Abstract Breast cancer originates from ducts and epithelial cells, and gradually develops from hyperplasia to atypical hyperplasia, in situ (adeno) carcinoma, to early and advanced invasive carcinoma. Traditional high-throughput sequencing mainly aims to identify candidate ‘driver genes’ attributable to development and progression of breast cancer, which has deficiencies in characterizing genomic structure alteration and subclone evolution, and thus ignores intratumoral, intertumoral or interpatient heterogeneity. The single-cell sequencing technology analyzes transcriptome (e.g., gene copy number and gene expression), explores cellular composition, differentiation and fate, fine-maps the tumor microenvironment, and provides supporting evidence for accurate stratification as well as personalized, precise therapy. At the same time, a complex relationship between breast cancer cells and T cells, macrophages and other immune cells can be revealed, thus facilitating discovery of new therapeutic targets and immune checkpoints. Here, we review state-of-the-art single-cell sequencing technologies and its application in breast cancer, in order to decipher multi-faceted alterations in the crosstalk/interactions between tumors and its microenvironments at the single-cell level, and provide a basis for better understanding of complicated pathogenesis and new avenues for immunotherapy. Keywords:single-cell sequencing;breast cancer;tumor microenvironment;immunotherapy
PDF (891KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文 本文引用格式 张强, 顾明亮. 单细胞测序技术及其在乳腺癌研究中的应用. 遗传[J], 2020, 42(3): 250-268 doi:10.16288/j.yczz.19-268 Qiang Zhang. Single-cell sequencing and its application in breast cancer. Hereditas(Beijing)[J], 2020, 42(3): 250-268 doi:10.16288/j.yczz.19-268
乳腺癌是乳腺组织中各级导管和乳腺上皮细胞恶性增生的一种癌症,其临床表现为乳房肿块、乳腺形状改变、皮肤上的酒窝、乳头溢出液或出现红色或有鳞的皮肤。乳腺癌分为原位癌和浸润性癌,一般原位癌并不致命,但由于癌细胞间的粘连性降低,癌细胞一旦脱落,会随血液或淋巴液扩散至全身形成转移,引发浸润性癌。据全球癌症监测台(global cancer observatory, GLOBOCAN)的数据显示,乳腺癌高居中国女性癌症发病率首位,严重威胁女性健康[1]。目前的研究表明,环境、遗传与生活方式和乳腺癌发生发展密切相关,营养干预、减轻体重已被证实是有效的一级预防措施[2]。近年来大规模的研究已较为系统地描绘了乳腺癌的致癌因素并勾勒出基因相互作用的网络,揭示了乳腺癌的生物学复杂性,扩大了病患早期诊断、分级治疗和生物标志物靶向治疗的途径,为乳腺癌的精准治疗提供了理论支持和潜在的靶点[3]。针对乳腺癌的靶向治疗,能有效阻断肿瘤细胞之间的信息传递,抑制肿瘤的生长,达到治疗的目的。然而,目前对乳腺癌的靶向治疗导致其产生耐药性已成为临床治疗中的普遍现象,因此亟需发现新的治疗模式以降低乳腺癌的耐药性和复发风险。
传统高通量测序技术主要被用于新癌症基因的发现和证明瘤内的异质性。乳腺癌的全基因组测序表明,体细胞突变类型中的“驱动”突变(driver mutations)会促进肿瘤的发展,而“搭车”突变(passenger mutations)可能是基因组不稳定的产物,但值得注意的是“驱动”突变和“搭车”突变之间在乳腺癌发展中会发生逆转[4,5]。Stephens等[6]发现在测序的100种乳腺癌中,有73种不同的癌症基因突变组合,其中的6种组合可以归纳到JUN激酶途径中。Shah等[7]在104例三阴性乳腺癌(triple-negative breast cancer, TNBC)病例中发现,确诊时TNBC表现出广泛而连续的基因组进化谱,并且大多数都包含数百个体细胞突变。通过对其进行基因组和转录组测序,发现常见的乳腺癌突变类型多数与p53信号通路、磷脂酰肌醇激酶PIK3和表皮生长因子受体酪氨酸激酶ERBB信号通路有关。虽然乳腺癌突变基因和突变组合存在多样性,但与其相关的信号通路却具有高度一致性。正是由于信号通路存在一致性,其乳腺癌表型可能相似。由于细胞间的异质性,即使相同表型的细胞也存在遗传信息的差异,很多分子信息会在整体分析中丢失。这意味着通过传统的高通量测序对乳腺癌进行更精确的分子分型会更加的困难。为了弥补传统测序技术的局限性,单细胞测序技术则应运而生。
单细胞测序技术是以单个细胞为起始材料,允许在单细胞水平上进行拷贝数和基因表达的分析,从而通过构建以非线性分支结构为特征的系统发育树,绘制癌细胞多样性和克隆进化的清晰图像[8,9,10,11]。癌症作为一种基因组疾病,从原发性肿瘤,经过循环肿瘤细胞,到转移性肿瘤,涉及到一系列基因组的变化[12,13,14]。在细胞水平上,癌细胞的异质性早已被染色体核型分析[5]和组织切片荧光原位杂交(fluorescence in situ hybridization, FISH)所证实[15]。乳腺癌常常表现出瘤内基因组的显著异质性,其克隆的多样性影响了临床的诊断和治疗[16]。单细胞测序技术的应用,为了解乳腺癌生态系统的发生和进化,以及免疫治疗机制提供了可能[17]。本文对单细胞测序的技术进行了介绍,并对其在乳腺癌研究中的应用和进展进行了综述,为绘制乳腺癌基因突变图谱和揭示其免疫治疗的机制提供参考。
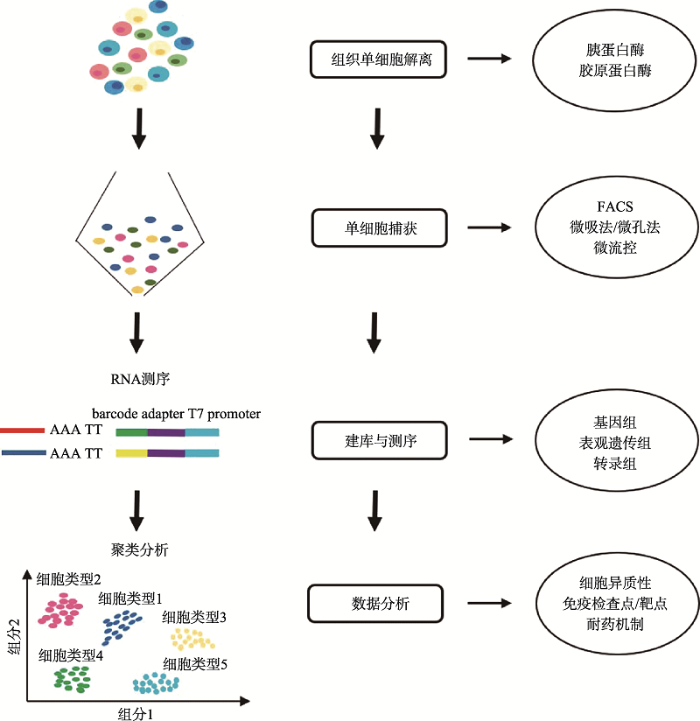
利用胰蛋白酶和/或胶原蛋白酶等对组织样本酶解消化,获得得单细胞悬液。然后,通过FACS、微流控等对单个细胞捕获。随后对其基因组、转录组或表观遗传组进行建库并测序,通过生物信息学分析解读测序数据。 Fig. 1Schematic of the single-cell sequencing technology
静息状态下,受体Ptc与SMO不会相互作用(黑色停止箭头),当Hh-N与Ptc结合后,会激活SMO,活化的SMO介导转录因子Gli1入核,驱动靶基因转录,激活癌相关成纤维细胞,使肿瘤细胞获得药物抗性的干细胞样表型(黑色箭头)。SMOi抑制SMO活性,Gli1无法入核(红色停止箭头),解除肿瘤细胞药物抗性,提高对多西紫杉醇的化疗敏感性(红色箭头)。 Fig. 2Chemoresistance mechanism of Hh-dependent signaling pathways in breast cancer
基于单细胞测序技术以全面剖析乳腺癌的特征,已取得相当大的进步。单细胞亚克隆异质性的研究为乳腺癌亚克隆频率及其演变提供了新的细节,基本描述了乳腺肿瘤生态系统复杂的细胞表型,以及肿瘤生态系统各组成部分之间的关系,为人们提供了较为全面的乳腺癌生态系统的图谱。针对乳腺癌的单细胞测序显示拷贝数的进化发生在肿瘤进化的早期,而点突变随着时间的推移逐渐进化,产生更广泛的克隆多样性,为乳腺癌肿瘤进化的交叉分枝模型提供了支持。在乳腺癌个体患者中,非浸润性导管原位癌与邻近浸润性导管癌病变之间存在直接的谱系关系,大多数的突变和拷贝数变异(copy number variations, CNVs)侵袭之前已在导管内完成,并具有多个突变和CNVs克隆体从导管逃逸,共同迁移到邻近组织,形成侵袭性癌。乳腺癌的耐药性是由选择罕见的预先存在的克隆引起的,还是通过诱导新的突变产生获得性耐药性引起的一直备受争议。新辅助化疗前后乳腺癌患者的纵向样本的单细胞DNA测序表明,耐药基因型是预先存在的,是化疗选择的。尽管可以识别出表达与化疗耐药性相关的单个基因的细胞亚群,但具有耐药程序的细胞在治疗前无法检测到。这些数据表明,乳腺癌从发生到治疗,不断进化(演化)过程中存在明显的异质性,为人们更好地理解乳腺癌的发生、转移和耐药提供了证据。
YuTL, CaiDL, ZhuGF, YeXJ, MinTS, ChenHY, LuDR, ChenHM . Effects of CSN4 knockdown on proliferation and apoptosis of breast cancer MDA-MB-231 cells Hereditas(Beijing), 2019,41(4):318-326. [本文引用: 1]
BoyleP, LevinB. World Cancer Report 2008. Lyon: IARC Press, 2008. [本文引用: 1]
YaoCB, ZhouX, ChenCS, LeiQY . The regulatory mechanisms and functional roles of the Hippo signaling pathway in breast cancer Hereditas(Beijing), 2017,39(7):617-629. [本文引用: 1]
BaslanT, KendallJ, RodgersL, CoxH, RiggsM, StepanskyA, TrogeJ, RaviK, EspositoD, LakshmiB, WiglerM, NavinN, HicksJ . Genome-wide copy number analysis of single cells Nat Protoc, 2012,7(6):1024-1041. [本文引用: 1]
ZongC, LuS, ChapmanAR, XieXS . Genome-wide detection of single-nucleotide and copy-number variations of a single human cell Science, 2012,338(6114):1622-1626. [本文引用: 2]
SethiN, KangY . Unravelling the complexity of metastasis—molecular understanding and targeted therapies Nat Rev Cancer, 2011,11(10):735-748. [本文引用: 1]
LeeW, JiangZ, LiuJ, HavertyPM, GuanY, StinsonJ, YueP, ZhangY, PantKP, BhattD, HaC, JohnsonS, KennemerMI, MohanS, NazarenkoI, WatanabeC, SparksAB, ShamesDS, Gentleman R, de Sauvage FJ, SternH, PanditaA, BallingerDG, DrmanacR, ModrusanZ, SeshagiriS, ZhangZ,. The mutation spectrum revealed by paired genome sequences from a lung cancer patient Nature, 2010,465(7297):473-477. [本文引用: 1]
ClarkJ, AttardG, JhavarS, FlohrP, ReidA, De-BonoJ, EelesR, ScardinoP, CuzickJ, FisherG, ParkerMD, FosterCS, BerneyD, KovacsG, CooperCS . Complex patterns of ETS gene alteration arise during cancer development in the human prostate Oncogene, 2008,27(14):1993-2003. [本文引用: 1]
WangY, WatersJ, LeungML, UnruhA, RohW, ShiX, ChenK, ScheetP, VattathilS, LiangH, MultaniA, ZhangH, ZhaoR, MichorF, Meric-BernstamF, NavinNE . Clonal evolution in breast cancer revealed by single nucleus genome sequencing Nature, 2014,512(7513):155-160. [本文引用: 1]
DerE, RanabothuS, SuryawanshiH, AkatKM, ClancyR, MorozovP, KustagiM, CzuppaM, IzmirlyP, BelmontHM, WangT, JordanN, BornkampN, NwaukoniJ, MartinezJ, GoilavB, BuyonJP, TuschlT, PuttermanC . Single cell RNA sequencing to dissect the molecular heterogeneity in lupus nephritis JCI Insight, 2017,2(9):93009. [本文引用: 2]
AutengruberA, GerekeM, HansenG, HennigC, BruderD . Impact of enzymatic tissue disintegration on the level of surface molecule expression and immune cell function Eur J Microbiol Immunol, 2012,2(2):112-120. [本文引用: 2]
BaronM, VeresA, WolockSL, FaustAL, GaujouxR, VetereA, RyuJH, WagnerBK, Shen-OrrSS, KleinAM, MeltonDA, Yanai I. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra- cell population structure . Cell Syst, 2016, 3(4): 346-360.e4. [本文引用: 2]
MuraroMJ, DharmadhikariG, GrünD, GroenN, DielenT, Jansen E, van Gurp L, EngelseMA, CarlottiF, de Koning EJ, van Oudenaarden A, . A single-cell transcriptome atlas of the human pancreas Cell Syst, 2016,3(4):385-394. [本文引用: 2]
WollnyD, ZhaoS, EverlienI, LunXK, BrunkenJ, BrüneD, ZiebellF, TabanskyI, WeichertW, Marciniak- CzochraA, Martin-VillalbaA . Single-cell analysis uncovers clonal acinar cell heterogeneity in the adult pancreas Dev Cell, 2016,39(3):289-301. [本文引用: 2]
LiD, PengSY, ZhangZW, FengRC, LiL, LiangJ, TaiS, TengCB . Complete disassociation of adult pancreas into viable single cells through cold trypsin-EDTA digestion J Zhejiang Univ Sci B, 2013,14(7):596-603. [本文引用: 2]
TreutleinB, BrownfieldDG, WuAR, NeffNF, MantalasGL, EspinozaFH, DesaiTJ, KrasnowMA, QuakeSR . Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq Nature, 2014,509(7500):371-375. [本文引用: 2]
ChapmanHA, LiX, AlexanderJP, BrumwellA, LorizioW, TanK, SonnenbergA, WeiY, VuTH . Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice J Clin Invest, 2011,121(7):2855-2862. [本文引用: 2]
LafziA, MoutinhoC, PicelliS, HeynH . Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies Nat Protoc, 2018,13(12):2742-2757. [本文引用: 4]
GrossA, SchoendubeJ, ZimmermannS, SteebM, ZengerleR, KoltayP . Technologies for single-cell isolation Int J Mol Sci, 2015,16(8):16897-16919. [本文引用: 1]
ZongC, LuS, ChapmanAR, XieXS . Genome-wide detection of single-nucleotide and copy-number variations of a single human cell Science, 2012,338(6114):1622-1626. [本文引用: 1]
GoleJ, GoreA, RichardsA, ChiuYJ, FungHL, BushmanD, ChiangHI, ChunJ, LoYH, ZhangK . Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells Nat Biotechnol, 2013,31(12):1126. [本文引用: 1]
GrünD, Van OudenaardenA . Design and analysis of single-cell sequencing experiments Cell, 2015,163(4):799-810. [本文引用: 1]
EllsworthDL, BlackburnHL, ShriverCD, RabizadehS, Soon-ShiongP, EllsworthRE . Single-cell sequencing and tumorigenesis: improved understanding of tumor evolution and metastasis Clin Transl Med, 2017,6(1):15. [本文引用: 1]
TrouttAB, McHeyzer-Williams MG, PulendranB, NossalGJ, . Ligation-anchored PCR: a simple amplification technique with single-sided specificity Proc Natl Acad Sci USA, 1992,89(20):9823-9825. [本文引用: 1]
ZhangL, CuiX, SchmittK, HubertR, NavidiW, ArnheimN . Whole genome amplification from a single cell: implications for genetic analysis Proc Natl Acad Sci USA, 1992,89(13):5847-5851. [本文引用: 1]
TeleniusH, CarterNP, BebbCE, Nordenskj?ldM, PonderBA, TunnacliffeA . Degenerate oligonucleotide- primed PCR: general amplification of target DNA by a single degenerate primer Genomics, 1992,13(3):718-725. [本文引用: 1]
CheungVG, NelsonSF . Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA Proc Natl Acad Sci USA, 1996,93(25):14676-14679. [本文引用: 1]
DeanFB, NelsonJR, GieslerTL, LaskenRS . Rapid amplification of plasmid and phage DNA using phi29 DNA polymerase and multiply-primed rolling circle amplification Genome Res, 2001,11(6):1095-1099. [本文引用: 1]
LaskenRS . Single-cell sequencing in its prime Nat Biotechnol, 2013,31(3):211-212. [本文引用: 1]
CohenAA, Geva-ZatorskyN, EdenE, Frenkel- MorgensternM, IssaevaI, SigalA, MiloR, Cohen- SaidonC, LironY, KamZ, CohenL, DanonT, PerzovN, AlonU . Dynamic proteomics of individual cancer cells in response to a drug Science, 2008,322(5907):1511-1516. [本文引用: 1]
RajA, van OudenaardenA . Single-molecule approaches to stochastic gene expression Annu Rev Biophys, 2009,38:255-270. [本文引用: 1]
TangFC, BarbacioruC, NordmanE, LiB, XuNL, BashkirovVI, LaoKQ, SuraniMA . RNA-Seq analysis to capture the transcriptome landscape of a single cell Nat Protoc, 2010,5(3):516-535. [本文引用: 1]
TangFC, BarbacioruC, WangYZ, NordmanE, LeeC, XuNL, WangXH, BodeauJ, TuchBB, SiddiquiA, LaoKQ , Surani MA. mRNA-Seq whole-transcriptome analysis of a single cell Nat Methods, 2009,6(5):377-382. [本文引用: 1]
CusanovichDA, DazaR, AdeyA, PlinerHA, ChristiansenL, GundersonKL, SteemersFJ, TrapnellC, ShendureJ . Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing Science, 2015,348(6237):910-914. [本文引用: 1]
St?hlPL, SalménF, VickovicS, LundmarkA, NavarroJF, MagnussonJ, GiacomelloS, AspM, WestholmJO, HussM, MollbrinkA, LinnarssonS, CodeluppiS, BorgA, PonténF, CosteaPI, SahlénP, MulderJ, BergmannO, LundebergJ, FrisénJ . Visualization and analysis of gene expression in tissue sections by spatial transcriptomics Science, 2016,353(6294):78-82. [本文引用: 1]
WangY, WatersJ, LeungML, UnruhA, RohW, ShiXQ, ChenK, ScheetP, VattathilS, LiangH, MultaniA, ZhangH, ZhaoR, MichorF, Meric-BernstamF, NavinNE . Clonal evolution in breast cancer revealed by single nucleus genome sequencing Nature, 2014,512(7513):155-160. [本文引用: 1]
GaoRL, KimC, SeiE, FoukakisT, CrosettoN, ChanLK, SrinivasanM, ZhangH, Meric-BernstamF, NavinN . Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer Nat Commun, 2017,8(1):228. [本文引用: 1]
ChungW, EumHH, LeeHO, LeeKM, LeeHB, KimKT, RyuHS, KimS, LeeJE, ParkYH, KanZ, HanW, ParkWY . Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer Nat Commun, 2017,8:15081. [本文引用: 1]
AllredDC, WuY, MaoS, NagtegaalID, LeeS, PerouCM, MohsinSK, O’Connell P, TsimelzonA, MedinaD, . Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution Clin Cancer Res, 2008,14(2):370-378. [本文引用: 1]
VirnigBA, TuttleTM, ShamliyanT, KaneRL . Ductal carcinoma in situ of the breast: a systematic review of incidence, treatment, and outcomes J Nati Cancer Inst, 2010,102(3):170-178. [本文引用: 1]
GaoRL, DavisA, McDonald TO, SeiE, ShiXQ, WangY, TsaiPC, CasasentA, WatersJ, ZhangH, Meric-Bernstam F, MichorF, Navin NE. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer Nat Genet, 2016,48(10):1119-1130. [本文引用: 1]
GerdesMJ, G?kmen-PolarY, SuiY, PangAS, LaPlante N, HarrisAL, TanPH, GintyF, BadveSS, . Single-cell heterogeneity in ductal carcinoma in situ of breast Mod Pathol, 2018,31(3):406-417. [本文引用: 1]
WagnerJ, RapsomanikiMA, ChevrierS, AnzenederT, LangwiederC, DykgersA, ReesM, RamaswamyA, MuenstS, SoysalSD, JacobsA, WindhagerJ, SilinaK, van den BroekM, DedesKJ, Rodríguez MartínezM, WeberWP, BodenmillerB, . A single-cell atlas of the tumor and immune ecosystem of human breast cancer Cell, 2019,177(5):1-16. [本文引用: 1]
AziziE, CarrAJ, PlitasG, CornishAE, KonopackiC, PrabhakaranS, NainysJ, WuK, KiseliovasV, SettyM, ChoiK, FrommeRM, DaoP, McKenneyPT, WastiRC, KadaveruK, MazutisL, RudenskyAY, Pe'er D. Single-Cell Map of diverse immune phenotypes in the breast tumor microenvironment Cell, 2018, 174(5): 1293-1308.e36. [本文引用: 1]
RuffellB, AuA, RugoHS, EssermanLJ, HwangES, CoussensLM . Leukocyte composition of human breast cancer Proc Natl Acad Sci USA, 2012,109(8):2796-2801. [本文引用: 1]
SavasP, VirassamyB, YeC, SalimA, MintoffCP, CaramiaF, SalgadoR, ByrneDJ, TeoZL, DushyanthenS, ByrneA, WeinL, LuenSJ, PolinessC, NightingaleSS, SkandarajahAS, GyorkiDE, ThorntonCM, BeavisPA, FoxSB, Kathleen, DarcyPK, Speed TP, MackayLK, NeesonPJ, LoiS . Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis Nat Med, 2018,24(7):986-993. [本文引用: 1]
NoyR, PollardJW . Tumor-associated macrophages: from mechanisms to therapy Immunity, 2014,41(1):49-61. [本文引用: 1]
PerrimonN, PitsouliC, ShiloBZ . Signaling mechanisms controlling cell fate and embryonic patterning Cold Spring Harb Perspect Biol, 2012,4(8):a005975. [本文引用: 1]
WisemanBS, WerbZ . Stromal effects on mammary gland development and breast cancer Science, 2002,296(5570):1046-1049. [本文引用: 1]
HuiM, CazetA, NairR, WatkinsDN, O'Toole SA, SwarbrickA, . The Hedgehog signalling pathway in breast development, carcinogenesis and cancer therapy Breast Cancer Res, 2013,15(2):203. [本文引用: 1]
AmakyeD, JaganiZ, DorschM . Unraveling the therapeutic potential of the Hedgehog pathway in cancer Nat Med, 2013,19(11):1410-1422. [本文引用: 1]
O'TooleSA, Machalek DA, ShearerRF, MillarEK, NairR, SchofieldP, McLeodD, CooperCL, McNeilCM, McFarlandA, NguyenA, OrmandyCJ, QiuMR, RabinovichB, MartelottoLG, VuD, HanniganGE, MusgroveEA, ChristD, SutherlandRL, WatkinsDN, SwarbrickA, . Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer Cancer Res, 2011,71(11):4002-4014. [本文引用: 1]
CazetAS, HuiMN, ElsworthBL, WuSZ, RodenD, ChanCL, SkhinasJN, CollotR, YangJ, HarveyK, JohanMZ, CooperC, NairR, HerrmannD, McFarland A, DengN, Ruiz-BorregoM, RojoF, TrigoJM, BezaresS, CaballeroR, LimE, TimpsonP, O'TooleS, WatkinsDN, CoxTR, SamuelMS, MartínM, SwarbrickA . Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer Nat Commun, 2018,9(1):2897. [本文引用: 1]
 ,聊城市人民医院,转化医学研究联合实验室,聊城 252000
,聊城市人民医院,转化医学研究联合实验室,聊城 252000
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT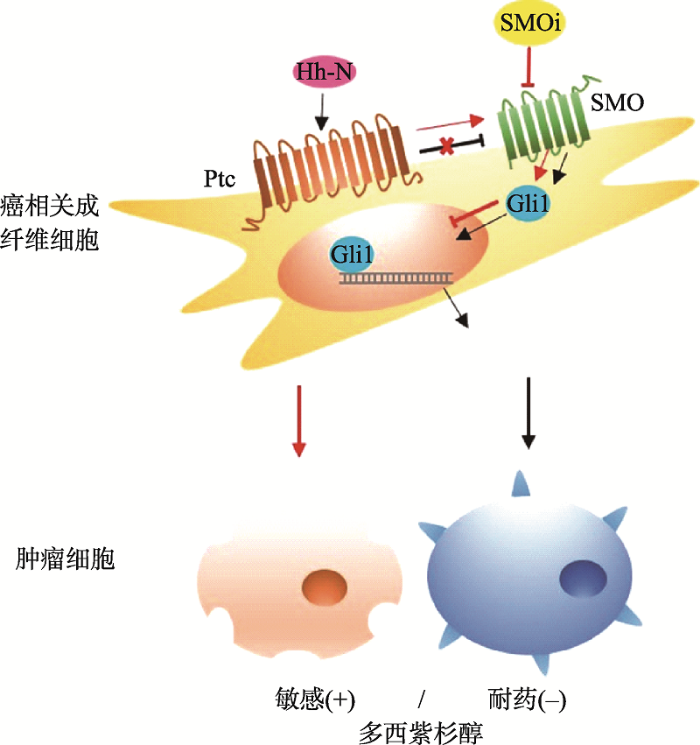 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT
{kind=link}
{kind=link}
{kind=link}
{kind=link}