白H3K4me3的结合通常依靠一个包含2~4个芳香族残基的笼子(aromatic cage)。在这个笼子中,K4me3由范德华力和π键稳定[5](图1A)。不同的PHD中aromatic cage氨基酸组成不一样,但是位于序列的相似位置,其中一个色氨酸(Trp)残基,在位点和残基类型上都完全保守,是识别结合甲基化赖氨酸的PHD中最特异的残基[17]。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1PHD结构域的晶体结构图
A:ING2的PHD结构域结合H3k4me3 (2g6q[23]);B:BHC80的PHD结构域结合未修饰的H3 (2puy[19]);C:ATRX的PHD结构域识别H3K9me3 (3ql9[24]);D:BPTF的PHD结构域结合H3K4和H3R2 (2fuu[25]);E:DPF3b的PHD1识别乙酰化修饰(5i3l[26],黄色代表结合K14ac的疏水性口袋);F:BRPF2的PHD2结构域(2lq6[27])。图中所用模型来自PDB数据库(Protein Data Bank),晶体结构展示软件为PyMOL (version 2.4)。
Fig. 1The crystal structures of PHD domain
在结合过程中,组蛋白H3的肽段形成第三条反向平行的β链,与PHD现存的双链β-sheet配对[5]。PHD蛋白的β1链和H3K4me3的R2-T6残基之间形成分子间氢键,稳定了结合复合体的特征骨架[18]。
1.2 识别未修饰的H3
研究表明,BHC80[19]的PHD结构域可以与未修饰的组蛋白H3的前8个残基结合(图1B),并且当K4位点发生甲基化时,这个结合会被阻止,但是K9和K14位点的甲基化不会对其造成影响[19]。类似的,AIRE[20]的PHD结构域也可以与未修饰的组蛋白H3氮端的前8个残基结合。与H3K4me3-PHD类似,未修饰的组蛋白H3尾巴也形成一个β链,与PHD的β1链配对。但是,这一类的PHD很明显缺失了芳香族的残基,由一大片的酸性残基作为替代,并且PHD的第1个与锌离子结合的半胱氨酸残基的N端会存在一个或者一对保守的酸性残基[19,20]。
1.3 识别H3K9me3
ATRX通过其ADD结构域识别H3K9me3。ADD (ATRX-DNMT3-DNMT3L)结构域富含半胱氨酸残基,是4个不同的蛋白ATRX、DNMT3A、DNMT3B和DNMT3L共同拥有的结构。ATRX的ADD结构域由1个N端类GATA锌指(GATA-like finger)、1个PHD结构域、1个长的C端螺旋(C-terminal helix),通过广泛的疏水性相互作用组装在一起,形成一个球状的结构域[21]。在ATRX的ADD结构域识别K9me3的过程中,涉及到一个非典型的极性口袋(polar pocket),该口袋只含有一个芳香族的酪氨酸(Tyr),并且富含极性的氨基酸(polar residues)[8]。除了分子表面的范德华力以外,K9me3的识别还涉及到了芳香族Tyr203与甲基化赖氨酸之间的阳离子π作用,以及Tyr203、Asp207、Gln219和Ala224与甲基化的赖氨酸之间的非传统的C-H…O氢键的相互作用[21](图1C)。
类似地,在TRIM33[7]和CHD4[9]中也存在PHD的一个芳香族残基与K9之间形成π键,以及非传统的氢键作用。对于TRIM33,它具有一个PHD-Bromo盒子(cassette),其Bromo结构域与H3的K14、K18、K23乙酰化修饰结合,PHD结构域与H3K9me3结合。在结合K9me3的过程中,K9me3与芳香族Trp889之间形成π键,与PHD的羰基氧之间形成非传统的C-H…O氢键[7];而在CHD4的PHD2中,K9me3与芳香族Phe451之间形成π键,与其他残基之间形成氢键,这样的模式稳定了PHD-H3K9me3的结构[9]。
1.4 识别H3R2
在许多蛋白(例如BPTF[5]、ING4[22]和UHRF[11]等)中,H3K4和H3R2的识别发生在同一个PHD的不同位点。在这个过程中,H3R2的侧链和H3K4me3分别进入到2个相邻的识别口袋,这两个口袋通过一个保守的Trp残基分隔开(图1D)。其中K4me3的结合口袋是芳香族笼子,通过π键,疏水相互作用和范德华力,稳定PHD与三甲基集团之间的结合。而R2口袋通常是酸性的,通过Asp、Glu和Gln等残基与R2之间形成盐桥或者氢键来进行配对[5]。UHRF1的PHD同样依赖于Asp347和Asp350与H3R2之间形成氢键进行结合[11]。1.5 识别乙酰化
识别乙酰化组蛋白的PHD通常是串联的两个,例如DPF3b[14]、MOZ[13]和MORF[28]的DPF模块可以与H3K14以及其他乙酰化的组蛋白尾巴相互作用。研究发现,DPF3b的2个串联的PHD可以形成一个组合的球状结构域。其中PHD1可以结合H3K14ac,PHD2可以结合H3K4me0和H3R2me0(图1E)。PHD2结合H3K4me0的口袋,与前述结合H3K4me0的PHD类似,拥有酸性和疏水性的残基。而PHD1结合H3K14ac的口袋是由Phe264、Leu296、Trp311和Ile314组成的疏水性口袋。PHD1在β2表面产生了一个窄的沟槽,当复合体形成,H3的T11和K14ac的侧链插入到这个沟槽中,由一系列氢键和范德华力稳定[14]。类似的,MOZ通过其Phe211、Leu242、Trp257和Ile260组成相应的疏水性口袋来识别K14ac[13]。
1.6 识别DNA
BRPF1/2[27]中的PHD可以结合DNA,它们都拥有2个PHD结构域,中间由一个锌原子连接。BRPF1/2的PHD1对于未修饰的H3有高度的特异性。PHD2呈鞍状结构,可以与DNA相互作用,与一般的PHD结构域不同,PHD2具有2个与锌离子配位的组氨酸残基,其序列特征为C4HC2H,并且还含有一个额外的β3-β4发夹结构(图1F)。PHD2对于DNA的结合能力,可能是由于PHD结构域中具有静电势(electrostatic potential)的表面的作用[27]。2 植物中PHD蛋白功能及作用机制
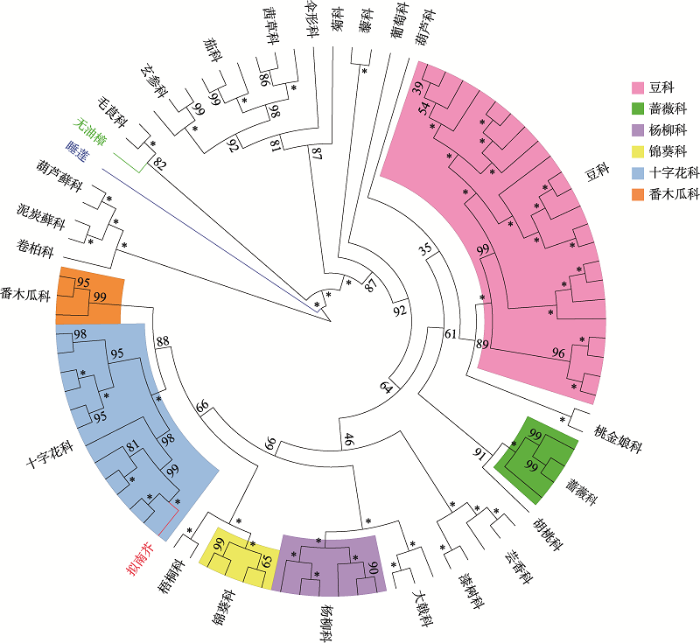
染色质对基因的转录调控主要依赖于两种作用:一种是激活因子,主要依赖于组蛋白的乙酰化修饰,使DNA结构变得松散,有利于RNA聚合酶等转录起始因子与DNA的结合;另一种是抑制因子,它们可以通过对组蛋白进行抑制型的甲基化修饰,以招募去乙酰化酶,去掉激活类修饰,进而增强染色质的屏障作用。含有PHD结构域的蛋白,以两种形式参与转录调控作用,或者本身即拥有组蛋白修饰酶活性,或者可以与各种组蛋白修饰酶相互作用。还有部分PHD结构域蛋白与DNA甲基化相关,具有E3泛素连接酶活性,或者可以作为染色质重塑因子,对各种植物的生长发育以及应对各种逆境胁迫过程着重要作用。PHD结构域蛋白在植物中广泛存在。在植物基因组网站Phytozome12中搜索可以发现,已注释的拟南芥PHD结构域基因有82个,水稻(Oryza sativa L.)有84个,无油樟(Amborella trichopoda)有67个。以拟南芥MMD1的同源蛋白构建系统发育树,发现该PHD结构域蛋白在植物中广泛存在,在十字花科和豆科中的同源蛋白数量最多,在蔷薇科、芸香科、杨柳科、番木瓜科等科目中也有存在,说明该蛋白在植物进化过程中具有保守性,并且可能在多种植物的生长发育过程中发挥了重要的作用(图2)。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2植物中部分PHD结构域蛋白MMD1的系统发育树
图中展示了部分拟南芥MMD1的同源蛋白的进化关系(序列来源于植物基因组网站Phytozome12)。使用IQ-TREE (version 1.6.12),以最大似然法(Maximum Likelihood, ML)构建系统发育树,选用的构树模型为JTT+R6,以葫芦藓科、泥炭藓科和卷柏科的MMD1同源蛋白定根。图中数字为分支的自展值,星号“*”代表自展值为100。拟南芥的MMD1分支标为红色,睡莲的MMD1同源蛋白分支标为蓝色,无油樟的MMD1同源蛋白分支标为绿色。图中标注了几个主要的科目,分别为豆科(粉色)、蔷薇科(绿色)、杨柳科(紫色)、锦葵科(黄色)、十字花科(蓝色)和番木瓜科(橙色)。
Fig. 2Phylogenetic tree of the PHD domain protein MMD1 in plants
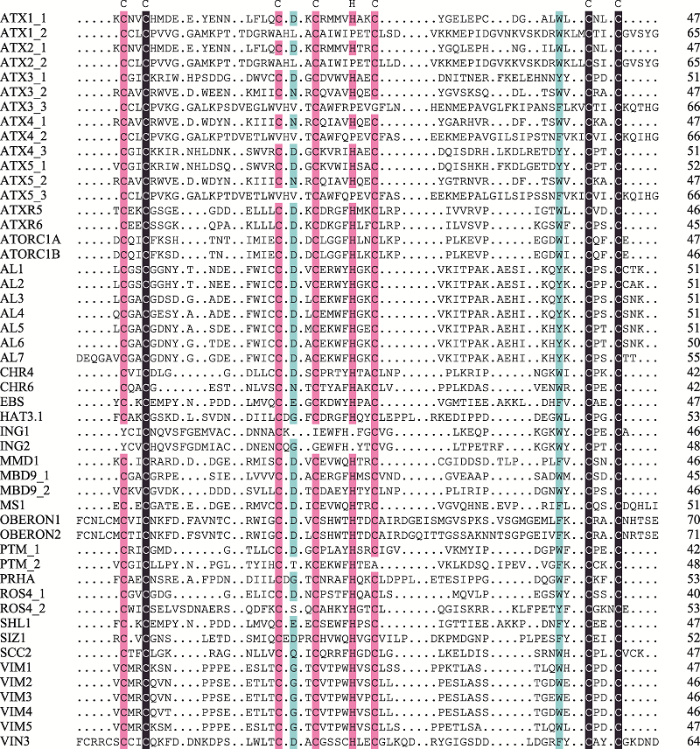
对拟南芥中已研究的PHD结构域蛋白进行序列比对,发现它们都具有保守的C4HC3序列特征(图3)。根据拟南芥中含有PHD结构域的蛋白发挥功能的调控机制不同,可以对其进行具体的分类(表1):主要包括本身具有组蛋白修饰酶活性(包括本身具有组蛋白甲基转移酶活性的ATX1/2/3/4/5和ATXR,本身具有组蛋白乙酰基转移酶活性的IDM1/ROS4);可以与组蛋白修饰酶相互作用(包括可以与组蛋白去乙酰化酶相互作用的EBS/SHL,与组蛋白甲基转移酶相互作用的MMD1、AL、VIN3),与DNA甲基化相关的MBD9和ORTH,具有E3泛素连接酶活性的SIZ1,以及可以作为染色质重塑因子的CHR4和PKL等。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3拟南芥中PHD结构域的序列比对
黑色表示同源性为100%,粉色表示同源性为75%,蓝色表示同源性为50%。可以看出PHD结构域保守的C4HC3氨基酸残基序列特征。图中各蛋白对应的TAIR基因号分别为:ATX1 (At2g31650)、TX2 (At1g05830)、ATX3 (At3g61740)、ATX4 (At4g27910)、ATX5 (At5g53430)、ATXR5 (At5g09790)、ATXR6 (At5g24330)、ATORC1A (At4g14700)、ATORC1B (At4g12620)、AL1 (At5g05610)、AL2 (At3g11200)、AL3 (At3g42790)、AL4 (At5g26210)、AL5 (At5g20510)、AL6 (At2g02470)、AL7 (At1g14510)、CHR4 (At5g44800)、CHR6 (At2g25170)、EBS (At4g22140)、HAT3.1 (At3g19510)、ING1 (At3g24010)、ING2 (At1g54390)、MMD1 (At1g66170)、MBD9 (At3g01460)、MS1 (At5g22260)、OBERON1 (At3g07780)、OBERON2 (At5g48160)、PTM (At5g35210)、PRHA (At4g29940)、ROS4 (At3g14980)、SHL1 (At4g39100)、SIZ1 (At5g60410)、SCC2 (At5g15540)、VIM1 (At1g57820)、VIM2 (At1g66050)、VIM3 (At5g39550)、VIM4 (At1g66040)、VIM5 (At1g57800)、VIN3 (At5g57380)。
Fig. 3Sequence alignment of PHD domains in Arabidopsis thaliana
Table 1
表1
表1拟南芥中PHD蛋白的特征与功能
Table 1
| 分类依据 | 蛋白名称 | 其他包含的结构域 | 识别的配体 | 作用特点 | 在植物中的功能 | 参考文献 | |
|---|---|---|---|---|---|---|---|
| 本身具有组蛋白修饰酶活性 | 本身具有组蛋白甲基转移酶活性 | ATX1 | ePHD结构域、SET结构域 | H3K4me3 | 使H3K4三甲基化 | 根系、叶片和花器官的发育以及一些逆境胁迫基因的转录调控 | [29~31] |
| ATX2 | ePHD结构域、SET结构域 | H3K4me2 | 使H3K4三甲基化 | 与ATX1拥有相似的序列,但是在调控基因转录方面具有非冗余的功能 | [32] | ||
| ATX3/4/5 | 编码了一个可能的H3K4甲基转移酶 | H3K4me2/3 | 是迄今为止在拟南芥基因组中发现的具有H3K4me2甲基转移酶活性的蛋白 | ATX3/4/5具有冗余的功能,可以调控一系列作用于营养生长和生殖生长的基因 | [33,34] | ||
| ATXR | SET结构域 | H3K4me0 | PHD结构域作用于SET结构域结合辅因子以及促进H3K27me1的过程 | 作用于植物中染色质结构、基因沉默和异染色质的DNA复制过程 | [35] | ||
| 本身具有组蛋白乙酰基转移酶活性 | IDM1/ ROS4 | MBD结构域、 乙酰基转移酶结构域 | H3K4me0 | PHD结构域影响IDM1乙酰转移酶的活性 | 对于DNA去甲基化具有负面调控,阻止高度同源的多拷贝基因和其他重复序列的DNA高度甲基化 | [36,37] | |
| 与组蛋白修饰酶相互作用 | 与组蛋白去乙酰化酶相互作用 | EBS/SHL | BAH结构域 | H3K4me2/3 | PHD结构域结合HDA6 | 作用于开花调控和种子 休眠 | [38,39] |
| 与组蛋白甲基转移酶相互作用 | MMD1 | MMD结构域 | H3K4me2/3 | 与组蛋白去甲基化酶JMJ16相互作用 | 植物减数分裂,调控浓缩等过程的蛋白 | [40~42] | |
| AL | 除AL3以外所有AL蛋白都结合H3K4me2/3 | 是植物中特有的一类转录因子,PHD结构域与PRC1蛋白相互作用,招募PRC2从而积累H3K27me3 | 调控植物的生长发育,以及应对低温、干旱、高盐等非生物胁迫 | [43~46] | |||
| VIN3 | H3K9me2和H3K4me2 | PHD结构域与PRC2的相互作用,PHD-PRC2复合体使H3K27me3水平升高 | 作用于春化作用所需的FLC表观遗传学基因沉默过程 | [47~51] | |||
| 与DNA甲基化相关 | 结合甲基化的DNA | MBD9 | MBD结构域 Bromo结构域 | DNA甲基化 | MBD结构域结合甲基化的DNA,Bromo结构域可能发挥了催化组蛋白乙酰化反应的作用 | 通过DNA甲基化和组蛋白乙酰基化,分别间接和直接调控基因的表达,影响拟南芥的生长发育 | [52,53] |
| ORTH | RING结构域、SRA结构域 | DNA甲基化 | SRA结构域作用于结合甲基化的DNA | 作用于调控DNA甲基化 | [54,55] | ||
| 具有E3泛素连接酶 活性 | SIZ1 | RING、SAP、SXS、PINIT 结构域 | H3R2me2和H3K4me3 | PHD结构域与染色质重塑复合体有关,也可能作为一个E3泛素连接酶 | 通过调控基因的表达,作用于植物的生长发育以及应对干旱、低盐的胁迫的过程 | [56~58] | |
| 是染色质重塑因子 | CHR4 | Chromodomain结构域 | 是依赖于ATP的染色质重塑因子 | 植物的生长发育和DNA损伤应答 | [59,60] | ||
| 是染色质重塑因子 | PKL | Chromodomain结构域 | 是依赖于ATP的染色质重塑因子 | DNA损伤应答,以及调控植物生长和响应胁迫基因的表达 | [61,62] | ||
| 与bHLH型的转录因子相互作用 | OBE | 可能结合 bHLH型的转录因子 | 促进依赖于转录因子MP的基因的激活表达 | 在生长素介导的调控发育过程中,作用于根系和顶端分生组织的维持和建立 | [63~65] | ||
| 其他 | SCC2 | 在陆地植物有PHD,动物和 真菌中没有 | 未修饰及甲基化的H3、H4和H2A | 作用于减数分裂过程,介导染色质黏连蛋白cohesin的招募过程 | [66] | ||
| ORC | 只有植物的ORC1中含有PHD结构域 | H3K4me3,更倾向于结合未修饰的H3 | 通过PHD结构域识别靶基因启动子区域的H3K4me3来激活基因的转录表达 | 作用于DNA复制的起始,在细胞周期中调控转录 过程 | [67,68] | ||
| MS1 | 调控作用于花粉外壁形成,花粉细胞溶质和绒毡层的基因的表达,对于减数分裂后的花粉和绒毡层的发育具有重要作用 | [69] | |||||
| PTM | DDT 结构域 | H3K4me3 | 结合到ABI4的启动子上,以激活ABI4基因的表达。 | 结合叶绿体被膜的转录调控因子,作用于将叶绿体信号传递到细胞膜 | [70] | ||
新窗口打开|下载CSV
2.1 本身具有组蛋白修饰酶活性
2.1.1 本身具有组蛋白甲基转移酶活性拟南芥ATX (ARABIDOPSIS TRITHORAX-LIKE)基因分为两个亚家族,分别为ATX1/2亚家族和ATX3/4/5亚家族。ATX1和ATX2蛋白包含ePHD结构域(extended plant homeodomain)和SET结构域。ePHD结构域包含1个N端pre-PHD (C2HC Zinc finger)、1个长的连接区域和1个PHD (C4HC3 Zinc finger),ePHD结构域可以结合双链DNA但是不能结合组蛋白。而SET结构域使ATX1/2具有甲基转移酶活性。ATX1作用于H3K4me3,而ATX2作用于H3K4me2,这两个组蛋白修饰都是活跃转录的标志。
ATX1参与根系、叶片和花器官的发育以及一些逆境胁迫基因的转录调控[29]。研究表明,ATX1在调控目标基因表达时具有两种不同的作用:一种是作为共同激活子参与前起始复合物PIC的形成,招募RNA聚合酶II和TATA结合蛋白;另一种是被磷酸化的RNA聚合酶II招募到转录起始位点下游行使H3K4三甲基化作用[31]。ATX1催化的H3K4me3不作用于转录起始,而是作用于活跃转录的延伸[30]。与其他的PHD结构域结合H3K4me3的功能不同,ATX1的ePHD结构域通过结合PI5P (phosphatidylinositol 5-phosphate),影响ATX1的亚细胞定位,从而调控一些特异依赖于ATX1的基因的表达[71]。
ATX2与ATX1拥有相似的序列,但是它们在调控基因转录方面具有非冗余的功能,在作用于同一个基因的调控时,有不同的靶向特异性,并且使用了不同的分子生物学机制[32]。例如,ATX1和ATX2都使XTH33的转录水平上调。在atx2突变体中,XTH33的H3K4me2的标记消失伴随着基因转录水平的降低,并且H3K4me3不是XTH33转录所需要的(在活跃转录的野生型XTH33中也没有H3K4me3)。
说明ATX2导致的H3K4me2累积对转录是必要的,而ATX1则并不对XTH33的核小体直接进行修饰,发挥了间接的调控作用[32]。
ATX3、ATX4、ATX5同样包含PHD结构域,并且可能编码了H3K4甲基转移酶,参与H3K4的二甲基化和三甲基化。ATX3/4/5具有冗余的功能,可以调控一系列作用于营养生长和生殖生长的基因。它们是迄今为止在拟南芥基因组中发现的唯一具有H3K4me2甲基转移酶活性的蛋白[34]。
ATXR5/6 (ARABIDOPSIS TRITHORAX-RELATED PROTEIN 5/6)是拟南芥中另一对含有PHD结构域的蛋白,具有H3K27me1甲基转移酶活性。ATXR5/6参与植物中染色质结构、基因沉默和异染色质的DNA复制过程[72]。atxr5和atxr6突变体表现出异染色质成分(heterochromatic elements)、转座子和重复序列的转录激活。ATXR5/6的PHD结构域可以识别未甲基化的H3K4,参与SET结构域结合辅因子以及促进H3K27me1的过程[35]。
除此之外,在酵母和人类的PHD蛋白中也有类似的例子。酿酒酵母(Saccharomyces cerevisiae)的Spp1是一个含有PHD结构域的蛋白,同时也是组蛋白H3K4甲基转移酶Set1(COMPASS)复合体的成员。Spp1的PHD结构域结合组蛋白H3K4,并且可以调控甲基转移酶COMPASS的活性[73]。在人类中的一些蛋白,包括PHF2、PHF8和KIAA1718等,除了包含PHD结构域以外,还包含1个具有组蛋白去甲基化酶活性的JmjC (Jumonji-C)结构域。这些蛋白的PHD结构域,在活跃转录基因的启动子区域,与含有H3K4me3的核小体结合。而JmjC结构域则可以移除与转录抑制相关的组蛋白H3K9、H3K27的单、双甲基化以及H4K20的单甲基化。这样的双重作用,确保了基因在被转录激活之后,组蛋白转录抑制标记能够恰当地移除[74]。
2.1.2 本身具有组蛋白乙酰基转移酶活性
IDM1 (INCREASED DNA METHYLATION1)是一个组蛋白H3乙酰基转移酶,具有1个MBD结构域(methyl-CpG-binding domain)、1个PHD结构域和1个组蛋白乙酰基转移酶结构域[37]。MBD结构域参与识别甲基化的DNA;PHD结构域可以识别未甲基化的H3K4。IDM1对于DNA甲基化具有负面调控,
阻止高度同源的多拷贝基因和其他重复序列的DNA高度甲基化,其抑制作用表现在两个方面:第一,IDM1的H3乙酰化活性可以抑制DNA高度甲基化,乙酰化的H3(H3K18Ac、H3K23Ac)创造了一个允许5-甲基胞嘧啶DNA糖苷酶(参与DNA脱甲基化)作用的染色质环境;第二,PHD结构域抑制DNA高度甲基化。一方面,PHD结构域的突变,会引起IDM1乙酰转移酶活性的丧失,这也说明了PHD结构域可以不依赖于其结合组蛋白H3的功能,去影响IDM1乙酰转移酶的活性;另一方面,PHD结构域只能识别未甲基化的H3K4,其与H3K4me0的结合会被H3K4me2/3抑制。因此,由于PHD结构域的存在,IDM1在H3K4me2/3水平高的位点与甲基化的DNA结合是被抑制的[75]。
在酿酒酵母中,Yng1蛋白包含PHD finger结构域,并且该蛋白是NuA3 HAT复合体(组蛋白乙酰基转移酶)的组成部分。Yng1通过PHD结构域识别H3K4me3,增强了NuA3 HAT复合体对组蛋白H3底物作用的活性,使H3K14乙酰化,从而激活基因的表达[76]。
2.2 与组蛋白修饰酶相互作用
2.2.1 与组蛋白去乙酰化酶相互作用SHL (SHORT LIFE)和EBS (EARLY BOLTING IN SHORT DAYS)是拟南芥中的两个旁系同源基因,都包含PHD结构域和BAH (bromo-adjacent homology)结构域,参与染色质介导的开花抑制和种子休眠。研究发现,SHL的过量表达还会引起植物育性的降低[77]。SHL和EBS在调控开花时间上发挥重要作用,主要通过其PHD结构域识别H3K4me2和H3K4me3,分别结合到SOC1和FT (SOC1和FT都是开花调节基因[78])的调控区域。这两种PHD蛋白通过结合组蛋白去乙酰化酶HDA6 (HISTONE DEACETYLASE 6),阻止高水平的H3乙酰化,维持了SOC1和FT的不活跃的染色质构象[79]。EBS与HDA6的相互作用,说明组蛋白甲基化和乙酰化对于开花时间的精确控制是一个关键的因素。另外,在EBS和SHL参与的种子休眠调控的过程中,组蛋白甲基化和乙酰化修饰共同协作对于种子休眠的精确调控也十分重要[39]。SHL和EBS在植物中高度保守,但是在其他真核生物中却没有,说明这些基因介导的调控方式是植物中的一种特有的机制。
2.2.2 与组蛋白甲基转移酶相互作用
拟南芥的MMD1 (MALE MEIOCYTE DEATH 1)对于植物减数分裂过程十分重要[40]。其PHD结构可以识别H3K4me2/3,通过结合目标基因启动子区域的组蛋白标记,招募其他的调控因子到相同的位点,从而调控基因的表达[41]。最近研究发现,MMD1拥有另一个保守的MMD结构域,其与JMJ16 (组蛋白去甲基化酶,具有H3K4me3去甲基酶活性[80])的FYR-C结构域相互作用,从而拓展了JMJ16的H3K9me3去甲基酶活性,使目标基因启动子区域的抑制基因表达的组蛋白修饰减少,从而促进了基因表达,包括编码浓缩蛋白CAP-D3的基因表达。在这个过程中,PHD结构域通过结合H3K4me3,使JMJ16能够准确定位到基因的启动子区域[42]。
拟南芥中AL (ALFIN-LIKE)蛋白是植物中特有的一类转录因子,对于植物的生长发育,以及应对低温、干旱、高盐等非生物胁迫具有重要的调控作用[44,46]。AL蛋白家族包括AL1、AL2、AL3、AL4、AL5、AL6和AL7,除AL3以外都能通过PHD结构域与H3K4me3和H3K4me2结合来调控目标基因的表达[45]。在种子萌发时,AL蛋白通过与PRC1蛋白相互作用,招募PRC2 (一个保守的H3K27甲基转移酶[81])从而促进H3K27me3的积累,将种子发育相关基因的活跃转录的染色质状态转变为不活跃状态,对于种子萌发和早期的幼苗生长十分重要[43]。
植物的春化作用中所需的FLC表观遗传学基因沉默过程,需要PRC2和2个PHD finger蛋白—VRN5和VIN3的参与。VIN3蛋白的PHD结构域与PRC2的相互作用被认为是PcG家族调控基因表达的保守机制[82],PHD结构域蛋白在增强PRC2活性上发挥了重要作用。高水平的H3K27me3能够稳定基因的沉默状态,PHD-PRC2复合体使FLC基因的H3K27me3水平升高,从而使其保持沉默[82]。
类似的调控过程在果蝇(Drosophila melanogaster)和哺乳动物中也存在。哺乳动物的PHF1蛋白拥有1个N端Tudor结构域和2个PHD结构域,是PRC2的组成部分。PHF1包含两个PHD finger结构域的区域,可以直接与PRC2的催化亚基EZH2 (甲基转移酶活性)相互作用[83]。在果蝇中,Pcl (PHF1的同源基因)和E(z) (EZH2的同源基因)也可以互相结合[84],说明PHF1的PHD finger功能的保守性[85]。此外,研究发现在没有H3K36me3的情况下,PHF1可以增强PRC2的甲基转移酶活性,这可能是由于PHD finger结构域与其他组蛋白翻译后修饰(例如未修饰的H3或H3K4me3,与H3K27me3没有空间冲突)相互作用[86]。
2.3 与DNA甲基化相关
AtMBD9 (METHYL-CPG BINDING DOMAIN 9)蛋白拥有5个与染色质结构修饰调控基因表达相关的结构域,分别为1个MBD结构域、2个PHD结构域、1个Bromo结构域、1个FYRN结构域(N-terminal phenylalanine/tyrosine-rich domain)和1个FYR结构域(C-terminal phenylalanine/tyrosine-rich domain)[53]。AtMBD9是一个转录调控因子,通过DNA甲基化和组蛋白乙酰基化这两种表观遗传途径,分别间接和直接调控基因的表达,从而影响拟南芥的生长发育[52]。研究表明,AtMBD9突变体出现DNA高度甲基化,并且AtMBD9通过调控H4乙酰化来影响开花调节基因FLC的表达,从而调节开花时间,其突变体表现出早开花和根系分支增多的表型[53]。MBD结构域可以结合甲基化的DNA,Bromo结构域可能发挥了催化组蛋白乙酰化反应的作用,因为该结构域在其他组蛋白乙酰基转移酶中经常存在[87],而PHD结构域的功能还没有具体的研究。拟南芥ORTH/VIM (ORTHRUS/VARIANT IN METHYLATION)基因家族有6个成员,是哺乳动物UHRF(UBIQUITIN-LIKE CONTAINING, PHD, RING FINGER)的直系同源基因群[55]。ORTH1-ORTH5编码的蛋白拥有1个PHD结构域、2个RING结构域和1个SRA(SET RING associated)结构域。研究表明,ORTH蛋白在体外拥有E3泛素连接酶活性,并且可以介导DNA甲基化。而第6个成员ORL1/VIM6 (ORTH LIKE-1/VARIANT IN METHYLATION6)只有1个RING结构域和1个SRA结构域,其SRA结构域作用于结合甲基化的DNA[88]。而在人的UHRF1中,PHD和SRA结构域共同作用,使其结合到甲基化的H3K9上[89]。
正确的甲基化对于基因调控十分重要。MET1 (DNA METHYLTRANSFERASE 1)作用于DNA的CG甲基化。研究发现,ORTH/VIM可以通过识别MET1建立的CG甲基化在相应的位点积累,成为MET1介导的DNA甲基化途径中关键的组成部分[54]。
此外,ORTH2/VIM1的PHD结构域可以与NtSET (SU(VAR)3-9蛋白,烟草(Nicotiana tabacum)中的一种甲基转移酶)相互作用[90]。因此,该PHD结构域是一个蛋白互作结构域,可能通过招募H3K9甲基转移酶,在DNA甲基化和H3K9组蛋白修饰之间建立联系。
在VIM突变体的靶基因上,活跃的染色质标记如H3K4me3和H3K9/K14ac明显增加,而抑制的染色质标记如H3K9me2和H3K27me3减少。此外,VIM的不足会引起异染色质染色中心(chromocenters) H3K9me2的明显减少[54]。因此,VIM蛋白通过调控激活和抑制的组蛋白修饰,使靶基因沉默,在协调组蛋白修饰和DNA甲基化状态的转变方面发挥了重要作用。
2.4 具有E3泛素连接酶活性
类泛素化(SUMOylation)在调控真核生物生长发育的多个方面发挥了重要的作用。Siz/PISA家族是一类具有SP-RING特征结构域的SUMO E3泛素连接酶,具有保守的特征结构域,包括SAP、PINIT和PHD finger结构域(只有植物成员具有PHD结构域,动物和酵母中没有)[91]。拟南芥AtSIZ1通过调控基因的表达,参与植物的生长发育以及应对干旱、低盐的胁迫的过程[57]。AtSIZ1包含了5个结构域,分别是SAP结构域、Siz/PIAS-RING结构域(作用于发挥SUMO E3连接酶活性)、PINIT结构域、SXS结构域(促进与SUMO的结合)以及PHD结构域。研究人员推测其PHD结构域与染色质重塑复合体有关,或者可能具有E3泛素连接酶活性[58]。
在水稻中OsSIZ1的SP-RING结构域主要发挥了类泛素化作用。OsSIZ1的PHD结构域可以识别H3R2me2和H3K4me3,研究指出PHD结构域可能和DNA结合结构域SAP一起协同作用,通过使一些效应因子(包括转录因子、组蛋白、HDAC组蛋白去乙酰化酶等)发生类泛素化,从而调控基因的表达[56]。
人类的KAP1也是一个含有PHD结构域且具有E3泛素连接酶活性的蛋白。KAP1的PHD结构域作为E3连接酶,介导了分子内的Bromo结构域的类泛素化,参与了基因沉默[92]。
2.5 是染色质重塑因子
依赖于ATP的染色质重塑因子主要包括:SWI/ SNF (SWITCH/SUCROSE NON-FERMENTING)、ISWI (IMITATION SWITCH)、INO80 (INOSITOL 80)和CHD (CHROMODOMAIN-HELICASE-DNA BINDING)。其中CHD是依赖于ATP的染色质重塑因子,在调控基因表达方面具有重要作用。CHD蛋白可以分为3个亚家族,分别为Chd1亚家族(也称为CHD1蛋白)、Mi-2亚家族(也称为CHD3蛋白)和CHD7亚家族[61]。拟南芥中Mi-2亚家族有3个成员(PKL、CHR4和CHR7),水稻中也有3个(CHR207、CHR729和CHR703)。这些蛋白除了CHR7以外,都拥有1个PHD结构域和2个chromo结构域。拟南芥PKL和CHR4作用于DNA损伤应答[59],与水稻CHR729一样在调控植物生长和响应胁迫基因的表达中发挥重要的作用[60]。
PKL对一些基因的表达具有抑制作用,这些基因位点上有H3K27me3富集,推测该抑制作用与H3K27me3有关。PKL还可以激活一些基因的表达,它出现在例如ACT7 (ACTIN7)和UBQ10 (POLYUBIQUITIN 10)这类不需要依赖PKL表达的基因的启动子区域,并且促进普遍的染色质重塑过程[62]。CHR7没有PHD结构域,但是与PKL在激活基因表达上拥有重叠的功能,这说明PHD结构域可能对激活转录表达是不必要的。
2.6 与bHLH型的转录因子相互作用
TIP3 (TDR interacting protein 3)是水稻中的一种雄性不育基因,包含PHD结构域,主要于花药发育期间在绒毡层和小孢子中表达[93]。TDR是一个bHLH型的转录因子,通过直接激活其靶基因的表达来调控绒毡层的发育和退化、脂质的代谢以及花粉的形成等过程[93]。研究发现,TIP3可以作为转录激活因子与TDR相互作用,从而影响与绒毡层程序性死亡和花粉壁发育相关基因的表达;并且酵母双杂交实验表明,TIP3的PHD结构域可以在在酵母中与3个bHLH型的转录因子TDR、EAT1和TIP2相互作用[93]。因此,含有PHD结构域的蛋白还可能是通过不依赖于组蛋白修饰的方式,直接与转录因子结合,将转录因子招募到基因的启动子区域,从而发挥基因表达调控的功能。
3 结语与展望
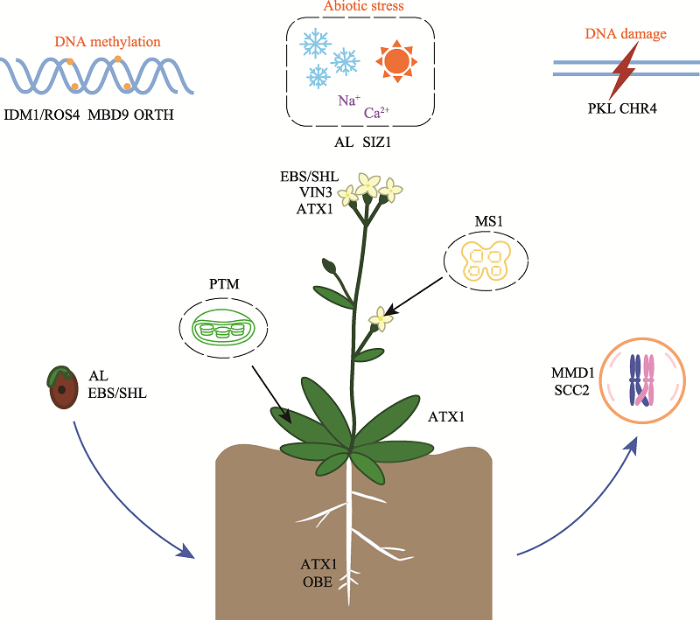
在植物中,含有PHD结构域的蛋白作为转录调控因子参与了对植物生长发育具有重要作用的基因的转录调控。正如拟南芥中的PHD蛋白,参与种子萌发、根系发育、发芽、开花、减数分裂,以及减数分裂后的花粉发育等过程(图4)。图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4拟南芥中PHD结构域蛋白的功能示意图
图中均为拟南芥的PHD结构域蛋白。可以看出,这些蛋白参与的生长发育相关的转录调控过程主要包括:种子萌发、根系发育、发芽、开花、减数分裂,以及减数分裂后的花粉发育等过程,并且还有蛋白与DNA甲基化相关。同时,对于植物的逆境胁迫和DNA损伤修复过程也发挥了重要的作用。
Fig. 4Schematic diagram of the role of PHD domain proteins in Arabidopsis thaliana
基因的转录调控与各种染色质修饰以及DNA甲基化密切相关。一般认为DNA甲基化与转录抑制相关,组蛋白乙酰化促进转录激活,而组蛋白甲基化则通过招募各种下游效应蛋白来发挥激活(H3K4me)或抑制(H3K9me)转录的修饰效应[94]。PHD结构域由于不同的氨基酸序列组成和结构特异性,对于不同的组蛋白乙酰化和甲基化修饰的结合也具有特定的偏好性。因此,对于PHD结构域结合特异性的精准研究,将会对其如何发挥转录调控功能具有十分重要的指示作用。目前已有研究在PHD结构域的结合特异性的基础上,开发相应的化学探针(chemical probes)以干扰其与组蛋白H3之间的相互作用[95],以及通过一些小分子抑制子(small molecule inhibitors)来竞争结合PHD结构域[96]。此外,开发具有E3泛素连接酶活性的PHD结构域配体以降解PHD蛋白也可能成为未来的一种研究思路[95]。
此外,含有PHD结构域的蛋白不仅在植物中保守存在,在人类基因组中也有约180多个含有PHD结构域的蛋白,这些蛋白同样具有转录调控、细胞周期和凋亡等方面的重要作用。对于植物PHD结构域蛋白的研究,可以更好地了解PHD结构域蛋白的作用机制,以运用于人类的肿瘤和疾病治疗。例如,在基因靶向治疗中,可以将外源性的PHD结构域蛋白导入人体,依靠PHD结构域对组蛋白密码的识别,从而靶向特定基因的转录激活和抑制,以达到抑制肿瘤基因表达等目的。
综上所述,在基因的转录调控过程中,含有PHD结构域的蛋白通过与不同的表观遗传效应蛋白(例如组蛋白甲基转移酶、组蛋白去甲基化酶、组蛋白乙酰基转移酶等)相互作用,以协调恰当的染色质修饰状态或者DNA甲基化状态,共同调控基因的转录表达。因此,研究含有PHD结构域的蛋白,对于植物生长发育和逆境胁迫等调控过程的解析具有十分重要的意义。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.4161/15592324.2014.993253URLPMID:26156103 [本文引用: 2]

Posttranslational modifications present in the amino-terminal tails of histones play a pivotal role in the chromatin-mediated regulation of gene expression patterns that control plant developmental transitions. Therefore, the function of protein domains that specifically recognize these histone covalent modifications and recruit chromatin remodeling complexes and the transcriptional machinery to modulate gene expression is essential for a proper control of plant development. Plant HomeoDomain (PHD) motifs act as effectors that can specifically bind a number of histone modifications and mediate the activation or repression of underlying genes. In this review we summarize recent findings that emphasize the crucial role of this versatile family of chromatin
DOI:10.1016/j.celrep.2017.12.099URLPMID:29386129 [本文引用: 1]
Histone post-translational modifications (PTMs) and their recognition by histone readers exert crucial functions in eukaryotes. Despite extensive studies, conservation and diversity of histone PTM regulation between animals and plants remain less explored because of a lack of systematic knowledge of histone readers in plants. Based on a high-throughput surface plasmon resonance imaging (SPRi) platform, we report the lab-on-chip profiling of interactions between 204 putative reader domains and 11 types of histone peptides in Arabidopsis thaliana. Eleven reader hits were then chosen for histone combinatorial readout pattern profiling. Systematic analysis of histone PTM recognition in Arabidopsis thaliana reveals that plant and human histone readers share conservation in domain types and recognition mechanisms. The differences in particular histone mark recognition by transcription regulator EML1 and DNA damage repair factor MSH6 indicate plant-specific histone PTMs function in Arabidopsis thaliana acquired during evolution.
DOI:10.1006/jmbi.2000.4308URLPMID:11124022 [本文引用: 1]
The PHD (plant homeo domain) is a approximately 50-residue motif found mainly in proteins involved in eukaryotic transcription regulation. The characteristic sequence feature is a conserved Cys(4)-HisCys(3) zinc binding motif. We have determined the solution structure of the PHD motif from the human Williams-Beuren syndrome transcription factor (WSTF) protein. The domain folds into an interleaved zinc finger which binds two Zn(2+) in a similar manner to that of the RING and FYVE domains. The structure reveals a conserved zinc-binding core, together with two variable loops that are likely candidates for interactions between the various PHD domains and their specific ligands.
DOI:10.1016/s0969-2126(03)00122-9URLPMID:12842043 [本文引用: 1]
The design of proteins with tailored functions remains a relatively elusive goal. Small size, a well-defined structure, and the ability to maintain structural integrity despite multiple mutations are all desirable properties for such designer proteins. Many zinc binding domains fit this description. We determined the structure of a PHD finger from the transcriptional cofactor Mi2beta and investigated the suitability of this domain as a scaffold for presenting selected binding functions. The two flexible loops in the structure were mutated extensively by either substitution or expansion, without affecting the overall fold of the domain. A binding site for the corepressor CtBP2 was also grafted onto the domain, creating a new PHD domain that can specifically bind CtBP2 both in vitro and in the context of a eukaryotic cell nucleus. These results represent a step toward designing new regulatory proteins for modulating aberrant gene expression in vivo.
DOI:10.1038/nature04802URLPMID:16728978 [本文引用: 5]
Mono-, di- and trimethylated states of particular histone lysine residues are selectively found in different regions of chromatin, thereby implying specialized biological functions for these marks ranging from heterochromatin formation to X-chromosome inactivation and transcriptional regulation. A major challenge in chromatin biology has centred on efforts to define the connection between specific methylation states and distinct biological read-outs impacting on function. For example, histone H3 trimethylated at lysine 4 (H3K4me3) is associated with transcription start sites of active genes, but the molecular 'effectors' involved in specific recognition of H3K4me3 tails remain poorly understood. Here we demonstrate the molecular basis for specific recognition of H3(1-15)K4me3 (residues 1-15 of histone H3 trimethylated at K4) by a plant homeodomain (PHD) finger of human BPTF (bromodomain and PHD domain transcription factor), the largest subunit of the ATP-dependent chromatin-remodelling complex, NURF (nucleosome remodelling factor). We report on crystallographic and NMR structures of the bromodomain-proximal PHD finger of BPTF in free and H3(1-15)K4me3-bound states. H3(1-15)K4me3 interacts through anti-parallel beta-sheet formation on the surface of the PHD finger, with the long side chains of arginine 2 (R2) and K4me3 fitting snugly in adjacent pre-formed surface pockets, and bracketing an invariant tryptophan. The observed stapling role by non-adjacent R2 and K4me3 provides a molecular explanation for H3K4me3 site specificity. Binding studies establish that the BPTF PHD finger exhibits a modest preference for K4me3- over K4me2-containing H3 peptides, and discriminates against monomethylated and unmodified counterparts. Furthermore, we identified key specificity-determining residues from binding studies of H3(1-15)K4me3 with PHD finger point mutants. Our findings call attention to the PHD finger as a previously uncharacterized chromatin-binding module found in a large number of chromatin-associated proteins.
DOI:10.2174/138945009788185040URLPMID:19442115 [本文引用: 1]
The Inhibitor of Growth (ING) tumor suppressors are implicated in oncogenesis, control of DNA damage repair, cellular senescence and apoptosis. All members of the ING family contain unique amino-terminal regions and a carboxy-terminal plant homeodomain (PHD) finger. While the amino-terminal domains associate with a number of protein effectors including distinct components of histone deacetylase (HDAC) and histone acetyltransferase (HAT) complexes, the PHD finger binds strongly and specifically to histone H3 trimethylated at lysine 4 (H3K4me3). In this review we describe the molecular mechanism of H3K4me3 recognition by the ING1-5 PHD fingers, analyze the determinants of the histone specificity and compare the biological activities and structures within subsets of PHD fingers. The atomic-resolution structures of the ING PHD fingers in complex with a H3K4me3 peptide reveal that the histone tail is bound in a large and deep binding site encompassing nearly one-third of the protein surface. An extensive network of intermolecular hydrogen bonds, hydrophobic and cation-pi contacts, and complementary surface interactions coordinate the first six residues of the H3K4me3 peptide. The trimethylated Lys4 occupies an elongated groove, formed by the highly conserved aromatic and hydrophobic residues of the PHD finger, whereas the adjacent groove accommodates Arg2. The two grooves are connected by a narrow channel, the small size of which defines the PHD finger's specificity, excluding interactions with other modified histone peptides. Binding of the ING PHD fingers to H3K4me3 plays a critical role in regulating chromatin acetylation. The ING proteins function as tethering molecules that physically link the HDAC and HAT enzymatic complexes to chromatin. In this review we also highlight progress recently made in understanding the molecular basis underlying biological and tumorigenic activities of the ING tumor suppressors.
DOI:10.1016/j.cell.2011.11.032URL [本文引用: 3]
Specific chromatin marks keep master regulators of differentiation silent yet poised for activation by extracellular signals. We report that nodal TGF-beta signals use the poised histone mark H3K9me3 to trigger differentiation of mammalian embryonic stem cells. Nodal receptors induce the formation of companion Smad4-Smad2/3 and TRIM33-Smad2/3 complexes. The PHD-Bromo cassette of TRIM33 facilitates binding of TRIM33-Smad2/3 to H3K9me3 and H3K18ac on the promoters of mesendoderm regulators Gsc and Mixl1. The crystal structure of this cassette, bound to histone H3 peptides, illustrates that PHD recognizes K9me3, and Bromo binds an adjacent K18ac. The interaction between TRIM33Smad2/3 and H3K9me3 displaces the chromatin-compacting factor HP1g, making nodal response elements accessible to Smad4-Smad2/3 for Pol II recruitment. In turn, Smad4 increases K18 acetylation to augment TRIM33-Smad2/3 binding. Thus, nodal effectors use the H3K9me3 mark as a platform to switch master regulators of stem cell differentiation from the poised to the active state.
DOI:10.1038/nsmb.2062URLPMID:21666679 [本文引用: 2]
ATR-X (alpha-thalassemia/mental retardation, X-linked) syndrome is a human congenital disorder that causes severe intellectual disabilities. Mutations in the ATRX gene, which encodes an ATP-dependent chromatin-remodeler, are responsible for the syndrome. Approximately 50% of the missense mutations in affected persons are clustered in a cysteine-rich domain termed ADD (ATRX-DNMT3-DNMT3L, ADD(ATRX)), whose function has remained elusive. Here we identify ADD(ATRX) as a previously unknown histone H3-binding module, whose binding is promoted by lysine 9 trimethylation (H3K9me3) but inhibited by lysine 4 trimethylation (H3K4me3). The cocrystal structure of ADD(ATRX) bound to H3(1-15)K9me3 peptide reveals an atypical composite H3K9me3-binding pocket, which is distinct from the conventional trimethyllysine-binding aromatic cage. Notably, H3K9me3-pocket mutants and ATR-X syndrome mutants are defective in both H3K9me3 binding and localization at pericentromeric heterochromatin; thus, we have discovered a unique histone-recognition mechanism underlying the ATR-X etiology.
DOI:10.1074/jbc.M110.208207URLPMID:21278251 [本文引用: 3]
A major challenge in chromatin biology is to understand the mechanisms by which chromatin is remodeled into active or inactive states as required during development and cell differentiation. One complex implicated in these processes is the nucleosome remodeling and histone deacetylase (NuRD) complex, which contains both histone deacetylase and nucleosome remodeling activities and has been implicated in the silencing of subsets of genes involved in various stages of cellular development. Chromodomain-helicase-DNA-binding protein 4 (CHD4) is a core component of the NuRD complex and contains a nucleosome remodeling ATPase domain along with two chromodomains and two plant homeodomain (PHD) fingers. We have previously demonstrated that the second PHD finger of CHD4 binds peptides corresponding to the N terminus of histone H3 methylated at Lys(9). Here, we determine the solution structure of PHD2 in complex with H3K9me3, revealing the molecular basis of histone recognition, including a cation-pi recognition mechanism for methylated Lys(9). Additionally, we demonstrate that the first PHD finger also exhibits binding to the N terminus of H3, and we establish the histone-binding surface of this domain. This is the first instance where histone binding ability has been demonstrated for two separate PHD modules within the one protein. These findings suggest that CHD4 could bind to two H3 N-terminal tails on the same nucleosome or on two separate nucleosomes simultaneously, presenting exciting implications for the mechanism by which CHD4 and the NuRD complex could direct chromatin remodeling.
DOI:10.1074/jbc.C600286200URLPMID:17142463 [本文引用: 1]
The PHD finger motif is a signature chromatin-associated motif that is found throughout eukaryotic proteomes. Here we have determined the histone methyl-lysine binding activity of the PHD fingers present within the Saccharomyces cerevisiae proteome. We provide evidence on the genomic scale that PHD fingers constitute a general class of effector modules for histone H3 trimethylated at lysine 4 (H3K4me3) and histone H3 trimethylated at lysine 36 (H3K36me3). Structural modeling of PHD fingers demonstrates a conserved mechanism for recognizing the trimethyl moiety and provides insight into the molecular basis of affinity for the different methyl-histone ligands. Together, our study suggests that a common function for PHD fingers is to transduce methyl-lysine events and sheds light on how a single histone modification can be linked to multiple biological outcomes.
DOI:10.1038/cr.2011.124URLPMID:21808300 [本文引用: 3]
DOI:10.1038/cr.2011.123URLPMID:21808299 [本文引用: 1]
DOI:10.1101/gad.188359.112URLPMID:22713874 [本文引用: 3]
Histone acetylation is a hallmark for gene transcription. As a histone acetyltransferase, MOZ (monocytic leukemia zinc finger protein) is important for HOX gene expression as well as embryo and postnatal development. In vivo, MOZ forms a tetrameric complex with other subunits, including several chromatin-binding modules with regulatory functions. Here we report the solution structure of the tandem PHD (plant homeodomain) finger (PHD12) of human MOZ in a free state and the 1.47 A crystal structure in complex with H3K14ac peptide, which reveals the structural basis for the recognition of unmodified R2 and acetylated K14 on histone H3. Moreover, the results of chromatin immunoprecipitation (ChIP) and RT-PCR assays indicate that PHD12 facilitates the localization of MOZ onto the promoter locus of the HOXA9 gene, thereby promoting the H3 acetylation around the promoter region and further up-regulating the HOXA9 mRNA level. Taken together, our findings suggest that the combinatorial readout of the H3R2/K14ac by PHD12 might represent an important epigenetic regulatory mechanism that governs transcription and also provide a clue of cross-talk between the MOZ complex and histone H3 modifications.
DOI:10.1038/nature09139URLPMID:20613843 [本文引用: 3]
Histone lysine acetylation and methylation have an important role during gene transcription in a chromatin context. Knowledge concerning the types of protein modules that can interact with acetyl-lysine has so far been limited to bromodomains. Recently, a tandem plant homeodomain (PHD) finger (PHD1-PHD2, or PHD12) of human DPF3b, which functions in association with the BAF chromatin remodelling complex to initiate gene transcription during heart and muscle development, was reported to bind histones H3 and H4 in an acetylation-sensitive manner, making it the first alternative to bromodomains for acetyl-lysine binding. Here we report the structural mechanism of acetylated histone binding by the double PHD fingers of DPF3b. Our three-dimensional solution structures and biochemical analysis of DPF3b highlight the molecular basis of the integrated tandem PHD finger, which acts as one functional unit in the sequence-specific recognition of lysine-14-acetylated histone H3 (H3K14ac). Whereas the interaction with H3 is promoted by acetylation at lysine 14, it is inhibited by methylation at lysine 4, and these opposing influences are important during transcriptional activation of the mouse DPF3b target genes Pitx2 and Jmjd1c. Binding of this tandem protein module to chromatin can thus be regulated by different histone modifications during the initiation of gene transcription.
DOI:10.1074/jbc.R111.219139URLPMID:21454653 [本文引用: 1]
The study of histone modifications and their interaction with effector modules/proteins has attracted increasing attention in recent years. Accumulating evidence indicates that epigenetic regulation, which involves post-translational modification on histones and DNAs or the participation of RNAs, plays an important role in many cellular processes. Histone modifications can function individually but are also capable of functioning combinatorially as a pattern. Recently, much more attention has focused on interpreting combined histone patterns by their downstream effectors. Structure/function-based studies on paired module-mediated histone cross-talk have greatly enhanced our understanding of the plasticity of the
[本文引用: 1]
DOI:10.1016/j.tibs.2011.03.005URLPMID:21514168 [本文引用: 1]
PHD (plant homeodomain) zinc fingers are structurally conserved modules found in proteins that modify chromatin as well as mediate molecular interactions in gene transcription. The original discovery of their role in gene transcription is attributed to the recognition of lysine-methylated histone H3. Recent studies show that PHD fingers have a sophisticated histone sequence reading capacity that is modulated by the interplay between different histone modifications. These studies underscore the functional versatility of PHD fingers as epigenome readers that control gene expression through molecular recruitment of multiprotein complexes of chromatin regulators and transcription factors. Moreover, they reinforce the concept that evolutionary changes in amino acids surrounding ligand binding sites on a conserved structural fold impart great functional diversity upon this family of proteins.
[本文引用: 1]
DOI:10.1038/nature06034URLPMID:17687328 [本文引用: 4]
Histone methylation is crucial for regulating chromatin structure, gene transcription and the epigenetic state of the cell. LSD1 is a lysine-specific histone demethylase that represses transcription by demethylating histone H3 on lysine 4 (ref. 1). The LSD1 complex contains a number of proteins, all of which have been assigned roles in events upstream of LSD1-mediated demethylation apart from BHC80 (also known as PHF21A), a plant homeodomain (PHD) finger-containing protein. Here we report that, in contrast to the PHD fingers of the bromodomain PHD finger transcription factor (BPTF) and inhibitor of growth family 2 (ING2), which bind methylated H3K4 (H3K4me3), the PHD finger of BHC80 binds unmethylated H3K4 (H3K4me0), and this interaction is specifically abrogated by methylation of H3K4. The crystal structure of the PHD finger of BHC80 bound to an unmodified H3 peptide has revealed the structural basis of the recognition of H3K4me0. Knockdown of BHC80 by RNA inhibition results in the de-repression of LSD1 target genes, and this repression is restored by the reintroduction of wild-type BHC80 but not by a PHD-finger mutant that cannot bind H3. Chromatin immunoprecipitation showed that BHC80 and LSD1 depend reciprocally on one another to associate with chromatin. These findings couple the function of BHC80 to that of LSD1, and indicate that unmodified H3K4 is part of the 'histone code'. They further raise the possibility that the generation and recognition of the unmodified state on histone tails in general might be just as crucial as post-translational modifications of histone for chromatin and transcriptional regulation.
DOI:10.1016/j.str.2009.02.017URL [本文引用: 2]
Summary
Human autoimmune regulator (AIRE) functions to control thymic expression of tissue-specific antigens via sequence-specific histone H3 recognition by its plant homeodomain (PHD) finger. Mutations in the AIRE PHD finger have been linked to autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). Here we report the three-dimensional solution structure of the first PHD finger of human AIRE bound to a histone H3 peptide. The structure reveals a detailed network of interactions between the protein and the amino-terminal residues of histone H3, and particularly key electrostatic interactions of a conserved aspartic acid 297 in AIRE with the unmodified lysine 4 of histone H3 (H3K4). NMR binding study with H3 peptides carrying known posttranslational modifications flanking H3K4 confirms that transcriptional regulation by AIRE through its interactions with histone H3 is confined to the first N-terminal eight residues in H3. Our study offers a molecular explanation for the APECED mutations and helps define a subclass of the PHD finger family proteins that recognize histone H3 in a sequence-specific manner.DOI:10.1073/pnas.0704057104URLPMID:17609377 [本文引用: 2]
The chromatin-associated protein ATRX was originally identified because mutations in the ATRX gene cause a severe form of syndromal X-linked mental retardation associated with alpha-thalassemia. Half of all of the disease-associated missense mutations cluster in a cysteine-rich region in the N terminus of ATRX. This region was named the ATRX-DNMT3-DNMT3L (ADD) domain, based on sequence homology with a family of DNA methyltransferases. Here, we report the solution structure of the ADD domain of ATRX, which consists of an N-terminal GATA-like zinc finger, a plant homeodomain finger, and a long C-terminal alpha-helix that pack together to form a single globular domain. Interestingly, the alpha-helix of the GATA-like finger is exposed and highly basic, suggesting a DNA-binding function for ATRX. The disease-causing mutations fall into two groups: the majority affect buried residues and hence affect the structural integrity of the ADD domain; another group affects a cluster of surface residues, and these are likely to perturb a potential protein interaction site. The effects of individual point mutations on the folding state and stability of the ADD domain correlate well with the levels of mutant ATRX protein in patients, providing insights into the molecular pathophysiology of ATR-X syndrome.
DOI:10.1016/j.molcel.2008.12.016URLPMID:19187765 [本文引用: 1]
Aberrations in chromatin dynamics play a fundamental role in tumorigenesis, yet relatively little is known of the molecular mechanisms linking histone lysine methylation to neoplastic disease. ING4 (Inhibitor of Growth 4) is a native subunit of an HBO1 histone acetyltransferase (HAT) complex and a tumor suppressor protein. Here we show a critical role for specific recognition of histone H3 trimethylated at lysine 4 (H3K4me3) by the ING4 PHD finger in mediating ING4 gene expression and tumor suppressor functions. The interaction between ING4 and H3K4me3 augments HBO1 acetylation activity on H3 tails and drives H3 acetylation at ING4 target promoters. Further, ING4 facilitates apoptosis in response to genotoxic stress and inhibits anchorage-independent cell growth, and these functions depend on ING4 interactions with H3K4me3. Together, our results demonstrate a mechanism for brokering crosstalk between H3K4 methylation and H3 acetylation and reveal a molecular link between chromatin modulation and tumor suppressor mechanisms.
DOI:10.1038/nature04814URLPMID:16728977 [本文引用: 1]
Covalent modifications of histone tails have a key role in regulating chromatin structure and controlling transcriptional activity. In eukaryotes, histone H3 trimethylated at lysine 4 (H3K4me3) is associated with active chromatin and gene expression. We recently found that plant homeodomain (PHD) finger of tumour suppressor ING2 (inhibitor of growth 2) binds H3K4me3 and represents a new family of modules that target this epigenetic mark. The molecular mechanism of H3K4me3 recognition, however, remains unknown. Here we report a 2.0 A resolution structure of the mouse ING2 PHD finger in complex with a histone H3 peptide trimethylated at lysine 4. The H3K4me3 tail is bound in an extended conformation in a deep and extensive binding site consisting of elements that are conserved among the ING family of proteins. The trimethylammonium group of Lys 4 is recognized by the aromatic side chains of Y215 and W238 residues, whereas the intermolecular hydrogen-bonding and complementary surface interactions, involving Ala 1, Arg 2, Thr 3 and Thr 6 of the peptide, account for the PHD finger's high specificity and affinity. Substitution of the binding site residues disrupts H3K4me3 interaction in vitro and impairs the ability of ING2 to induce apoptosis in vivo. Strong binding of other ING and YNG PHD fingers suggests that the recognition of H3K4me3 histone code is a general feature of the ING/YNG proteins. Elucidation of the mechanisms underlying this novel function of PHD fingers provides a basis for deciphering the role of the ING family of tumour suppressors in chromatin regulation and signalling.
DOI:10.1038/nsmb.2062URLPMID:21666679 [本文引用: 1]
ATR-X (alpha-thalassemia/mental retardation, X-linked) syndrome is a human congenital disorder that causes severe intellectual disabilities. Mutations in the ATRX gene, which encodes an ATP-dependent chromatin-remodeler, are responsible for the syndrome. Approximately 50% of the missense mutations in affected persons are clustered in a cysteine-rich domain termed ADD (ATRX-DNMT3-DNMT3L, ADD(ATRX)), whose function has remained elusive. Here we identify ADD(ATRX) as a previously unknown histone H3-binding module, whose binding is promoted by lysine 9 trimethylation (H3K9me3) but inhibited by lysine 4 trimethylation (H3K4me3). The cocrystal structure of ADD(ATRX) bound to H3(1-15)K9me3 peptide reveals an atypical composite H3K9me3-binding pocket, which is distinct from the conventional trimethyllysine-binding aromatic cage. Notably, H3K9me3-pocket mutants and ATR-X syndrome mutants are defective in both H3K9me3 binding and localization at pericentromeric heterochromatin; thus, we have discovered a unique histone-recognition mechanism underlying the ATR-X etiology.
DOI:10.1038/nature04802URLPMID:16728978 [本文引用: 1]
Mono-, di- and trimethylated states of particular histone lysine residues are selectively found in different regions of chromatin, thereby implying specialized biological functions for these marks ranging from heterochromatin formation to X-chromosome inactivation and transcriptional regulation. A major challenge in chromatin biology has centred on efforts to define the connection between specific methylation states and distinct biological read-outs impacting on function. For example, histone H3 trimethylated at lysine 4 (H3K4me3) is associated with transcription start sites of active genes, but the molecular 'effectors' involved in specific recognition of H3K4me3 tails remain poorly understood. Here we demonstrate the molecular basis for specific recognition of H3(1-15)K4me3 (residues 1-15 of histone H3 trimethylated at K4) by a plant homeodomain (PHD) finger of human BPTF (bromodomain and PHD domain transcription factor), the largest subunit of the ATP-dependent chromatin-remodelling complex, NURF (nucleosome remodelling factor). We report on crystallographic and NMR structures of the bromodomain-proximal PHD finger of BPTF in free and H3(1-15)K4me3-bound states. H3(1-15)K4me3 interacts through anti-parallel beta-sheet formation on the surface of the PHD finger, with the long side chains of arginine 2 (R2) and K4me3 fitting snugly in adjacent pre-formed surface pockets, and bracketing an invariant tryptophan. The observed stapling role by non-adjacent R2 and K4me3 provides a molecular explanation for H3K4me3 site specificity. Binding studies establish that the BPTF PHD finger exhibits a modest preference for K4me3- over K4me2-containing H3 peptides, and discriminates against monomethylated and unmodified counterparts. Furthermore, we identified key specificity-determining residues from binding studies of H3(1-15)K4me3 with PHD finger point mutants. Our findings call attention to the PHD finger as a previously uncharacterized chromatin-binding module found in a large number of chromatin-associated proteins.
DOI:10.1016/j.jsb.2016.07.001URLPMID:27402533 [本文引用: 1]
Histone acetylation plays an important role in chromatin dynamics and is associated with active gene transcription. This modification is written by acetyltransferases, erased by histone deacetylases and read out by bromodomain containing proteins, and others such as tandem PHD fingers of DPF3b. Here we report the high resolution crystal structure of the tandem PHD fingers of DPF3b in complex with an H3K14ac peptide. In the complex structure, the histone peptide adopts an alpha-helical conformation, unlike previously observed by NMR, but similar to a previously reported MOZ-H3K14ac complex structure. Our crystal structure adds to existing evidence that points to the alpha-helix as a natural conformation of histone tails as they interact with histone-associated proteins.
DOI:10.1016/j.jsb.2012.06.014URL [本文引用: 3]
Plant homeodomain (PHD) finger is found to be a versatile reader that functions in recruiting transcription factors and chromatin modification complexes. Bromodomain- and PHD finger-containing (BRPF) proteins are identified as scaffold component in a couple of histone acetyltransferase (HATS) complexes but the biological function of PHD fingers, composing the motif called PZPM (PHD/Zn-knuckle/PHD Motif), in BRPF proteins is far from being well understood. Here we report the three-dimensional solution structure of the second PHD finger of PZPM in human BRPF2. According to the structure, BRPF2 PHD2 possesses a two-strand p sheet which is different from any other PHD fingers. Functionally, this PHD finger can potentially bind DNA non-specifically with an evolutionarily conserved and positively charged surface. We provide the structural and interaction information of this atypical PHD finger and categorize this BRPF2 PHD2 into a new subset of PHD finger. Moreover our work also shed light on the functional aspect of the PZPM. (C) 2012 Elsevier Inc.
DOI:10.1074/jbc.M111.244400URLPMID:21880731 [本文引用: 1]
MOZ (monocytic leukemic zinc-finger protein) and MORF (MOZ-related factor) are histone acetyltransferases important for HOX gene expression as well as embryo and postnatal development. They form complexes with other regulatory subunits through the scaffold proteins BRPF1/2/3 (bromodomain-PHD (plant homeodomain) finger proteins 1, 2, or 3). BRPF proteins have multiple domains, including two PHD fingers, for potential interactions with histones. Here we show that the first PHD finger of BRPF2 specifically recognizes the N-terminal tail of unmodified histone H3 (unH3) and report the solution structures of this PHD finger both free and in complex with the unH3 peptide. Structural analysis revealed that the unH3 peptide forms a third antiparallel beta-strand that pairs with the PHD1 two-stranded antiparallel beta-sheet. The binding specificity was determined primarily through the recognition of arginine 2 and lysine 4 of the unH3 by conserved aspartic acids of PHD1 and of threonine 6 of the unH3 by a conserved asparagine. Isothermal titration calorimetry and NMR assays showed that post-translational modifications such as H3R2me2as, H3T3ph, H3K4me, H3K4ac, and H3T6ph antagonized the interaction between histone H3 and PHD1. Furthermore, histone binding by PHD1 was important for BRPF2 to localize to the HOXA9 locus in vivo. PHD1 is highly conserved in yeast NuA3 and other histone acetyltransferase complexes, so the results reported here also shed light on the function and regulation of these complexes.
DOI:10.1093/jxb/eru355URL [本文引用: 2]
ARABIDOPSIS HOMOLOG of TRITHORAX1 (ATX1/SDG27), a known regulator of flower development, encodes a H3K4-histone methyltransferase that maintains a number of genes in an active state. In this study, the role of ATX1 in root development was evaluated. The loss-of-function mutant atx1-1 was impaired in primary root growth. The data suggest that ATX1 controls root growth by regulating cell cycle duration, cell production, and the transition from cell proliferation in the root apical meristem (RAM) to cell elongation. In atx1-1, the quiescent centre (QC) cells were irregular in shape and more expanded than those of the wild type. This feature, together with the atypical distribution of T-divisions, the presence of oblique divisions, and the abnormal cell patterning in the RAM, suggests a lack of coordination between cell division and cell growth in the mutant. The expression domain of QC-specific markers was expanded both in the primary RAM and in the developing lateral root primordia of atx1-1 plants. These abnormalities were independent of auxin-response gradients. ATX1 was also found to be required for lateral root initiation, morphogenesis, and emergence. The time from lateral root initiation to emergence was significantly extended in the atx1-1 mutant. Overall, these data suggest that ATX1 is involved in the timing of root development, stem cell niche maintenance, and cell patterning during primary and lateral root development. Thus, ATX1 emerges as an important player in root system architecture.
DOI:10.1371/journal.pgen.1003111URLPMID:23284292 [本文引用: 2]
Tri-methylated H3 lysine 4 (H3K4me3) is associated with transcriptionally active genes, but its function in the transcription process is still unclear. Point mutations in the catalytic domain of ATX1 (ARABIDOPSIS TRITHORAX1), a H3K4 methyltransferase, and RNAi knockdowns of subunits of the AtCOMPASS-like (Arabidopsis Complex Proteins Associated with Set) were used to address this question. We demonstrate that both ATX1 and AtCOMPASS-like are required for high level accumulation of TBP (TATA-binding protein) and Pol II at promoters and that this requirement is independent of the catalytic histone modifying activity. However, the catalytic function is critically required for transcription as H3K4me3 levels determine the efficiency of transcription elongation. The roles of H3K4me3, ATX1, and AtCOMPASS-like may be of a general relevance for transcription of Trithorax-activated eukaryotic genes.
DOI:10.1105/tpc.110.080150URLPMID:21266657 [本文引用: 2]
The Arabidopsis thaliana trithorax-like protein, ATX1, shares common structural domains, has similar histone methyltransferase (HMT) activity, and belongs in the same phylogenetic subgroup as its animal counterparts. Most of our knowledge of the role of HMTs in trimethylating lysine 4 of histone H3 (H3K4me3) in transcriptional regulation comes from studies of yeast and mammalian homologs. Little is known about the mechanism by which ATX1, or any other HMT of plant origin, affects transcription. Here, we provide insights into how ATX1 influences transcription at regulated genes, playing two distinct roles. At promoters, ATX1 is required for TATA binding protein (TBP) and RNA Polymerase II (Pol II) recruitment. In a subsequent event, ATX1 is recruited by a phosphorylated form of Pol II to the +300-bp region of transcribed sequences, where it trimethylates nucleosomes. In support of this model, inhibition of phosphorylation of the C-terminal domain of Pol II reduced the amounts of H3K4me3 and ATX1 bound at the +300-nucleotide region. Importantly, these changes did not reduce the occupancy of ATX1, TBP, or Pol II at promoters. Our results indicate that ATX1 affects transcription at target genes by a mechanism distinct from its ability to trimethylate H3K4 within genes.
DOI:10.1105/tpc.107.056614URLPMID:18375658 [本文引用: 3]
Gene duplication followed by functional specialization is a potent force in the evolution of biological diversity. A comparative study of two highly conserved duplicated genes, ARABIDOPSIS TRITHORAX-LIKE PROTEIN1 (ATX1) and ATX2, revealed features of both partial redundancy and of functional divergence. Although structurally similar, their regulatory sequences have diverged, resulting in distinct temporal and spatial patterns of expression of the ATX1 and ATX2 genes. We found that ATX2 methylates only a limited fraction of nucleosomes and that ATX1 and ATX2 influence the expression of largely nonoverlapping gene sets. Even when coregulating shared targets, ATX1 and ATX2 may employ different mechanisms. Most remarkable is the divergence of their biochemical activities: both proteins methylate K4 of histone H3, but while ATX1 trimethylates it, ATX2 dimethylates it. ATX2 and ATX1 provide an example of separated K4 di from K4 trimethyltransferase activity.
DOI:10.1111/nph.14933URLPMID:29250818 [本文引用: 1]
Trithorax-group proteins (TrxGs) play essential regulatory roles in chromatin modification to activate transcription. Although TrxGs have been shown to be extensively involved in the activation of developmental genes, how the specific TrxGs function in the dehydration and abscisic acid (ABA)-mediated modulation of downstream gene expression remains unknown. Here, we report that two evolutionarily conserved Arabidopsis thaliana TrxGs, ARABIDOPSIS TRITHORAX4 (ATX4) and ATX5, play essential roles in the drought stress response. atx4 and atx5 single loss-of-function mutants showed drought stress-tolerant and ABA-hypersensitive phenotypes during seed germination and seedling development, while the atx4 atx5 double mutant displayed further exacerbation of the phenotypes. Genome-wide RNA-sequencing analyses showed that ATX4 and ATX5 regulate the expression of genes functioning in dehydration stress. Intriguingly, ABA-HYPERSENSITIVE GERMINATION 3 (AHG3), an essential negative regulator of ABA signaling, acts genetically downstream of ATX4 and ATX5 in response to ABA. ATX4 and ATX5 directly bind to the AHG3 locus and trimethylate histone H3 of Lys 4 (H3K4). Moreover, ATX4 and ATX5 occupancies at AHG3 are dramatically increased under ABA treatment, and are also essential for RNA polymerase II (RNAPII) occupancies. Our findings reveal novel molecular functions of A. thaliana TrxGs in dehydration stress and ABA responses.
DOI:10.1104/pp.16.01944URLPMID:28550207 [本文引用: 2]
Methylation of Lys residues in the tail of the H3 histone is a key regulator of chromatin state and gene expression, conferred by a large family of enzymes containing an evolutionarily conserved SET domain. One of the main types of SET domain proteins are those controlling H3K4 di- and trimethylation. The genome of Arabidopsis (Arabidopsis thaliana) encodes 12 such proteins, including five ARABIDOPSIS TRITHORAX (ATX) proteins and seven ATX-Related proteins. Here, we examined three until-now-unexplored ATX proteins, ATX3, ATX4, and ATX5. We found that they exhibit similar domain structures and expression patterns and are redundantly required for vegetative and reproductive development. Concurrent disruption of the ATX3, ATX4, and ATX5 genes caused marked reduction in H3K4me2 and H3K4me3 levels genome-wide and resulted in thousands of genes expressed ectopically. Furthermore, atx3/atx4/atx5 triple mutants resulted in exaggerated phenotypes when combined with the atx2 mutant but not with atx1 Together, we conclude that ATX3, ATX4, and ATX5 are redundantly required for H3K4 di- and trimethylation at thousands of sites located across the genome, and genomic features associated with targeted regions are different from the ATXR3/SDG2-controlled sites in Arabidopsis.
DOI:10.1038/nsmb.1611URLPMID:19503079 [本文引用: 2]
Constitutive heterochromatin in Arabidopsis thaliana is marked by repressive chromatin modifications, including DNA methylation, histone H3 dimethylation at Lys9 (H3K9me2) and monomethylation at Lys27 (H3K27me1). The enzymes catalyzing DNA methylation and H3K9me2 have been identified; alterations in these proteins lead to reactivation of silenced heterochromatic elements. The enzymes responsible for heterochromatic H3K27me1, in contrast, remain unknown. Here we show that the divergent SET-domain proteins ARABIDOPSIS TRITHORAX-RELATED PROTEIN 5 (ATXR5) and ATXR6 have H3K27 monomethyltransferase activity, and atxr5 atxr6 double mutants have reduced H3K27me1 in vivo and show partial heterochromatin decondensation. Mutations in atxr5 and atxr6 also lead to transcriptional activation of repressed heterochromatic elements. Notably, H3K9me2 and DNA methylation are unaffected in double mutants. These results indicate that ATXR5 and ATXR6 form a new class of H3K27 methyltransferases and that H3K27me1 represents a previously uncharacterized pathway required for transcriptional repression in Arabidopsis.
DOI:10.1371/journal.pgen.1005210URLPMID:25933434 [本文引用: 1]
Active DNA demethylation plays crucial roles in the regulation of gene expression in both plants and animals. In Arabidopsis thaliana, active DNA demethylation is initiated by the ROS1 subfamily of 5-methylcytosine-specific DNA glycosylases via a base excision repair mechanism. Recently, IDM1 and IDM2 were shown to be required for the recruitment of ROS1 to some of its target loci. However, the mechanism(s) by which IDM1 is targeted to specific genomic loci remains to be determined. Affinity purification of IDM1- and IDM2- associating proteins demonstrated that IDM1 and IDM2 copurify together with two novel components, methyl-CpG-binding domain protein 7 (MBD7) and IDM2-like protein 1 (IDL1). IDL1 encodes an alpha-crystallin domain protein that shows high sequence similarity with IDM2. MBD7 interacts with IDM2 and IDL1 in vitro and in vivo and they form a protein complex associating with IDM1 in vivo. MBD7 directly binds to the target loci and is required for the H3K18 and H3K23 acetylation in planta. MBD7 dysfunction causes DNA hypermethylation and silencing of reporter genes and a subset of endogenous genes. Our results suggest that a histone acetyltransferase complex functions in active DNA demethylation and in suppression of gene silencing at some loci in Arabidopsis.
DOI:10.1016/j.molcel.2014.06.008URLPMID:25002145 [本文引用: 2]
DNA methylation patterns are dynamically controlled by DNA methylation and active DNA demethylation, but the mechanisms of regulation of active DNA demethylation are not well understood. Through forward genetic screens for Arabidopsis mutants showing DNA hypermethylation at specific loci and increased silencing of reporter genes, we identified IDM2 (increased DNA methylation 2) as a regulator of DNA demethylation and gene silencing. IDM2 dysfunction causes DNA hypermethylation and silencing of reporter genes and some endogenous genes. These effects of idm2 mutations are similar to those of mutations in IDM1, a regulator of active DNA demethylation. IDM2 encodes an alpha-crystallin domain protein in the nucleus. IDM2 and IDM1 interact physically and partially colocalize at discrete subnuclear foci. IDM2 is required for the full activity of H3K18 acetylation but not H3K23 acetylation of IDM1 in planta. Our results suggest that IDM2 functions in active DNA demethylation and in antisilencing by regulating IDM1.
DOI:10.1038/s41467-018-04836-yURLPMID:29930355 [本文引用: 1]
The ability of a cell to dynamically switch its chromatin between different functional states constitutes a key mechanism regulating gene expression. Histone mark
DOI:10.1111/pce.13046URLPMID:28770581 [本文引用: 2]
The Arabidopsis protein EARLY BOLTING IN SHORT DAYS (EBS), a plant-specific transcriptional regulator, is involved in the control of flowering time by repressing the floral integrator FT. The EBS protein binds the H3K4me3 histone mark and interacts with histone deacetylases to modulate gene expression. Here, we show that EBS also participates in the regulation of seed dormancy. ebs mutations cause a reduction in seed dormancy, and the concurrent loss of function of the EBS homologue SHORT LIFE (SHL) enhances this dormancy alteration. Transcriptomic analyses in ebs mutant seeds uncovered the misregulation of several regulators of seed dormancy including the MADS box gene AGAMOUS-LIKE67 (AGL67). AGL67 interacts genetically with EBS in seed dormancy regulation, indicating that both loci act in the same pathway. Interestingly, EBS functions independently of the master regulator gene of dormancy DELAY OF GERMINATION 1 (DOG1) and other genes encoding chromatin remodelling factors involved in the control of seed dormancy. Altogether, these data show that EBS is a central repressor of germination during seed dormancy and that SHL acts redundantly with EBS in the control of this developmental process. Our observations suggest that a tightly regulated crosstalk among histone modifications is necessary for a proper control of seed dormancy.
URLPMID:12782723 [本文引用: 2]
In plants, reproductive development requires normal meiosis, which involved several highly coordinated events. Such meiotic events are regulated in a number of ways in yeast and animal systems, including transcriptional and checkpoint control mechanisms. Although a number of mutations that affect different aspects of meiosis have been characterized in plants, very little is known about the regulation of plant meiosis at the molecular level. In particular, no meiosis-specific transcriptional regulators have been identified in plants, and checkpoint control has not been observed during plant meiosis. We report here the isolation and characterization of a new Arabidopsis male-sterile mutant that exhibits meiotic defects. Meiocytes from mutant plants appeared normal up to diakinesis, when they exhibited signs of apoptosis, including defects in chromosome behavior, cytoplasmic shrinkage, and chromatin fragmentation, followed by cell death before cytokinesis. Therefore, the mutant was named male meiocyte death1 (mmd1). The MMD1 gene was cloned using Dissociation transposon tagging and encodes a plant homeo domain domain-containing protein. MMD1 is expressed preferentially during male meiosis. Our results suggest that MMD1 may be involved in the regulation of gene expression during meiosis and that the mmd1 mutation triggers cell death in male meiocytes.
DOI:10.1105/tpc.16.00040URLPMID:27385818 [本文引用: 2]
Chromosome condensation, a process mediated by the condensin complex, is essential for proper chromosome segregation during cell division. Unlike rapid mitotic chromosome condensation, meiotic chromosome condensation occurs over a relatively long prophase I and is unusually complex due to the coordination with chromosome axis formation and homolog interaction. The molecular mechanisms that regulate meiotic chromosome condensation progression from prophase I to metaphase I are unclear. Here, we show that the Arabidopsis thaliana meiotic PHD-finger protein MMD1/DUET is required for progressive compaction of prophase I chromosomes to metaphase I bivalents. The MMD1 PHD domain is required for its function in chromosome condensation and binds to methylated histone tails. Transcriptome analysis and qRT-PCR showed that several condensin genes exhibit significantly reduced expression in mmd1 meiocytes. Furthermore, MMD1 specifically binds to the promoter region of the condensin subunit gene CAP-D3 to enhance its expression. Moreover, cap-d3 mutants exhibit similar chromosome condensation defects, revealing an MMD1-dependent mechanism for regulating meiotic chromosome condensation, which functions in part by promoting condensin gene expression. Together, these discoveries provide strong evidence that the histone reader MMD1/DUET defines an important step for regulating the progression of meiotic prophase I chromosome condensation.
DOI:10.1038/s41477-020-0697-0URLPMID:32572214 [本文引用: 2]
Histone demethylation is crucial for proper chromatin structure and to ensure normal development, and requires the large family of Jumonji C (JmjC)-containing demethylases; however, the molecular mechanisms that regulate the substrate specificity of these JmjC-containing demethylases remain largely unknown. Here, we show that the substrate specificity of the Arabidopsis histone demethylase JMJ16 is broadened from Lys 4 of histone H3 (H3K4) alone in somatic cells to both H3K4 and H3K9 when it binds to the meiocyte-specific histone reader MMD1. Consistent with this, the JMJ16 catalytic domain exhibits both H3K4 and H3K9 demethylation activities. Moreover, the JMJ16 C-terminal FYR domain interacts with the JMJ16 catalytic domain and probably restricts its substrate specificity. By contrast, MMD1 can compete with the N-terminal catalytic domain of JMJ16 for binding to the FYR-C domain, thereby expanding the substrate specificity of JMJ16 by preventing the FYR domain from binding to the catalytic domain. We propose that MMD1 and JMJ16 together in male meiocytes promote gene expression in an H3K9me3-dependent manner and thereby contribute to meiotic chromosome condensation.
DOI:10.1371/journal.pgen.1004091URLPMID:24465219 [本文引用: 2]
Seed germination and subsequent seedling growth define crucial steps for entry into the plant life cycle. For those events to take place properly, seed developmental genes need to be silenced whereas vegetative growth genes are activated. Chromatin structure is generally known to play crucial roles in gene transcription control. However, the transition between active and repressive chromatin states during seed germination is still poorly characterized and the underlying molecular mechanisms remain largely unknown. Here we identified the Arabidopsis PHD-domain H3K4me3-binding ALFIN1-like proteins (ALs) as novel interactors of the Polycomb Repressive Complex 1 (PRC1) core components AtBMI1b and AtRING1a. The interactions were confirmed by diverse in vitro and in vivo assays and were shown to require the AL6 N-terminus containing PAL domain conserved in the AL family proteins and the AtRING1a C-terminus containing RAWUL domain conserved in animal and plant PRC1 ring-finger proteins (including AtRNIG1a/b and AtBMI1a/b). By T-DNA insertion mutant analysis, we found that simultaneous loss of AL6 and AL7 as well as loss of AtBMI1a and AtBMI1b retards seed germination and causes transcriptional derepression and a delayed chromatin state switch from H3K4me3 to H3K27me3 enrichment of several seed developmental genes (e.g. ABI3, DOG1, CRU3, CHO1). We found that AL6 and the PRC1 H3K27me3-reader component LHP1 directly bind at ABI3 and DOG1 loci. In light of these data, we propose that AL PHD-PRC1 complexes, built around H3K4me3, lead to a switch from the H3K4me3-associated active to the H3K27me3-associated repressive transcription state of seed developmental genes during seed germination. Our finding of physical interactions between PHD-domain proteins and PRC1 is striking and has important implications for understanding the connection between the two functionally opposite chromatin marks: H3K4me3 in activation and H3K27me3 in repression of gene transcription.
DOI:10.1111/nph.12194URL [本文引用: 2]
Phosphate (Pi) starvation in plants induces dense and elongated root hairs, which increase the absorptive surface area of the roots and play a critical role in Pi uptake. The molecular mechanism underlying these changes remains unclear. Forward and reverse genetic approaches were employed to identify novel genes involved in root hair formation on Pi starvation. The mutant per2, with defects in root hair elongation specifically under low Pi conditions, was identified in a large-scale genetic screen of T-DNA insertion lines. The phenotype was caused by a mutation in the homeodomain protein ALFIN-LIKE 6 (AL6). From a screen of mutants defective in genes that showed lower transcript abundance in per2 relative to wild-type roots on low Pi medium, we identified four putative downstream targets of AL6, namely ETC1, NPC4, SQD2 and PS2, all of which were critical in root hair elongation of Pi-deficient plants. The results further indicate that AL6 is involved in the control of growth and several key responses to Pi starvation. Our findings demonstrate that AL6 controls the transcription of a suite of genes critical for root hair elongation under low Pi conditions, suggesting a novel physiological function for an Alfin gene in Arabidopsis.
DOI:10.1111/j.1365-313X.2009.03795.xURLPMID:19154204 [本文引用: 1]
In yeast and animals, tri- and dimethylation of histone H3 at lysine 4 (H3K4me3/2) are markers of transcriptionally active genes that have recently been shown to be primary ligands for the plant homeodomain (PHD) finger. However, PHD fingers able to bind to H3K4me3/2 have not been identified in plants. Here, we identify 83 canonical PHD fingers in the Arabidopsis proteome database that are supported by both SMART and Pfam prediction. Among these, we focus on PHD fingers in ING (inhibitor of growth) homologues (AtING) and Alfin1-like (AL) proteins, which are highly similar to those in human ING2 and bromodomain PHD finger transcription factor (BPTF), based on predicted tertiary structures. ING proteins are found in yeast, animals and plants, whereas AL proteins exist only in plants. In vitro binding experiments indicated that PHD fingers in AtING and AL proteins in Arabidopsis can bind to H3K4me3, and, to a lesser extent, to H3K4me2. In addition, mutational analysis confirmed that a predicted aromatic cage and a specific conserved acidic residue are both crucial for binding to H3K4me3/2. Finally, we demonstrate that AtING and AL proteins are nuclear proteins that are expressed in various tissues of the Arabidopsis plant. Thus, we propose that ING and AL proteins are nuclear proteins that are involved in chromatin regulation by binding to H3K4me3/2, the active histone markers, in plants.
DOI:10.1111/tpj.12773URLPMID:25619813 [本文引用: 2]
Plant homeodomain (PHD) finger proteins affect processes of growth and development by changing transcription and reading epigenetic histone modifications, but their functions in abiotic stress responses remain largely unclear. Here we characterized seven Arabidopsis thaliana Alfin1-like PHD finger proteins (ALs) in terms of the responses to abiotic stresses. ALs localized to the nucleus and repressed transcription. Except AL6, all the ALs bound to G-rich elements. Mutations of the amino acids at positions 34 and 35 in AL6 caused loss of ability to bind to G-rich elements. Expression of the AL genes responded differentially to osmotic stress, salt, cold and abscisic acid treatments. AL5-over-expressing plants showed higher tolerance to salt, drought and freezing stress than Col-0. Consistently, al5 mutants showed reduced stress tolerance. We used ChIP-Seq assays to identify eight direct targets of AL5, and found that AL5 binds to the promoter regions of these genes. Knockout mutants of five of these target genes exhibited varying tolerances to stresses. These results indicate that AL5 inhibits multiple signaling pathways to confer stress tolerance. Our study sheds light on mechanisms of AL5-mediated signaling in abiotic stress responses, and provides tools for improvement of stress tolerance in crop plants.
DOI:10.1105/tpc.112.104760URL [本文引用: 1]
Vernalization is an environmentally induced epigenetic switch in which winter cold triggers epigenetic silencing of floral repressors and thus provides competence to flower in spring. Vernalization triggers the recruitment of chromatin-modifying complexes to a clade of flowering repressors that are epigenetically silenced via chromatin modifications. In Arabidopsis thaliana, VERNALIZATION INSENSITIVE3 (VIN3) and its related plant homeodomain finger proteins act together with Polycomb Repressive Complex 2 to increase repressive histone marks at floral repressor loci, including FLOWERING LOCUS C (FLC) and its related genes, by vernalization. Here, we show that VIN3 family of proteins nonredundantly functions to repress different subsets of the FLC gene family during the course of vernalization. Each VIN3 family protein binds to modified histone peptides in vitro and directly associates with specific sets of FLC gene family chromatins in vivo to mediate epigenetic silencing. In addition, members of the FLC gene family are also differentially regulated during the course of vernalization to mediate proper vernalization response. Our results show that these two gene families cooperated during the course of evolution to ensure proper vernalization response through epigenetic changes.
DOI:10.1104/pp.16.01320URLPMID:27999085 [本文引用: 1]
Vernalization is a response to winter cold to initiate flowering in spring. VERNALIZATION INSENSITIVE3 (VIN3) is induced by winter cold and is essential to vernalization response in Arabidopsis (Arabidopsis thaliana). VIN3 encodes a PHD-finger domain that binds to modified histones in vitro. An alteration in the binding specificity of the PHD-finger domain of VIN3 results in a hypervernalization response. The hypervernalization response is achieved by increased enrichments of VIN3 and trimethylation of Histone H3 Lys 27 at the FLC locus without invoking the increased enrichment of Polycomb Repressive Complex 2. Our result shows that the binding specificity of the PHD-finger domain of VIN3 plays a role in mediating a proper vernalization response in Arabidopsis.
DOI:10.1038/nature02195URLPMID:14712276
In biennials and winter annuals, flowering is typically blocked in the first growing season. Exposure to the prolonged cold of winter, through a process called vernalization, is required to alleviate this block and permit flowering in the second growing season. In winter-annual types of Arabidopsis thaliana, a flowering repressor, FLOWERING LOCUS C (FLC), is expressed at levels that inhibit flowering in the first growing season. Vernalization promotes flowering by causing a repression of FLC that is mitotically stable after return to warm growing conditions. Here we identify a gene with a function in the measurement of the duration of cold exposure and in the establishment of the vernalized state. We show that this silencing involves changes in the modification of histones in FLC chromatin.
DOI:10.1111/j.1365-313X.2009.03891.xURLPMID:19392705
VERNALIZATION INSENSITIVE 3 (VIN3), which is required for the vernalization-mediated epigenetic repression of FLOWERING LOCUS C (FLC) in Arabidopsis thaliana, is quantitatively induced in response to low temperatures. We found that hypoxic conditions also induce VIN3 in a quantitative manner but high salt, high temperatures and osmotic stress do not. Inhibition of mitochondrial respiration did not induce VIN3 expression, consistent with the lack of VIN3 induction in response to other stresses that affect the rate of mitochondrial respiration. De novo protein synthesis is required for VIN3 induction during hypoxic conditions; this situation is not the case for VIN3 induction by low temperatures, indicating that different mechanisms act to induce VIN3 expression in response to cold and hypoxic conditions. Without VIN3 activity, fewer seedlings survived following a 72-h period of hypoxic treatment, indicating that VIN3 is required for the survival of Arabidopsis thaliana in response to hypoxic stress. Complementation of the vin3 mutant with a VIN3 transgene restored the wild-type response to low oxygen and confirmed the role of VIN3 in protecting both shoots and roots during low oxygen conditions. Loss of VIN3 protein did not affect the transcriptional regulation of genes known to be important in the response to low oxygen stress, which suggests that there is a novel mechanism to combat hypoxia that involves VIN3. This mechanism is likely to involve chromatin remodelling and may be similar to the role of VIN3 in the epigenetic repression of FLC during the vernalization response.
DOI:10.1104/pp.110.161083URLPMID:20671111 [本文引用: 1]
VERNALIZATION INSENSITIVE3 (VIN3) induction by vernalization is one of the earliest events in the vernalization response of Arabidopsis (Arabidopsis thaliana). However, the mechanism responsible for vernalization-mediated VIN3 induction is poorly understood. Here, we show that the constitutive repression of VIN3 in the absence of the cold is due to multiple repressive components, including a transposable element-derived sequence, LIKE-HETEROCHROMATIN PROTEIN1 and POLYCOMB REPRESSION COMPLEX2. Furthermore, the full extent of VIN3 induction by vernalization requires activating complex components, including EARLY FLOWERING7 and EARLY FLOWERING IN SHORT DAYS. In addition, we observed dynamic changes in the histone modifications present at VIN3 chromatin during the course of vernalization. Our results show that the induction of VIN3 includes dynamic changes at the level of chromatin triggered by long-term cold exposure.
DOI:10.1111/j.1365-313X.2009.03860.xURLPMID:19419532 [本文引用: 2]
Mutations within the Arabidopsis METHYL-CpG BINDING DOMAIN 9 gene (AtMBD9) cause pleotropic phenotypes including early flowering and multiple lateral branches. Early flowering was previously attributed to the repression of flowering locus C (FLC) due to a reduction in histone acetylation. However, the reasons for other phenotypic variations remained obscure. Recent studies suggest an important functional correlation between DNA methylation and histone modifications. By investigating this relationship, we found that the global genomic DNA of atmbd9 was over-methylated, including the FLC gene region. Recombinant AtMBD9 does not have detectable DNA demethylation activity in vitro, but instead has histone acetylation activity. Ectopic over-expression of AtMBD9 and transient DNA demethylation promotes flowering and causes partial recovery of the normal branching phenotype. Co-immunoprecipitation assays suggest that AtMBD9 interacts in vivo with some regions of the FLC gene and binds to histone 4 (H4). Gene expression profile analysis revealed earlier up-regulation of some flower-specific transcriptional factors and alteration of potential hormonal and signal transducer axillary branching regulatory genes. In accordance with this result, AtMBD9 itself was found to be localized in the nucleus and expressed in the flower and axillary buds. Together, these results suggest that AtMBD9 controls flowering time and axillary branching by modulating gene expression through DNA methylation and histone acetylation, and reveal another component of the epigenetic mechanism controlling gene expression.
DOI:10.1111/j.1365-313X.2006.02691.xURLPMID:16623890 [本文引用: 3]
The functional characterization of mammalian proteins containing a methyl-CpG-binding domain (MBD) has revealed that MBD proteins can decipher the epigenetic information encoded by DNA methylation, and integrate DNA methylation, modification of chromatin structure and repression of gene expression. The Arabidopsis genome has 13 putative genes encoding MBD proteins, and no specific biological function has been defined for any AtMBD genes. In this study, we identified three T-DNA insertion mutant alleles at the AtMBD9 locus, and found that all of them exhibited obvious developmental abnormalities. First, the atmbd9 mutants flowered significantly earlier than wild-type plants. The expression of FLOWERING LOCUS C (FLC), a major repressor of Arabidopsis flowering, was markedly attenuated by the AtMBD9 mutations. This FLC transcription reduction was associated with a significant decrease in the acetylation level in histone H3 and H4 of FLC chromatin in the atmbd9 mutants. Secondly, the atmbd9 mutants produced more shoot branches by increasing the outgrowth of axillary buds when compared with wild-type plants. The two known major factors controlling the outgrowth of axillary buds in Arabidopsis, auxin and the more axillary growth (MAX) pathway, were found not to be involved in producing this enhanced shoot branching phenotype in atmbd9 mutants, indicating that AtMBD9 may regulate a novel pathway to control shoot branching. This pathway is not related to FLC expression as over-expression of FLC in atmbd9-2 restored its flowering time to one similar to that of the wild type, but did not alter the shoot branching phenotype.
DOI:10.1093/mp/ssu079URL [本文引用: 3]
Methylcytosine-binding proteins containing SRA (SET-and RING-Associated) domain are required for the establishment and/or maintenance of DNA methylation in both plants and animals. We previously proposed that Arabidopsis VIM/ORTH proteins with an SRA domain maintain DNA methylation and epigenetic gene silencing in heterochromatic regions. However, their endogenous targets of epigenetic gene silencing have not been analyzed globally and the mechanisms by which VIM proteins coordinate DNA methylation and epigenetic silencing are largely unknown. In this study, a genome-wide transcript profiling analysis revealed 544 derepressed genes in a vim1/2/3 triple mutant, including 133 known genes. VIM1 bound to promoter and transcribed regions of the up-regulated genes in vim1/2/3 and VIM deficiency caused severe DNA hypomethylation in all sequence contexts at direct VIM1 targets. We found a drastic loss of H3K9me2 at heterochromatic chromocenters in vim1/2/3 nuclei. Furthermore, aberrant changes in transcriptionally active and repressive histone modifications were observed at VIM1 targets in vim1/2/3. VIM1-binding capacity to target genes was significantly reduced in the met1 background, indicating that VIM1 primarily recognizes CG methylation deposited by MET1. Overall, our data indicate that VIM proteins regulate genome-wide epigenetic gene silencing through coordinated modulation of DNA methylation and histone modification status in collaboration with MET1.
DOI:10.1111/j.1365-313X.2008.03631.xURLPMID:18643997 [本文引用: 2]
Appropriate methylation of genomes is essential for gene regulation. Here, we describe the six-member ORTHRUS (ORTH) gene family of Arabidopsis thaliana that plays a role in DNA methylation in vivo. ORTH1- ORTH5 are predicted to encode proteins that contain one plant homeodomain (PHD), two really interesting new gene (RING) domains, and one set ring associated (SRA) domain, whereas ORTHlike-1 encodes a protein with only one RING and SRA domain. cDNAs for ORTH1, ORTH2, ORTH5 and ORTHlike-1 were isolated, and when expressed as glutathione-S-transferase (GST) fusion proteins, were capable of promoting ubiquitylation in vitro with the E2 AtUBC11. ORTH1 promotes ubiquitylation when paired with additional AtUBC8 family members. ORTH1 proteins with substitutions in metal-ligand binding residues in each ORTH1 RING domain individually, and ORTH1 truncation derivatives lacking one or both RING domains, were tested for their ability to catalyze ubiquitylation in vitro. In these assays, either ORTH1 RING domain is capable of promoting ubiquitylation. The PHD alone is not active as an E3 ligase, nor is it required for ligase activity. GFP-ORTH1 and GFP-ORTH2 are nuclear-localized in transgenic Arabidopsis plants. Overexpression of ORTH1 or ORTH2 in Arabidopsis leads to an altered flowering time. Inspection of DNA methylation at FWA and Cen180 repeats revealed hypomethylation when ORTH proteins were overexpressed. Once initiated, a late-flowering phenotype persisted in the absence of the ORTH transgene, consistent with epigenetic effects at FWA. We conclude that ORTH proteins are E3 ligases mediating DNA methylation status in vivo.
DOI:10.1016/j.febslet.2012.04.063URL [本文引用: 2]
We determined the three-dimensional structure of the PHD finger of the rice Siz/PIAS-type SUMO ligase, OsSiz1, by NMR spectroscopy and investigated binding ability for a variety of methylated histone H3 tails, showing that OsSiz1-PHD primarily recognizes dimethylated Arg2 of the histone H3 and that methylations at Arg2 and Lys4 reveal synergy effect on binding to OsSiz1-PHD. The K4 cage of OsSiz1-PHD for trimethylated Lys4 of H3K4me3 was similar to that of the BPTF-PHD finger, while the R2 pocket for Arg2 was different. It is intriguing that the PHD module of Siz/PIAS plays an important role, with collaboration with the DNA binding domain SAP, in gene regulation through SUMOy-lation of a variety of effectors associated with the methylated arginine-riched chromatin domains. (C) 2012 Federation of European Biochemical Societies. Published by Elsevier B. V.
DOI:10.1105/tpc.106.049981URLPMID:17905899 [本文引用: 2]
Posttranslational modifications of proteins by small ubiquitin-like modifiers (SUMOs) regulate protein degradation and localization, protein-protein interaction, and transcriptional activity. SUMO E3 ligase functions are executed by SIZ1/SIZ2 and Mms21 in yeast, the PIAS family members RanBP2, and Pc2 in human. The Arabidopsis thaliana genome contains only one gene, SIZ1, that is orthologous to the yeast SIZ1/SIZ2. Here, we show that Arabidopsis SIZ1 is expressed in all plant tissues. Compared with the wild type, the null mutant siz1-3 is smaller in stature because of reduced expression of genes involved in brassinosteroid biosynthesis and signaling. Drought stress induces the accumulation of SUMO-protein conjugates, which is in part dependent on SIZ1 but not on abscisic acid (ABA). Mutant plants of siz1-3 have significantly lower tolerance to drought stress. A genome-wide expression analysis identified approximately 1700 Arabidopsis genes that are induced by drought, with SIZ1 mediating the expression of 300 of them by a pathway independent of DREB2A and ABA. SIZ1-dependent, drought-responsive genes include those encoding enzymes of the anthocyanin synthesis pathway and jasmonate response. From these results, we conclude that SIZ1 regulates Arabidopsis growth and that this SUMO E3 ligase plays a role in drought stress response likely through the regulation of gene expression.
DOI:10.1073/pnas.0500778102URLPMID:15894620 [本文引用: 2]
Plants sense phosphate (Pi) deficiency and initiate signaling that controls adaptive responses necessary for Pi acquisition. Herein, evidence establishes that AtSIZ1 is a plant small ubiquitin-like modifier (SUMO) E3 ligase and is a focal controller of Pi starvation-dependent responses. T-DNA insertional mutated alleles of AtSIZ1 (At5g60410) cause Arabidopsis to exhibit exaggerated prototypical Pi starvation responses, including cessation of primary root growth, extensive lateral root and root hair development, increase in root/shoot mass ratio, and greater anthocyanin accumulation, even though intracellular Pi levels in siz1 plants were similar to wild type. AtSIZ1 has SUMO E3 ligase activity in vitro, and immunoblot analysis revealed that the protein sumoylation profile is impaired in siz1 plants. AtSIZ1-GFP was localized to nuclear foci. Steadystate transcript abundances of Pi starvation-responsive genes AtPT2, AtPS2, and AtPS3 were moderate but clearly greater in siz1 seedlings than in wild type, where Pi is sufficient. Pi starvation induced the expression of these genes to the same extent in siz1 and wild-type seedlings. However, two other Pi starvation-responsive genes, AtIPS1 and AtRNS1, are induced more slowly in siz1 seedlings by Pi limitation. PHR1, a MYB transcriptional activator of AtIPS1 and AtRNS1, is an AtSIZ1 sumoylation target. These results indicate that AtSIZ1 is a SUMO E3 ligase and that sumoylation is a control mechanism that acts both negatively and positively on different Pi deficiency responses.
DOI:10.1534/genetics.105.051664URLPMID:16547115 [本文引用: 2]
The genome of plants, like that of other eukaryotes, is organized into chromatin, a compact structure that reduces the accessibility of DNA to machineries such as transcription, replication, and DNA recombination and repair. Plant genes, which contain the characteristic ATPase/helicase motifs of the chromatin remodeling Swi2/Snf2 family of proteins, have been thoroughly studied, but their role in homologous recombination or DNA repair has received limited attention. We have searched for homologs of the yeast RAD54 gene, whose role in recombination and repair and in chromatin remodeling is well established. Forty Arabidopsis SWI2/SNF2 genes were identified and the function of a selected group of 14 was analyzed. Mutant analysis and/or RNAi-mediated silencing showed that 11 of the 14 genes tested played a role in response to DNA damage. Two of the 14 genes were involved in homologous recombination between inverted repeats. The putative ortholog of RAD54 and close homologs of ERCC6/RAD26 were involved in DNA damage response, suggesting functional conservation across kingdoms. In addition, genes known for their role in development, such as PICKLE/GYMNOS and PIE1, or in silencing, such as DDM1, turned out to also be involved in DNA damage response. A comparison of ddm1 and met1 mutants suggests that DNA damage response is affected essentially by chromatin structure and that cytosine methylation is less critical. These results emphasize the broad involvement of the SWI2/SNF2 family, and thus of chromatin remodeling, in genome maintenance and the link between epigenetic and genetic processes.
DOI:10.3389/fpls.2014.00223URLPMID:24904618 [本文引用: 2]
Chromodomain-Helicase-DNA (CHD)-binding proteins have been characterized in various species as important transcription regulators by their chromatin remodeling activity. However, in plant the function of these proteins has hardly been analyzed before except that Arabidopsis PIKLE and rice CHR729 are identified to play critical roles in the regulation of series of genes involved in developmental or stress responding process. In this review we focus on how plant CHD proteins regulate gene expression and the role of these proteins in controlling plant development and stress response.
DOI:10.4161/trns.2.6.17840URLPMID:22223048 [本文引用: 2]
It is well established that ATP-dependent chromatin remodelers modulate DNA access of transcription factors and RNA polymerases by
DOI:10.1104/pp.112.194878URL [本文引用: 2]
In Arabidopsis (Arabidopsis thaliana), the ATP-dependent chromatin remodeler PICKLE (PKL) determines expression of genes associated with developmental identity. PKL promotes the epigenetic mark trimethylation of histone H3 lysine 27 (H3K27me3) that facilitates repression of tissue-specific genes in plants. It has previously been proposed that PKL acts indirectly to promote H3K27me3 by promoting expression of the POLYCOMB REPRESSIVE COMPLEX2 complex that generates H3K27me3. We undertook expression and chromatin immunoprecipitation analyses to further characterize the contribution of PKL to gene expression and developmental identity. Our expression data support a critical and specific role for PKL in expression of H3K27me3-enriched loci but do not support a role for PKL in expression of POLYCOMB REPRESSIVE COMPLEX2. Moreover, our chromatin immunoprecipitation data reveal that PKL protein is present at the promoter region of multiple H3K27me3-enriched loci, indicating that PKL directly acts on these loci. In particular, we find that PKL is present at LEAFY COTYLEDON1 and LEAFY COTYLEDON2 during germination, which is when PKL acts to repress these master regulators of embryonic identity. Surprisingly, we also find that PKL is present at the promoters of actively transcribed genes that are ubiquitously expressed such as ACTIN7 and POLYUBIQUITIN10 that do not exhibit PKL-dependent expression. Taken together, our data contravene the previous model of PKL action and instead support a direct role for PKL in determining levels of H3K27me3 at repressed loci. Our data also raise the possibility that PKL facilitates a common chromatin remodeling process that is not restricted to H3K27me3-enriched regions.
DOI:10.1111/j.1365-313X.2009.03874.xURLPMID:19392692 [本文引用: 1]
In Arabidopsis thaliana, auxin is a key regulator of tissue patterning in the developing embryo. We have identified a group of proteins that act downstream of auxin accumulation in auxin-mediated root and vascular development in the embryo. Combined mutations in OBERON1 (OBE1) and OBERON2 (OBE2) give rise to obe1 obe2 double mutant seedlings that closely phenocopy the monopteros (mp) mutant phenotype, with an absence of roots and defective development of the vasculature. We show that, in contrast to the situation in mp mutants, obe1 obe2 double mutant embryos show auxin maxima at the root pole and in the provascular region, and that the SCF(TIR1) pathway, which translates auxin accumulation into transcriptional activation of auxin-responsive genes, remains intact. Although we focus on the impact of obe mutations on aspects of embryo development, the effect of such mutations on a broad range of auxin-related gene expression and the tissue expression patterns of OBE genes in seedlings suggest that OBE proteins have a wider role to play in growth and development. We suggest that OBE1 and OBE2 most likely control the transcription of genes required for auxin responses through the action of their PHD finger domains.
DOI:10.1242/dev.074492URLPMID:22378640 [本文引用: 1]
Plant growth is directed by the activity of stem cells within meristems. The first meristems are established during early embryogenesis, and this process involves the specification of both stem cells and their organizer cells. One of the earliest events in root meristem initiation is marked by re-specification of the uppermost suspensor cell as hypophysis, the precursor of the organizer. The transcription factor MONOPTEROS (MP) is a key regulator of hypophysis specification, and does so in part by promoting the transport of the plant hormone auxin and by activating the expression of TARGET OF MP (TMO) transcription factors, both of which are required for hypophysis specification. The mechanisms leading to the activation of these genes by MP in a chromatin context are not understood. Here, we show that the PHD-finger proteins OBERON (OBE) and TITANIA (TTA) are essential for MP-dependent embryonic root meristem initiation. TTA1 and TTA2 are functionally redundant and function in the same pathway as OBE1 and OBE2. These PHD-finger proteins interact with each other, and genetic analysis shows that OBE-TTA heterotypic protein complexes promote embryonic root meristem initiation. Furthermore, while MP expression is unaffected by mutations in OBE/TTA genes, expression of MP targets TMO5 and TMO7 is locally lost in obe1 obe2 embryos. PHD-finger proteins have been shown to act in initiation of transcription by interacting with nucleosomes. Indeed, we found that OBE1 binds to chromatin at the TMO7 locus, suggesting a role in its MP-dependent activation. Our data indicate that PHD-finger protein complexes are crucial for the activation of MP-dependent gene expression during embryonic root meristem initiation, and provide a starting point for studying the mechanisms of developmental gene activation within a chromatin context in plants.
DOI:10.1242/dev.014993URLPMID:18403411 [本文引用: 1]
Maintenance of the stem cell population located at the apical meristems is essential for repetitive organ initiation during the development of higher plants. Here, we have characterized the roles of OBERON1 (OBE1) and its paralog OBERON2 (OBE2), which encode plant homeodomain finger proteins, in the maintenance and/or establishment of the meristems in Arabidopsis. Although the obe1 and obe2 single mutants were indistinguishable from wild-type plants, the obe1 obe2 double mutant displayed premature termination of the shoot meristem, suggesting that OBE1 and OBE2 function redundantly. Further analyses revealed that OBE1 and OBE2 allow the plant cells to acquire meristematic activity via the WUSCHEL-CLAVATA pathway, which is required for the maintenance of the stem cell population, and they function parallel to the SHOOT MERISTEMLESS gene, which is required for preventing cell differentiation in the shoot meristem. In addition, obe1 obe2 mutants failed to establish the root apical meristem, lacking both the initial cells and the quiescent center. In situ hybridization revealed that expression of PLETHORA and SCARECROW, which are required for stem cell specification and maintenance in the root meristem, was lost from obe1 obe2 mutant embryos. Taken together, these data suggest that the OBE1 and OBE2 genes are functionally redundant and crucial for the maintenance and/or establishment of both the shoot and root meristems.
DOI:10.1371/journal.pgen.1008849URLPMID:32516352 [本文引用: 1]
Cohesin, a multisubunit protein complex, is required for holding sister chromatids together during mitosis and meiosis. The recruitment of cohesin by the sister chromatid cohesion 2/4 (SCC2/4) complex has been extensively studied in Saccharomyces cerevisiae mitosis, but its role in mitosis and meiosis remains poorly understood in multicellular organisms, because complete loss-of-function of either gene causes embryonic lethality. Here, we identified a weak allele of Atscc2 (Atscc2-5) that has only minor defects in vegetative development but exhibits a significant reduction in fertility. Cytological analyses of Atscc2-5 reveal multiple meiotic phenotypes including defects in chromosomal axis formation, meiosis-specific cohesin loading, homolog pairing and synapsis, and AtSPO11-1-dependent double strand break repair. Surprisingly, even though AtSCC2 interacts with AtSCC4 in vitro and in vivo, meiosis-specific knockdown of AtSCC4 expression does not cause any meiotic defect, suggesting that the SCC2-SCC4 complex has divergent roles in mitosis and meiosis. SCC2 homologs from land plants have a unique plant homeodomain (PHD) motif not found in other species. We show that the AtSCC2 PHD domain can bind to the N terminus of histones and is required for meiosis but not mitosis. Taken together, our results provide evidence that unlike SCC2 in other organisms, SCC2 requires a functional PHD domain during meiosis in land plants.
DOI:10.1073/pnas.0811093106URLPMID:19171893 [本文引用: 1]
Control of gene expression depends on a complex and delicate balance of various posttranslational modifications of histones. However, the relevance of specific combinations of histone modifications is not fully defined. Downstream effector proteins recognize particular histone modifications and transduce this information into gene expression patterns. Methylation of histone H3 at lysine 4 (H3K4me) is a landmark of gene expression control in eukaryotes. Its recognition depends on the presence in the effector protein of a motif termed plant homeodomain (PHD) that specifically binds to H3K4me3. Here, we establish that Arabidopsis ORC1, the large subunit of the origin recognition complex involved in defining origins of DNA replication, functions as a transcriptional activator of a subset of genes, the promoters of which are preferentially bound by ORC1. Arabidopsis ORC1 contains a PHD and binds to H3K4me3. In addition to H4 acetylation, ORC1 binding correlates with increased H4K20me3 in the proximal promoter region of ORC1 targets. This suggests that H4K20me3, unlike in animal cells, is associated with transcriptional activation in Arabidopsis. Thus, our data provide a molecular basis for the opposite role of ORC1 in transcriptional activation in plants and repression in animals. Since only ORC1 proteins of plant species contain a PHD, we propose that plant ORC1 constitutes a novel class of H3K4me3 effector proteins characteristic of the plant kingdom.
DOI:10.1016/j.str.2016.01.004URLPMID:26876097 [本文引用: 1]
DNA replication initiation relies on the formation of the origin recognition complex (ORC). The plant ORC subunit 1 (ORC1) protein possesses a conserved N-terminal BAH domain with an embedded plant-specific PHD finger, whose function may be potentially regulated by an epigenetic mechanism. Here, we report structural and biochemical studies on the Arabidopsis thaliana ORC1b BAH-PHD cassette which specifically recognizes the unmodified H3 tail. The crystal structure of ORC1b BAH-PHD cassette in complex with an H3(1-15) peptide reveals a strict requirement for the unmodified state of R2, T3, and K4 on the H3 tail and a novel multivalent BAH and PHD readout mode for H3 peptide recognition. Such recognition may contribute to epigenetic regulation of the initiation of DNA replication.
DOI:10.1105/tpc.107.054536URLPMID:18032630 [本文引用: 1]
The Arabidopsis thaliana MALE STERILITY1 (MS1) gene encodes a nuclear protein with Leu zipper-like and PHD-finger motifs and is important for postmeiotic pollen development. Here, we examined MS1 function using both cell biological and molecular biological approaches. We introduced a fusion construct of MS1 and a transcriptional repression domain (MS1-SRDX) into wild-type Arabidopsis, and the transgenic plants showed a semisterile phenotype similar to that of ms1. Since the repression domain can convert various kinds of transcriptional activators to dominant repressors, this suggested that MS1 functioned as a transcriptional activator. The Leu zipper-like region and the PHD motif were required for the MS1 function. Phenotypic analysis of the ms1 mutant and the MS1-SRDX transgenic Arabidopsis indicated that MS1 was involved in formation of pollen exine and pollen cytosolic components as well as tapetum development. Next, we searched for MS1 downstream genes by analyzing publicly available microarray data and identified 95 genes affected by MS1. Using a transgenic ms1 plant showing dexamethasone-inducible recovery of fertility, we further examined whether these genes were immediately downstream of MS1. From these results, we discuss a role of MS1 in pollen and tapetum development and the conservation of MS1 function in flowering plants.
DOI:10.1038/ncomms1486URLPMID:21934661 [本文引用: 1]
Chloroplast development, maintenance and function depend on the coordinated expression of chloroplast and nuclear genes. The retrograde chloroplast signals are essential in coordinating nuclear gene expression. Although the sources of signals in chloroplasts have been identified and the associated transcription factors in the nucleus extensively studied, the molecular mechanism that relays chloroplast signals to the nucleus remains a mystery. Here we show that PTM, a chloroplast envelope-bound plant homeodomain (PHD) transcription factor with transmembrane domains, functions in multiple retrograde signal pathways. The proteolytic cleavage of PTM occurs in response to retrograde signals and amino-terminal PTM accumulates in the nucleus, where it activates ABI4 transcription in a PHD-dependent manner associated with histone modifications. These results provide a molecular basis for the critical function of PTM in retrograde chloroplast signaling and shed new light on the mechanism whereby chloroplast signals are transmitted to the nucleus through the cytosol.
DOI:10.1073/pnas.0600944103URLPMID:16585509 [本文引用: 1]
The Arabidopsis homolog of trithorax, ATX1, regulates numerous functions in Arabidopsis beyond the homeotic genes. Here, we identified genome-wide targets of ATX1 and showed that ATX1 is a receptor for a lipid messenger, phosphatidylinositol 5-phosphate, PI5P. PI5P negatively affects ATX1 activity, suggesting a regulatory pathway connecting lipid-signaling with nuclear functions. We propose a model to illustrate how plants may respond to stimuli (external or internal) that elevate cellular PI5P levels by altering expression of ATX1-controlled genes.
DOI:10.1038/nature09290URLPMID:20631708 [本文引用: 1]
Multiple pathways prevent DNA replication from occurring more than once per cell cycle. These pathways block re-replication by strictly controlling the activity of pre-replication complexes, which assemble at specific sites in the genome called origins. Here we show that mutations in the homologous histone 3 lysine 27 (H3K27) monomethyltransferases, ARABIDOPSIS TRITHORAX-RELATED PROTEIN5 (ATXR5) and ATXR6, lead to re-replication of specific genomic locations. Most of these locations correspond to transposons and other repetitive and silent elements of the Arabidopsis genome. These sites also correspond to high levels of H3K27 monomethylation, and mutation of the catalytic SET domain is sufficient to cause the re-replication defect. Mutation of ATXR5 and ATXR6 also causes upregulation of transposon expression and has pleiotropic effects on plant development. These results uncover a novel pathway that prevents over-replication of heterochromatin in Arabidopsis.
DOI:10.1042/BCJ20190091URLPMID:31253666 [本文引用: 1]
Saccharomyces cerevisiae Spp1, a plant homeodomain (PHD) finger containing protein, is a critical subunit of the histone H3K4 methyltransferase complex of proteins associated with Set1 (COMPASS). The chromatin binding affinity of the PHD finger of Spp1 has been proposed to modulate COMPASS activity. During meiosis, Spp1 plays another role in promoting programmed double-strand break (DSB) formation by binding H3K4me3 via its PHD finger and interacting with a DSB protein, Mer2. However, how the Spp1 PHD finger performs site-specific readout of H3K4me3 is still not fully understood. In the present study, we determined the crystal structure of the highly conserved Spp1 N-terminal domain (Sc_Spp1NTD) in complex with the H3K4me3 peptide. The structure shows that Sc_Spp1NTD comprises a PHD finger responsible for methylated H3K4 recognition and a C3H-type zinc finger necessary to ensure the overall structural stability. Our isothermal titration calorimetry results show that binding of H3K4me3 to Sc_Spp1NTD is mildly inhibited by H3R2 methylation, weakened by H3T6 phosphorylation, and abrogated by H3T3 phosphorylation. This histone modification cross-talk, which is conserved in the Saccharomyces pombe and mammalian orthologs of Sc_Spp1 in vitro, can be rationalized structurally and might contribute to the roles of Spp1 in COMPASS activity regulation and meiotic recombination.
DOI:10.4161/epi.6.1.13297URL [本文引用: 1]
Several recent publications demonstrate a co-activator function for a subgroup of plant homeodomain fingers, which, in humans, comprises PHF2, PHF8 and KIAA1718. Besides an N-terminal plant homeodomain (PHD), these proteins also harbor an enzymatically active Jumonji-C domain (JmjC). While they have been shown to bind via their PHDs to H3K4me3-bearing nucleosomes at active gene promoters, their JmjC-domains are able to remove mono-and dimethyl-lysine 9 or 27 on histone H3 or monomethyl-lysine 20 on histone H4, chromatin modifications that correlate with transcriptional repression. Such dual histone crosstalk ensures the proper removal of repressive histone marks following transcriptional activation by RNA polymerases I and II. Mutations in the PHF8 gene lead to X-linked mental retardation (XLMR) and knockdown of KIAA1718 and PHF8 homologs in zebrafish causes brain defects. Thus, the co-activator function of this new class of chromatin-modifying enzymes has important functional roles in neuronal development. To continue with the nomenclature for histone demethylases, we propose the usage of KDM7A, -B and -C for KIAA1718, PHF8 and PHF2 proteins, respectively.
DOI:10.1126/science.1219416URLPMID:22700931 [本文引用: 1]
Active DNA demethylation is an important part of epigenetic regulation in plants and animals. How active DNA demethylation is regulated and its relationship with histone modification patterns are unclear. Here, we report the discovery of IDM1, a regulator of DNA demethylation in Arabidopsis. IDM1 is required for preventing DNA hypermethylation of highly homologous multicopy genes and other repetitive sequences that are normally targeted for active DNA demethylation by Repressor of Silencing 1 and related 5-methylcytosine DNA glycosylases. IDM1 binds methylated DNA at chromatin sites lacking histone H3K4 di- or trimethylation and acetylates H3 to create a chromatin environment permissible for 5-methylcytosine DNA glycosylases to function. Our study reveals how some genes are indicated by multiple epigenetic marks for active DNA demethylation and protection from silencing.
DOI:10.1016/j.molcel.2006.10.026URLPMID:17157260 [本文引用: 1]
Posttranslational histone modifications participate in modulating the structure and function of chromatin. Promoters of transcribed genes are enriched with K4 trimethylation and hyperacetylation on the N-terminal tail of histone H3. Recently, PHD finger proteins, like Yng1 in the NuA3 HAT complex, were shown to interact with H3K4me3, indicating a biochemical link between K4 methylation and hyperacetylation. By using a combination of mass spectrometry, biochemistry, and NMR, we detail the Yng1 PHD-H3K4me3 interaction and the importance of NuA3-dependent acetylation at K14. Furthermore, genome-wide ChIP-Chip analysis demonstrates colocalization of Yng1 and H3K4me3 in vivo. Disrupting the K4me3 binding of Yng1 altered K14ac and transcription at certain genes, thereby demonstrating direct in vivo evidence of sequential trimethyl binding, acetyltransferase activity, and gene regulation by NuA3. Our data support a general mechanism of transcriptional control through which histone acetylation upstream of gene activation is promoted partially through availability of H3K4me3,
DOI:10.1007/s004380000313URLPMID:11129039 [本文引用: 1]
The SHL gene from Arabidopsis thaliana encodes a small nuclear protein that contains a BAH domain and a PHD finger. Both domains are found in numerous (putative) transcriptional regulators and chromatin-remodeling factors. Different sets of transgenic lines were established to analyze the physiological relevance of SHL. SHL expression driven by the CaMV 35S promoter results in reduced growth, early flowering, early senescence, and impaired flower and seed formation. Antisense inhibition of SHL expression gives rise to dwarfism and delayed development. In-frame N-terminal fusion of the SHL protein to beta-glucuronidase (GUS) directs GUS to the nucleus of stably transformed Arabidopsis plants. Thus, SHL encodes a novel putative regulator of gene expression, which directly or indirectly influences a broad range of developmental processes.
DOI:10.1016/j.cell.2010.04.024URLPMID:20434991 [本文引用: 1]
DOI:10.1105/tpc.114.130781URL [本文引用: 1]
The interplay among histone modifications modulates the expression of master regulatory genes in development. Chromatin effector proteins bind histone modifications and translate the epigenetic status into gene expression patterns that control development. Here, we show that two Arabidopsis thaliana paralogs encoding plant-specific proteins with a plant homeodomain (PHD) motif, SHORT LIFE (SHL) and EARLY BOLTING IN SHORT DAYS (EBS), function in the chromatin-mediated repression of floral initiation and play independent roles in the control of genes regulating flowering. Previous results showed that repression of the floral integrator FLOWERING LOCUS T (FT) requires EBS. We establish that SHL is necessary to negatively regulate the expression of SUPPRESSOR OF OVEREXPRESSION OF CO1 (SOC1), another floral integrator. SHL and EBS recognize di-and trimethylated histone H3 at lysine 4 and bind regulatory regions of SOC1 and FT, respectively. These PHD proteins maintain an inactive chromatin conformation in SOC1 and FT by preventing high levels of H3 acetylation, bind HISTONE DEACETYLASE6, and play a central role in regulating flowering time. SHL and EBS are widely conserved in plants but are absent in other eukaryotes, suggesting that the regulatory module mediated by these proteins could represent a distinct mechanism for gene expression control in plants.
DOI:10.1105/tpc.18.00693URLPMID:30712008 [本文引用: 1]
Leaf senescence is governed by a complex regulatory network involving the dynamic reprogramming of gene expression. Age-dependent induction of senescence-associated genes (SAGs) is associated with increased levels of trimethylation of histone H3 at Lys4 (H3K4me3), but the regulatory mechanism remains elusive. Here, we found that JMJ16, an Arabidopsis (Arabidopsis thaliana) JmjC-domain containing protein, is a specific H3K4 demethylase that negatively regulates leaf senescence through its enzymatic activity. Genome-wide analysis revealed a widespread coordinated upregulation of gene expression and hypermethylation of H3K4me3 at JMJ16 binding genes associated with leaf senescence in the loss-of-function jmj16 mutant as compared with the wild type. Genetic analysis indicated that JMJ16 negatively regulates leaf senescence, at least partly through repressing the expression of positive regulators of leaf senescence, WRKY53 and SAG201 JMJ16 associates with WRKY53 and SAG201 and represses their precocious expression in mature leaves by reducing H3K4me3 levels at these loci. The protein abundance of JMJ16 gradually decreases during aging, which is correlated with increased H3K4me3 levels at WRKY53 and SAG201, suggesting that the age-dependent downregulation of JMJ16 is required for the precise transcriptional activation of SAGs during leaf senescence. Thus, JMJ16 is an important regulator of leaf senescence that demethylates H3K4 at SAGs in an age-dependent manner.
DOI:10.1016/j.molcel.2019.03.011URLPMID:30951652 [本文引用: 1]
The polycomb repressive complex 2 (PRC2) is a chromatin-associated methyltransferase catalyzing mono-, di-, and trimethylation of lysine 27 on histone H3 (H3K27). This activity is required for normal organismal development and maintenance of gene expression patterns to uphold cell identity. PRC2 function is often deregulated in disease and is a promising candidate for therapeutic targeting in cancer. In this review, we discuss the molecular mechanisms proposed to take part in modulating PRC2 recruitment and shaping H3K27 methylation patterns across the genome. This includes consideration of factors influencing PRC2 residence time on chromatin and PRC2 catalytic activity with a focus on the mechanisms giving rise to regional preferences and differential deposition of H3K27 methylation. We further discuss existing evidence for functional diversity between distinct subsets of PRC2 complexes with the aim of extracting key concepts and highlighting major open questions toward a more complete understanding of PRC2 function.
DOI:10.1073/pnas.0808687105URLPMID:18854416 [本文引用: 2]
Vernalization, the acceleration of flowering by winter, involves cold-induced epigenetic silencing of Arabidopsis FLC. This process has been shown to require conserved Polycomb Repressive Complex 2 (PRC2) components including the Su(z)12 homologue, VRN2, and two plant homeodomain (PHD) finger proteins, VRN5 and VIN3. However, the sequence of events leading to FLC repression was unclear. Here we show that, contrary to expectations, VRN2 associates throughout the FLC locus independently of cold. The vernalization-induced silencing is triggered by the cold-dependent association of the PHD finger protein VRN5 to a specific domain in FLC intron 1, and this association is dependent on the cold-induced PHD protein VIN3. In plants returned to warm conditions, VRN5 distribution changes, and it associates more broadly over FLC, coincident with significant increases in H3K27me3. Biochemical purification of a VRN5 complex showed that during prolonged cold a PHD-PRC2 complex forms composed of core PRC2 components (VRN2, SWINGER [an E(Z) HMTase homologue], FIE [an ESC homologue], MSI1 [p55 homologue]), and three related PHD finger proteins, VRN5, VIN3, and VEL1. The PHD-PRC2 activity increases H3K27me3 throughout the locus to levels sufficient for stable silencing. Arabidopsis PHD-PRC2 thus seems to act similarly to Pcl-PRC2 of Drosophila and PHF1-PRC2 of mammals. These data show FLC silencing involves changed composition and dynamic redistribution of Polycomb complexes at different stages of the vernalization process, a mechanism with greater parallels to Polycomb silencing of certain mammalian loci than the classic Drosophila Polycomb targets.
DOI:10.1128/MCB.02017-07URLPMID:18285464 [本文引用: 1]
The mammalian Polycomblike protein PHF1 was previously shown to interact with the Polycomb group (PcG) protein Ezh2, a histone methyltransferase whose activity is pivotal in sustaining gene repression during development and in adulthood. As Ezh2 is active only when part of the Polycomb Repressive Complexes (PRC2-PRC4), we examined the functional role of its interaction with PHF1. Chromatin immunoprecipitation experiments revealed that PHF1 resides along with Ezh2 at Ezh2-regulated genes such as the HoxA loci and the non-Hox MYT1 and WNT1 genes. Knockdown of PHF1 or of Ezh2 led to up-regulated HoxA gene expression. Interestingly, depletion of PHF1 did correlate with reduced occupancy of Bmi-1, a PRC1 component. As expected, knockdown of Ezh2 led to reduced levels of its catalytic products H3K27me2/H3K27me3. However, reduced levels of PHF1 also led to decreased global levels of H3K27me3. Notably, the levels of H3K27me3 decreased while those of H3K27me2 increased at the up-regulated HoxA loci tested. Consistent with this, the addition of PHF1 specifically stimulated the ability of Ezh2 to catalyze H3K27me3 but not H3K27me1/H3K27me2 in vitro. We conclude that PHF1 modulates the activity of Ezh2 in favor of the repressive H3K27me3 mark. Thus, we propose that PHF1 is a determinant in PcG-mediated gene repression.
DOI:10.1038/sj.emboj.7601837URLPMID:17762866 [本文引用: 1]
PRC2 is thought to be the histone methyltransferase (HMTase) responsible for H3-K27 trimethylation at Polycomb target genes. Here we report the biochemical purification and characterization of a distinct form of Drosophila PRC2 that contains the Polycomb group protein polycomblike (Pcl). Like PRC2, Pcl-PRC2 is an H3-K27-specific HMTase that mono-, di- and trimethylates H3-K27 in nucleosomes in vitro. Analysis of Drosophila mutants that lack Pcl unexpectedly reveals that Pcl-PRC2 is required to generate high levels of H3-K27 trimethylation at Polycomb target genes but is dispensable for the genome-wide H3-K27 mono- and dimethylation that is generated by PRC2. In Pcl mutants, Polycomb target genes become derepressed even though H3-K27 trimethylation at these genes is only reduced and not abolished, and even though targeting of the Polycomb protein complexes PhoRC and PRC1 to Polycomb response elements is not affected. Pcl-PRC2 is thus the HMTase that generates the high levels of H3-K27 trimethylation in Polycomb target genes that are needed to maintain a Polycomb-repressed chromatin state.
DOI:10.1074/jbc.M104294200URLPMID:11571280 [本文引用: 1]
The products of Polycomb group (PcG) genes are required for the epigenetic repression of a number of important developmental regulatory genes, including homeotic genes. Enhancer of zeste (E(Z)) is a Drosophila PcG protein that previously has been shown to bind directly to another PcG protein, Extra Sex Combs (ESC), and is present along with ESC in a 600-kDa complex in Drosophila embryos. Using yeast two-hybrid and in vitro binding assays, we show that E(Z) binds directly to another PcG protein, Polycomblike (PCL). PCL.E(Z) interaction is shown to be mediated by the plant homeodomain (PHD) fingers domain of PCL, providing evidence that this motif can act as an independent protein interaction domain. An association was also observed between PHF1 and EZH2, human homologs of PCL and E(Z), respectively, demonstrating the evolutionary conservation of this interaction. E(Z) was found to not interact with the PHD domains of three Drosophila trithorax group (trxG) proteins, which function to maintain the transcriptional activity of homeotic genes, providing evidence for the specificity of the interaction of E(Z) with the PCL PHD domain. Coimmunoprecipitation and gel filtration experiments demonstrate in vivo association of PCL with E(Z) and ESC in Drosophila embryos. We discuss the implications of PCL association with ESC.E(Z) complexes and the possibility that PCL may either be a subunit of a subset of ESC.E(Z) complexes or a subunit of a separate complex that interacts with ESC.E(Z) complexes.
DOI:10.1038/nsmb.2435URL [本文引用: 1]
The PHD finger protein 1 (PHF1) is essential in epigenetic regulation and genome maintenance. Here we show that the Tudor domain of human PHF1 binds to histone H3 trimethylated at Lys36 (H3K36me3). We report a 1.9-angstrom resolution crystal structure of the Tudor domain in complex with H3K36me3 and describe the molecular mechanism of H3K36me3 recognition using NMR. Binding of PHF1 to H3K36me3 inhibits the ability of the Polycomb PRC2 complex to methylate Lys27 of histone H3 in vitro and in vivo. Laser microirradiation data show that PHF1 is transiently recruited to DNA double-strand breaks, and PHF1 mutants impaired in the H3K36me3 interaction exhibit reduced retention at double-strand break sites. Together, our findings suggest that PHF1 can mediate deposition of the repressive H3K27me3 mark and acts as a cofactor in early DNA-damage response.
DOI:10.1074/jbc.M602851200URLPMID:16648632 [本文引用: 1]
The SWI/SNF and SAGA chromatin-modifying complexes contain bromodomains that help anchor these complexes to acetylated promoter nucleosomes. To study the importance of bromodomains in these complexes, we have compared the chromatin-remodeling and octamer-transfer activity of the SWI/SNF complex to a mutant complex that lacks the Swi2/Snf2 bromodomain. Here we show that the SWI/SNF complex can remodel or transfer SAGA-acetylated nucleosomes more efficiently than the Swi2/Snf2 bromodomain-deleted complex. These results demonstrate that the Swi2/Snf2 bromodomain is important for the remodeling as well as for the octamer-transfer activity of the complex on H3-acetylated nucleosomes. Moreover, we show that, although the wild-type SWI/SNF complex displaces SAGA that is bound to acetylated nucleosomes, the bromodomain mutant SWI/SNF complex is less efficient in SAGA displacement. Thus, the Swi2/Snf2 bromodomain is required for the full functional activity of SWI/SNF on acetylated nucleosomes and is important for the displacement of SAGA from acetylated promoter nucleosomes.
DOI:10.1016/j.cub.2007.01.009URL [本文引用: 1]
Summary
Epigenetic gene silencing suppresses transposon activity and is critical for normal development [1] and [2]. Two common epigenetic gene-silencing marks are DNA methylation and histone H3 lysine 9 dimethylation (H3K9me2) [1]. In Arabidopsis thaliana, H3K9me2, catalyzed by the methyltransferase KRYPTONITE (KYP/SUVH4), is required for maintenance of DNA methylation outside of the standard CG sequence context [3] and [4]. Additionally, loss of DNA methylation in the met1 mutant correlates with a loss of H3K9me2 [5] and [6]. Here we show that KYP-dependent H3K9me2 is found at non-CG methylation sites in addition to those rich in CG methylation. Furthermore, we show that the SRA domain of KYP binds directly to methylated DNA, and SRA domains with missense mutations found in loss-of-function kyp mutants have reduced binding to methylated DNA in vitro. These data suggest that DNA methylation is required for the recruitment or activity of KYP and suggest a self-reinforcing loop between histone and DNA methylation. Lastly, we found that SRA domains from two Arabidopsis SRA-RING proteins also bind methylated DNA and that the SRAdomains from KYP and SRA-RING proteins prefer methylcytosines in different sequence contexts. Hence, unlike the methyl-binding domain (MBD), which binds only methylated-CpG sequences, the SRA domain is a versatile new methyl-DNA-binding motif.DOI:10.18632/oncotarget.25425URLPMID:29899856 [本文引用: 1]
Ubiquitin-like containing PHD Ring Finger 1 (UHRF1) is a multi-domain protein with a methyl-DNA binding SRA (SET and RING-associated) domain, required for maintenance DNA methylation mediated by DNMT1. Primarily expressed in proliferating cells, UHRF1 is a cell-cycle regulated protein that is required for S phase entry. Furthermore, UHRF1 participates in transcriptional gene regulation by connecting DNA methylation to histone modifications. Upregulation of UHRF1 may serve as a biomarker for a variety of cancers; including breast, gastric, prostate, lung and colorectal carcinoma. To this end, overexpression of UHRF1 promotes cancer metastasis by triggering aberrant patterns of DNA methylation, and subsequently, silencing tumor suppressor genes. Various small molecule effectors of UHRF1 have been reported in the literature, although the mechanism of action may not be fully characterized. Small molecules that potentially bind to the SRA domain may affect the ability of UHRF1 to bind hemimethylated DNA; thereby reducing aberrant DNA methylation. Therefore, in a subset of cancers, small molecule UHRF1 inhibitors may restore normal gene expression and serve as useful anti-cancer therapeutics.
DOI:10.1111/j.1365-313X.2007.03286.xURLPMID:17892444 [本文引用: 1]
The heterochromatin of many eukaryotes is marked by both histone H3 lysine 9 (H3K9) methylation and DNA cytosine methylation. Several studies have revealed links between these two epigenetic markers. The molecular mechanisms involved in establishment of these links, however, remain largely unknown. In plants, H3K9 methylation is primarily carried out by a highly conserved family of proteins that contain SET and SRA (SET- and RING-associated) domains. Here, we show that the SRA-SET domain H3K9 methyltransferase NtSET1, as well as LIKE HETEROCHROMATIN PROTEIN1, binds heterochromatin DNA repeats. In the yeast two-hybrid assay, NtSET1 binds the DNA methylcytosine-binding protein VARIANT IN METHYLATION1 (VIM1), which contains conserved PHD, SRA and RING domains. This binding requires either the N-terminus of NtSET1 containing the SRA domain or the C-terminus of NtSET1 containing the SET domain and the PHD domain of VIM1. Consistent with a role in the establishment/maintenance of chromatin structure during cell division, VIM1 transcripts are abundant in actively dividing cells and the VIM1 protein is localized in the nucleus. While null vim1 mutant plants show a normal growth phenotype, transgenic Arabidopsis plants over-expressing VIM1 show inhibition in root growth and delay in flowering. We propose that SRA-SET domain H3K9 methyltransferases associate with the PHD-SRA-RING domain protein VIM1, mutually reinforcing H3K9 and DNA methylation in heterochromatinization.
DOI:10.4161/epi.4.7.9807URLPMID:19829068 [本文引用: 1]
Post-translational modification of many transcription factors and cofactors by the small ubiquitin-related modifier SUMO has been correlated with transcriptional repression. Recent investigations of the molecular mechanisms underlying SUMO-dependent repression have identified diverse chromatin modifying enzymes and chromatin associated proteins as effectors of SUMO-dependent changes in chromatin structure and gene expression. A surprising diversity of proteins has been identified to be recruited to promoters in a SUMO-dependent manner, including the histone deacetylase HDAC2, the histone demethylase LSD1, the histone methyltransferase SETDB1, the nucleosome remodeling ATPase Mi-2, and chromatin-associated proteins HP1 and L3MBTL1 and L3MBTL2. These findings suggest that SUMOylation plays a central role in coordinating histone modifications and chromatin structure important for regulation of gene expression.
DOI:10.1038/nsmb.1416URLPMID:18488044 [本文引用: 1]
The tandem PHD finger-bromodomain, found in many chromatin-associated proteins, has an important role in gene silencing by the human co-repressor KRAB-associated protein 1 (KAP1). Here we report the three-dimensional solution structure of the tandem PHD finger-bromodomain of KAP1. The structure reveals a distinct scaffold unifying the two protein modules, in which the first helix, alpha(Z), of an atypical bromodomain forms the central hydrophobic core that anchors the other three helices of the bromodomain on one side and the zinc binding PHD finger on the other. A comprehensive mutation-based structure-function analysis correlating transcriptional repression, ubiquitin-conjugating enzyme 9 (UBC9) binding and SUMOylation shows that the PHD finger and the bromodomain of KAP1 cooperate as one functional unit to facilitate lysine SUMOylation, which is required for KAP1 co-repressor activity in gene silencing. These results demonstrate a previously unknown unified function for the tandem PHD finger-bromodomain as an intramolecular small ubiquitin-like modifier (SUMO) E3 ligase for transcriptional silencing.
DOI:10.1111/tpj.14365URLPMID:31021015 [本文引用: 3]
Male reproductive development involves a complex series of biological events and precise transcriptional regulation is essential for this biological process in flowering plants. Several transcriptional factors have been reported to regulate tapetum and pollen development, however the transcriptional mechanism underlying Ubisch bodies and pollen wall formation remains less understood. Here, we characterized and isolated a male sterility mutant of TDR INTERACTING PROTEIN 3 (TIP3) in rice. The tip3 mutant displayed smaller and pale yellow anthers without mature pollen grains, abnormal Ubisch body morphology, no pollen wall formation, as well as delayed tapetum degeneration. Map-based cloning demonstrated that TIP3 encodes a conserved PHD-finger protein and further study confirmed that TIP3 functioned as a transcription factor with transcriptional activation activity. TIP3 is preferentially expressed in the tapetum and microspores during anther development. Moreover, TIP3 can physically interact with TDR, which is a key component of the transcriptional cascade in regulating tapetum development and pollen wall formation. Furthermore, disruption of TIP3 changed the expression of several genes involved in tapetum development and degradation, biosynthesis and transport of lipid monomers of sporopollenin in tip3 mutant. Taken together, our results revealed an unprecedented role for TIP3 in regulating Ubisch bodies and pollen exine formation, and presents a potential tool to manipulate male fertility for hybrid rice breeding.
DOI:10.1038/nrm1761URLPMID:16261189 [本文引用: 1]
Covalent modifications of histone tails have fundamental roles in chromatin structure and function. One such modification, lysine methylation, has important functions in many biological processes that include heterochromatin formation, X-chromosome inactivation and transcriptional regulation. Here, we summarize recent advances in our understanding of how lysine methylation functions in these diverse biological processes, and raise questions that need to be addressed in the future.
DOI:10.1021/acschembio.7b01093URLPMID:29529862 [本文引用: 2]
Plant homeodomain (PHD) zinc fingers are histone reader domains that are often associated with human diseases. Despite this, they constitute a poorly targeted class of readers, suggesting low ligandability. Here, we describe a successful fragment-based campaign targeting PHD fingers from the proteins BAZ2A and BAZ2B as model systems. We validated a pool of in silico fragments both biophysically and structurally and solved the first crystal structures of PHD zinc fingers in complex with fragments bound to an anchoring pocket at the histone binding site. The best-validated hits were found to displace a histone H3 tail peptide in competition assays. This work identifies new chemical scaffolds that provide suitable starting points for future ligand optimization using structure-guided approaches. The demonstrated ligandability of the PHD reader domains could pave the way for the development of chemical probes to drug this family of epigenetic readers.
DOI:10.1021/bi3009278URL [本文引用: 1]
A number of histone-binding domains are implicated in cancer through improper binding of chromatin. In a clinically reported case of acute myeloid leukemia (AML), a genetic fusion protein between nucleoporin 98 and the third plant homeodomain (PHD) finger of JARD1A drives an oncogenic transcriptional program that is dependent on histone binding by the PHD finger. By exploiting the requirement for chromatin binding in oncogenesis, therapeutics targeting histone readers may represent a new paradigm in drug development. In this study, we developed a novel small molecule screening strategy that utilizes HaloTag technology to identify several small molecules that disrupt binding of the JARIDIA PHD finger to histone peptides. Small molecule inhibitors were validated biochemically through affinity pull downs, fluorescence polarization, and histone reader specificity studies. One compound was modified through medicinal chemistry to improve its potency while retaining histone reader selectivity. Molecular modeling and site-directed mutagenesis of JARID1A PHD3 provided insights into the biochemical basis of competitive inhibition.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}