Generation of Chimeric RNAs by cis-splicing of adjacent genes (cis-SAGe) in mammals
Jianshu Zhuo, Xiaoyan Jing, Xin Du, Xiuqin YangCollege of Animal Science and Technology, Northeast Agricultural University, Harbin 150030, China编委: 吴东东
收稿日期:2017-05-31修回日期:2017-07-31网络出版日期:2018-01-22
| 基金资助: |
Received:2017-05-31Revised:2017-07-31Online:2018-01-22
| Fund supported: |
作者简介 About authors
作者简介:禚建树,硕士研究生,专业方向:分子遗传学与动物育种E-mail:
通讯作者:杨秀芹,博士,博士生导师,研究方向:分子遗传学与动物育种E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (333KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
禚建树, 荆晓燕, 杜欣, 杨秀芹. 哺乳动物嵌合RNAs产生机制:cis-SAGe. 遗传[J], 2018, 40(2): 145-154 doi:10.16288/j.yczz.17-197
Jianshu Zhuo, Xiaoyan Jing, Xin Du, Xiuqin Yang.
嵌合RNAs(chimeric RNAs)是指由两个或两个以上独立基因(即亲本基因)融合产生的新RNAs。最初发现的嵌合RNAs都是染色体重排的结果,已有研究表明染色体易位、倒位和中间缺失都能形成嵌合RNAs[1,2,3]。染色体重排首先在基因组DNA水平上形成融合基因(fusion gene),再通过转录、剪接形成嵌合RNAs。融合基因大多与癌症密切相关,可以作为临床诊断的分子标记和治疗靶标。BCR-ABL在慢性粒细胞白血病患者中的发生率高达95%[4],已经被广泛用于该病的早期诊断,临床上利用靶向该嵌合分子的酪氨酸激酶抑制剂,取得了较好的治疗效果。PAX3-FOXO1在大多数(60%~70%)腺泡状横纹肌肉瘤患者中均表达,直接导致了患者5年生存率显著低于不携带该融合基因者[5],可用作患者危险分层和预后评价的分子标记[6,7,8],因此有****建议从分子和临床特征上将横纹肌肉瘤分为两种不同的类型:含有该融合基因型和其他类型[9]。目前,已经鉴定了多种与恶性肿瘤密切相关的融合基因,如分泌型乳腺癌中ETV6-NTRK3[10]、腺样囊性癌中MYB-NFIB[11]、肺癌中EML4-ALK[12,13]、前列腺癌中TMPRSS2与ETS(E-twenty six)转录因子家族基因的融合[14,15,16]等。
嵌合RNAs在癌症发生中的重要作用引起了研究者们的广泛兴趣。新一代高通量测序技术和生物信息学的发展,也为嵌合RNAs鉴定提供了有力的工具。随着研究的深入,人们发现除了染色体重排,不同基因间在转录或者转录后的加工过程中也能形成嵌合RNAs,包括反式剪接(trans-splicing)、相邻基因间的顺式剪接(cis-splicing of adjacent genes, cis-SAGe)以及短同源序列(short homologous sequence, SHS)介导的转录滑动。传统的RNA加工都属于顺式剪接(cis-splicing),即成熟mRNA的所有外显子都来源于同一个前体mRNA序列;反式剪接的外显子则来源于不同前体mRNA:包括同一前体mRNA的两个拷贝、同一个双螺旋DNA的两条链,以及不同基因的前体mRNA[17]。cis-SAGe是同一条染色体上位置相邻的基因间发生转录通读[18,19,20],由此产生的嵌合RNAs虽然含有不同亲本基因的外显子,但这些外显子均来自同一个mRNA前体,因此也被归为顺式剪接范畴。SHS介导的转录滑动是指在转录过程中,亲本基因间以“短同源序列互补”的方式引起“模板转换”,形成嵌合RNA[21](图1)。本文对cis-SAGe的剪接模式、表达特性、产生机制及在生命活动中的作用进行了综述。
1 cis-SAGe产生条件及别名
cis-SAGe是同一条染色体上位置相邻、转录方向相同的基因间发生转录通读,即RNA聚合酶Ⅱ在催化转录时,越过基因边界,直到下游基因的转录终止位点结束,由此产生的初始转录本经过加工,就形成了含有不同亲本基因序列的嵌合RNAs,一些研究者将其称为通读转录本(read-through transcript)[18,19,20]。发生cis-SAGe的亲本基因间距离较短,平均为8.5 kb[22]或10 kb[23],远远小于人基因组各相邻基因间的平均距离;并且相邻基因间的距离越近,越容易形成通读转录本。此外,发生cis-SAGe的转录单元(从上游基因的启动子开始到下游基因的终止子结束)长度短于RNA聚合酶Ⅱ催化的经典转录单元平均长度,如SCNN1A-TNFRSF1A为48 kb,CTSD-IFITM10为31 kb,而人基因组单个基因转录单元的平均长度为56 kb[23]。在近年发表的文献里,cis-SAGe及其产物也被称作通读基因融合子(read-through gene fusions)[24]、基因间共转录和剪接(cotranscription and intergenic splicing)[25]、伴随着基因间剪接的相邻基因共转录(cotranscription of adjacent genes coupled with intergenic splicing, CoTIS)[25]、连体基因(cojoined gene)[23]、通读融合转录本(read-through fusion transcript)[26]、转录诱导的嵌合子(transcription-induced chimeras, TIC)[22,27]、相邻基因间顺式剪接形成的嵌合转录本(chimeric transcript generated by cis-splicing of adjacent genes)[28]、串联RNA嵌合子(tandem RNA chimera)[29]等等。如此繁多的名称,表明此类嵌合RNAs的研究尚处于初级阶段,虽然研究者对其产生方式达成了共识,但还没有一个广泛认同的名称,这影响了不同研究者间共享信息,在一定程度上阻碍了其研究进程。
图1
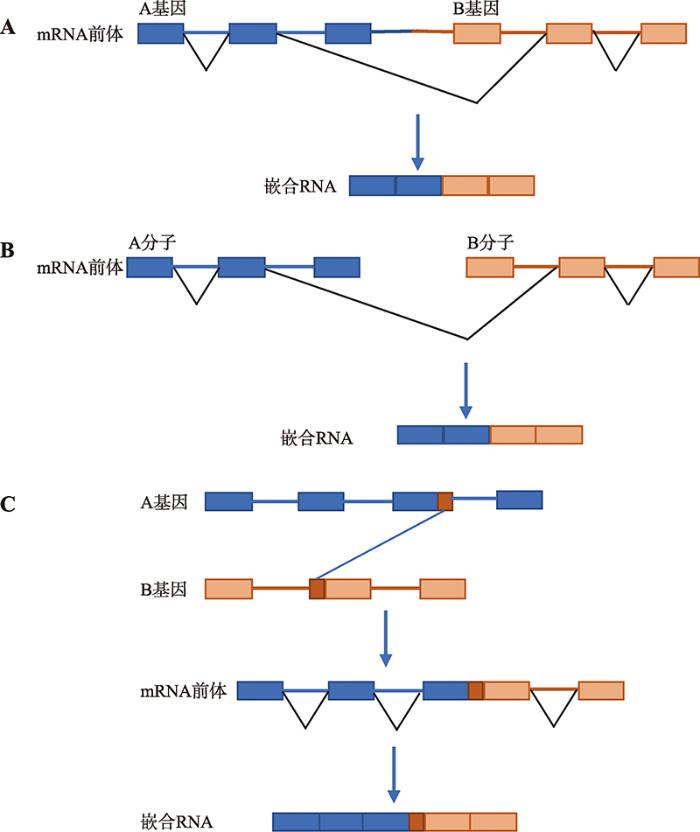 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1转录或转录后加工过程中嵌合RNA的产生方式
A:相邻基因间的顺式剪接;B:反式剪接;C:短同源序列介导的转录滑动。
Fig. 1Chimeric RNAs generated in transcription and post-transcriptional processing
2 cis-SAGe剪接特性
cis-SAGe主要存在2种剪接模式:(1)利用两个亲本的原有剪接位点进行剪接,所形成的嵌合子包括原有外显子的各种组合;(2)利用新剪接位点进行剪接,从而改变了原有外显子的长度,或者引入一个新的外显子[19,22]。不管以哪种方式进行剪接,形成的嵌合RNAs几乎都不含有上游基因的末尾外显子和下游基因的初始外显子,或者仅仅含有二者的截短序列,临近基因间隔区的部分则被剪接掉,并且绝大多数剪接位点都遵循GT-AG规则[23,30]。
cis-SAGe形成的新外显子主要是基因间序列剪接形成的,来自亲本内含子的则较少。Kim等[31]对UCSC数据库注释的人833个通读转录本分析发现,共计形成697个新外显子,其中482个(69%)来自基因间隔区(intergenic region)、215个(31%)来自亲本基因内部(intragenic region)——包括内含子和侧翼
序列,说明基因间区是通读转录本形成新外显子的热点区域。SINE(short interspersed nuclear element)、LINE(long interspersed nuclear element)和LTR(long terminal repeat)等转座元件在这些新外显子的形成中发挥了重要作用,约1/3的新外显子均来源于转座元件[31]。内含子序列(尤其是其中的重复序列)经外显子化(exonization)形成新外显子,在人、小鼠和狗等动物基因组中广泛存在,这种外显子从头(de novo)出现事件在经典mRNA的选择性剪接(alternative splicing, AS)中频繁发生,常常形成新的、稀有变异剪接体(transcript variant, TV)。在不影响原有基因组成、功能基础上,在进化上对新TV进行检验,是具有功能的新基因形成的一种进化策略[32]。从这个角度看,转座子序列可能在通读转录本的形成过程中具有极其重要的作用。
AS是指一个mRNA前体通过不同的剪接方式,产生多种成熟mRNA的现象。AS可以改变经典mRNA的转录量,从而对基因表达进行调控;也可以改变编码蛋白质的序列、结构,从而影响其功能,如改变了蛋白质的氨基端或羧基端、产生截短蛋白质或者增删了某个功能区域等[33]。通过AS,一个前体mRNA可产生多个TVs,人类95%~100%的多外显子(multiexon)基因均受到AS的影响[34,35]。因此,AS是真核生物转录组和蛋白质组多样性的重要机制。AS包括可变5°剪接位点、可变3°剪接位点、盒式外显子、内含子保留和互斥外显子5种基本形式。选择性剪接也广泛地存在于cis-SAGe中,Prakash等[23]对人基因组生物信息学分析结果进行RT-PCR和测序鉴定,发现至少63%的cis-SAGe存在AS。Kim等[31]利用RT-PCR和克隆、测序方法对人ZNF343- SNPRB、COX17-POPDC2、RBM7-REXO2、PRKRIP1- ORAI2和ATP5F1-C1orf162等5个通读转录本进行鉴定,各转录本分别获得了20、23、4、6和4个TVs。基于序列组成的结构分析表明,除互斥外显子外,这57个TVs的形成涉及了上述4种AS形式,序列中包括新外显子形成、外显子跳读/截短、外显子延长、序列缺失等情况。同时进行的外显子保守性分析表明,靠近两个亲本基因间隔区的序列保守性相对较低,是AS发生的密集区,如COX17- POPDC2的上、下游基因分别含有3和4个外显子,COX17的外显子1和2、POPDC2的外显子4组成型出现在所有的TVs中;在其他可变剪接外显子中,POPDC2基因外显子3的保守性远远高于外显子2,这种保守性降低的趋势越靠近基因间隔区越明显。
剪接后,两个基因的外显子融合点(exon junction)可以位于5°亲本的开放读码框(open reading frame, ORF)内部或者外侧,嵌合分子的ORF主要存在着如下几种情况[22,36]:(1)含有两个亲本ORF的框内(in-frame)融合序列,具有编码产生融合蛋白的潜在能力;(2)上游基因主要提供5°UTR给嵌合子,下游基因也可能只提供3°UTR,因此在转录和翻译水平上对另一个亲本基因进行表达调控;(3)形成移码突变(frame shift)或者提前终止密码子(premature termination codon, PTC),移码突变可能会改变5°亲本蛋白的羧基端[37],PTC则会通过无义突变介导的mRNA降解机制,抑制上游基因的表达[38]。此外,也有些外显子融合点存在于已知基因和非编码RNA之间[19]。
3 cis-SAGe表达特性
有些通读转录本仅在肿瘤细胞中表达,而正常组织的细胞中没有,如在霍普金淋巴瘤H/RS细胞中表达的LY75-CD302[39]、在乳腺癌样品中检测到的CTSD-IFITM10[26]等。Kim等[31]利用RT-PCR方法分析通读转录本在15种癌症和对应的正常组织中的表达,发现其具有明显的肿瘤偏好性:在肿瘤组织中的表达量明显高于正常组织,并且肿瘤组织表达更多种TVs,在所检测的5个嵌合分子共计57个TVs中,35个(61.4%)仅在肿瘤组织中表达。由此,作者推测肿瘤基因组更适于发生cis-SAGe。但也有许多通读转录本普遍表达于所检测的各种组织中,如NME1-NME2在心脏、骨骼肌、脾、肾、脑、淋巴结、白细胞等多种组织和细胞中都表达[22];在乳腺癌中鉴定的SIDT2-TAGLN和CTBS- GNG5嵌合分子以较高的丰度表达于多种组织中[26]。通读转录本DUS4L-BCAP29不仅在胃癌和前列腺癌中表达,也能在心脏、睾丸、肝脏、骨骼肌、肺、皮肤、肾上腺、子宫内膜等正常组织和多种非肿瘤细胞系中表达,并且其表达量在肿瘤和非肿瘤组织(细胞)间不存在差异[40]。
尽管通读转录本能编码产生功能性的融合蛋白,如嵌合分子LY75-CD302[39]、CTSD-IFITM10和SCNN1A-TNFRSF1A[26]等。但研究者认为大多数嵌合分子通过非编码RNA的形式发挥作用[19,23],如SLC45A3-ELK4[41]、BC039389-GATM和KLK4-KRSP1[19]。这种现象也可能是由于转录通读形成的嵌合分子在体内的表达量非常低,现有方法很难在蛋白水平上对其进行检测。
Akiva等[22]利用EST数据库鉴定RNAs时发现,约2%的人类基因会发生cis-SAGe。由于ESTs数据只是转录组的一个样本[42],不能代表所有的转录本,通读的实际发生率应该高于2%。不依赖ESTs的cis-SAGe分析表明,人类5%基因的编码区发生了转录通读[43]。利用计算机分析(in silico analysis)和高通量测序鉴定的嵌合RNAs中,约有30%可能是cis-SAGe形成的[22,24,44],Qin等[36]通过实验确定至少其中的25%是真实存在的通读转录本。因此,不管最终能否产生新蛋白质,在人类基因组中广泛存在的cis-SAGe事件增加了转录组和蛋白质组的多样性和复杂性。
4 cis-SAGe产物功能
4.1 参与癌症发生、发展
通读转录本与癌症发生、发展存在着密切关系,可以作为肿瘤诊断和预后的生物标记。BC039389- GATM在肾癌中表达量高于癌旁正常组织,KLK4- KRSP1只在肾癌组织中表达,其中所有的嫌色细胞癌、50%的2型乳头状肾细胞癌、20%肾透明细胞性癌、13%的1型乳头状肾细胞癌患者表达KLK4- KRSP1可变剪接体1(KKv1)。进一步研究发现,KKv1的表达与肿瘤大小、分级高低、组织学亚型存在着显著相关,并且在肾透明细胞性癌患者中,表达KKv1嵌合分子的患者总生存率显著低于不表达者[19]。SLC45A3-ELK4在前列腺癌细胞系(LNCaP和PC3)、组织样本中的表达量明显高于正常对照组,其表达水平与目前广泛接受的前列腺癌组织学分级指标——Gleason分数呈显著正相关,在前列腺癌转移患者中的表达量达到最高[28]。D2HGDH-GAL3ST2[45]和SYT8-TNNI2[46]分别以较高的频率存在于前列腺癌和尿路上皮癌组织中,在正常的对照组织中检测不到,并且在晚期癌症患者中D2HGDH- GAL3ST2的检出率增加。这些都表明通读转录本与癌症发生、发展存在着密切关系,可以作为肿瘤诊断和预后的生物标记。
4.2 调控基因表达
通读转录本存在如下几种可能:(1)翻译产生不同于亲本的新蛋白质多肽链;(2)只编码产生一个亲本的完整蛋白,另一个亲本发生了移码突变或者只作为UTR调控嵌合分子的表达;(3)形成非编码RNA。不管结果如何,都至少改变了一个亲本基因的表达水平。小鼠Ankhd1-Eif4ebp3通读转录本和Ankhd1基因具有相同的表达模式,这些组织内Eif4ebp3的表达量却显著降低[37];嵌合分子INS-IGF2、ZFP91- CNTF和MUTED-TXNDC5的表达和亲本基因间也存在着类似关系[23]。在多种癌细胞或者癌组织中均能检测到JMJD7- PLA2G4B[20,47],调控一系列基因的表达:敲低鳞状细胞癌和乳腺癌细胞中的该嵌合分子后,SKP2、TOP2A、PLK4、ASPM、BUB1、HMMR和L1CAM等基因的转录量显著降低;同时SKP蛋白显著降低并伴随着其抑制蛋白的高表达,敲低JMJD7却没有该现象[20]。
4.3 调控细胞活力和生长
持续不断的细胞增殖和改变的细胞周期都是癌症的标志[48],研究表明在癌细胞中特异或者高表达的通读转录本都对细胞活力和生长产生一定的影响。如JMJD7-PLA2G4B能够促进头颈部鳞状细胞癌细胞增殖、抑制饥饿诱导的细胞凋亡,并调控细胞周期进程[20];沉默D2HGDH-GAL3ST2,导致细胞增殖速度大幅下降[45];TSNAX-DISC1的表达与细胞从G1期向S期转变有关[49];在胃癌细胞中敲低和过表达DUS4L-BCAP29促进细胞增殖[50]等等。对于非肿瘤细胞,通读转录本也具有调控细胞活力和生长的作用。针对DUS4L-BCAP29设计的短干扰RNA(small interfering RNA, siRNA)(siDUS4L- BCAP29),能同时敲低嵌合分子和亲本基因DUS4L的表达,为此研究者又设计了特异干扰DUS4L基因的siRNA(siDUS4L),将这两种siRNA序列分别转染非肿瘤细胞系(GES-1和RWPE-1),发现二者都能抑制细胞增殖和迁移。通过基因芯片分析发现,siDUS4L-BCAP29和siDUS4L两种干扰序列处理GES-1细胞后,siDUS4L-BCAP29处理组具有特异富集的Gene Ontology条目,说明嵌合分子拥有不同于亲本基因DUS4L的独特功能。过表达分析进一步证实了DUS4L-BCAP29促进GES-1和RWPE-1细胞的增殖和运动[40]。
4.4 通读转录本和亲本基因的关系
相对于亲本基因,通读转录本在功能上具有两种可能:第一,和亲本功能相近。位于人11号染色体上的P2Y11基因编码产生一种ATP受体,嵌合分子P2Y11-SSF1普遍表达于所检测的11种组织中,并在粒细胞分化过程中上调表达。稳定过表达P2Y11-SSF1和P2Y11的细胞能分别对ATP刺激产生几乎完全相同的cAMP信号反应,但P2Y11-SSF1的表达量明显低于野生型的P2Y11[25];第二,改变了亲本的功能。通读转录本BC039389-GATM和KLK4-KRSP1以一种与其各自亲本基因相反的方式调控基因表达、影响细胞迁移和侵袭特性。如干扰BC039389-GATM能够促进IL8表达,而干扰野生型GATM基因,则抑制IL8表达;在A704和ACHN细胞中,干扰KLK4-KRSP1促进细胞迁移和侵袭,而干扰野生型KLK4则产生相反的效果[19]。在雄性激素依赖和非依赖型前列腺癌细胞中,干扰SLC45A3-ELK4均能促进细胞增殖,而干扰野生型的ELK4却没有这种作用[28,41]。5 cis-SAGe产生机制
5.1 CTCF与cis-SAGe
转录因子CTCF在通读形成嵌合RNAs的过程中发挥着重要作用。CTCF是脊椎动物体内主要的绝缘子结合蛋白,与绝缘子的活性密切相关[51,52]。TSNAX-DISC1两个亲本基因的间隔区存在着3个能与CTCF结合的绝缘子,敲低CTCF,TSNAX-DISC1的表达升高,两个亲本基因的表达则随之降低,说明CTCF与转录通读存在着一定的相关性。与TSNAX和DISC1间隔区对应的另一条链能转录形成一个基因间长链非编码RNA—linc-NR_034307,含有CTCF结合位点,并且能特异结合CTCF蛋白。TSNAX-DISC1和linc-NR_034307的表达存在着正相关:过表达linc-NR_034307导致TSNAX-DISC1表达量升高,亲本基因TSNAX和DISC1的表达量则随之降低;反之,敲低linc-NR_034307,TSNAX-DISC1表达量降低,同时伴随着亲本基因的表达量升高。鉴于该非编码RNA与TSNAX-DISC1亲本基因间隔区的绝缘子存在着较近的位置关系,研究者认为linc-NR_034307通过竞争性结合CTCF,抑制CTCF结合在TSNAX和DISC1基因间的绝缘子上,从而促进转录通读,产生更多的TSNAX-DISC1嵌合子[49]。SLC45A3和ELK4基因间隔区存在着两个CTCF结合位点[28,53],在LNCaP细胞内的分析表明,该嵌合分子的表达与CTCF的结合能力存在着负相关;但在雄性激素R1881的作用下,干扰CTCF却没有对嵌合子的表达量产生影响,可能是R1881本身具有阻止CTCF与靶位点结合的作用[28]。为了更深入研究SLC45A3-ELK4的产生机制,Qin等[54]对癌细胞和正常细胞在内的多种细胞(Hela、RWPE-1、LNCaP、PC3、293T、A2780和HCT116)进行了研究。相对于其他细胞,293T和LNCaP细胞内SLC45A3-ELK4和CTCF的表达水平及CTCF与绝缘子的结合能力都比较高,不同细胞间SLC45A3-ELK4和CTCF的表达(或CTCF在绝缘子上的结合能力)不存在着负相关关系。对11种前列腺癌临床样本的分析也得到了相似的结果:在SLC45A3-ELK4表达量高的样本内,CTCF的表达(或结合能力)也都比较高。然而,在前列腺癌细胞系(LNCaP、PC3和C4-2)内,人为添加雄性激素或血清导致了SLC45A3-ELK4表达量变化,并且这种变化与CTCF在绝缘子上的结合存在着负相关。进一步研究发现,雄性激素受体能与CTCF竞争性结合绝缘子,诱导嵌合子表达。这些结果表明,CTCF的表达量或与绝缘子的结合能力不是决定着不同细胞系(临床样本)间转录通读水平的主要因素,它可能是同种细胞对环境因素的一种适应性策略,即环境因素导致它发生变化,从而影响嵌合RNAs的产生。归根结底,SLC45A3-ELK4表达量变化是由环境因素决定的[54]。
5.2 Poly(A)信号与cis-SAGe
Poly(A)信号(poly(A) signals, PAS)在转录终止过程中发挥重要作用,一般认为PAS越强,其终止转录的效率越高[55]。因此,Akiva等[22]推测cis-SAGe与上游基因PAS的强弱有关,上游基因PAS越弱,RNA聚合酶顺利通过其转录终止位点、产生通读转录本的可能性越高。一半以上的人类基因具有多个poly(A)位点[56,57],可变聚腺苷酸化(alternative polyadenylation, Alt-PA)能够在转录后水平调控基因表达,也能够改变蛋白质多肽链的氨基酸组成。Alt-PA模式受组织和细胞的影响[58],睾丸、子宫和前列腺等生殖组织具有明显的poly(A)位点偏好性[59],而通读转录本在这些组织中的表达量较高[23]。与不能进行cis-SAGe的基因相比,通读基因具有较弱的PAS。Vilborg等[60]分析基因下游5 kb范围内正义链和反义链中AAUAAA出现的比例,发现通读基因的比值低于不能通读基因(分别为0.8和1.1),即通读基因的正义链中存在较少的AAUAAA。这些结果表明,上游基因的PAS影响转录终止效率和相应的cis- SAGe发生水平。Kim等[31]研究发现,人类基因组中所有的通读转录本及其TVs,都不含有上游基因的PAS,这种现象不仅存在于2个亲本基因形成的通读转录本中,在2个以上亲本基因构成的转录本中也存在。鉴于此,研究者认为上游基因PAS的全部移除是转录机器(transcriptional machinery)继续对基因间区和下游基因进行不间断转录的前提条件[31]。单细胞基因组测序发现,在同一组织的不同细胞间,基因组序列组成上存在着微小的差异[61],在没有癌变的正常组织中,也有少量细胞发生体细胞突变[62],在癌演进过程中,基因组的稳定性降低,体细胞突变频率增加[63]。同时考虑到转录通读本在正常组织中表达量极低[23,31,64]但却具有强烈的肿瘤组织偏好性[31],Kim等[31]认为在正常和肿瘤组织内,均有极少一部分细胞的基因组含有益于cis-SAGe发生的改变(上游基因PAS丢失)。这类细胞在正常组织内的含量很少,
以至于无法检测到,也不会对机体的正常功能产生影响,但是由于肿瘤组织内体细胞发生突变的比例升高,其含量也随之增多。因此,转录通读本的形成机制是上游基因的PAS发生改变,于是RNA聚合酶不能在上游基因的末尾处脱落,而是越过基因间区,直到下游基因的PAS附近。
6 结 语
综上所述,cis-SAGe可能是哺乳动物在进化过程中产生新基因的方式,其产物在一系列生理、病理活动中发挥着重要作用,鉴定通读转录本、揭示其功能及形成机制,具有非常重要的理论意义。由于RNA测序技术的日趋成熟和测序成本不断降低,转录组测序得到了越来越广泛的应用,伴随着多款嵌合RNA分析软件的成功开发与不断改进,许多嵌合RNAs(包括通读转录本)得到鉴定。然而,就研究水平来看,多数工作停留在生物信息学鉴定和实验验证层面,缺乏研究深度。目前,实验验证的主要方法是利用跨嵌合位点引物,对候选嵌合子进行RT-PCR扩增。由于在逆转录构建cDNA文库和测序反应中,都可能存在着模板转换现象,从而导致假阳性结果出现,一些研究者对嵌合RNA的鉴定结果持怀疑态度。RNA酶保护实验、Northern blotting等不依赖于逆转录的验证方法,虽然有助于消除上述问题,但检验的灵敏性降低。随着实验技术的不断完善,嵌合RNA分析将会成为遗传学和基因组学的一个重要研究内容。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
URLPMID:21593131 [本文引用: 1]

Next generation sequencing technology generates high-throughput data, which allows us to detect fusion genes at both transcript and genomic levels. To detect fusion genes, the current bioinformatics tools heavily rely on paired-end approaches and overlook the importance of reads that span fusion junctions. Thus there is a need to develop an efficient aligner to detect fusion events by accurate mapping of these junction-spanning single reads, particularly when the read gets longer with the improvement in sequencing technology.We present a novel method, FusionMap, which aligns fusion reads directly to the genome without prior knowledge of potential fusion regions. FusionMap can detect fusion events in both single- and paired-end datasets from either RNA-Seq or gDNA-Seq studies and characterize fusion junctions at base-pair resolution. We showed that FusionMap achieved high sensitivity and specificity in fusion detection on two simulated RNA-Seq datasets, which contained 75 nt paired-end reads. FusionMap achieved substantially higher sensitivity and specificity than the paired-end approach when the inner distance between read pairs was small. Using FusionMap to characterize fusion genes in K562 chronic myeloid leukemia cell line, we further demonstrated its accuracy in fusion detection in both single-end RNA-Seq and gDNA-Seq datasets. These combined results show that FusionMap provides an accurate and systematic solution to detecting fusion events through junction-spanning reads.FusionMap includes reference indexing, read filtering, fusion alignment and reporting in one package. The software is free for noncommercial use at (http://www.omicsoft.com/fusionmap).
[本文引用: 1]
URLPMID:19741701 [本文引用: 1]
Deep sequencing of 'transcriptomes'--the collection of all RNA transcripts produced at a given time--from worms to humans reveals that some transcripts are composed of sequence segments that are not co-linear, with pieces of sequence coming from distant regions of DNA, even different chromosomes. Some of these 'chimaeric' transcripts are formed by genetic rearrangements, but others arise during post-transcriptional events. The 'trans-splicing' process in lower eukaryotes is well understood, but events in higher eukaryotes are not. The existence of such chimaeric RNAs has far-reaching implications for the potential information content of genomes and the way it is arranged.
URLPMID:4126434 [本文引用: 1]
CELLS from nine consecutive patients with chronic myelogenous leukaemia (CML) have been analysed with quinacrine fluorescence and various Giemsa staining techniques. The Philadelphia (Ph) chromosome in all nine patients represents a deletion of the long arm of chromosome 22 (22q-). An unsuspected abnormality in all cells from the nine patients has been detected with these new staining techniques. It consists of the addition of dully fluorescing material to the end of the long arm of one chromosome 9 (9q+). In Giemsa-stained preparations, this material appears as an additional faint terminal band in one chromosome 9. The amount of additional material is approximately equal to the amount missing from the Ph(22q-) chromosome, suggesting that there may be a hitherto undetected translocation between the long arm of 22 and the long arm of 9, producing the 9q+ chromosome.
URLPMID:23526739 [本文引用: 1]
Abstract BACKGROUND: Rhabdomyosarcoma (RMS) is divided into two major histological subtypes: alveolar (ARMS) and embryonal (ERMS), with most ARMS expressing one of two oncogenic genes fusing PAX3 or PAX7 with FOXO1 (P3F and P7F, respectively). The Children's Oncology Group (COG) carried out a multi-institutional clinical trial to evaluate the prognostic value of PAX-FOXO1 fusion status. METHODS: Study participants were treated on COG protocol D9803 for intermediate risk ARMS or ERMS using multi-agent chemotherapy, radiotherapy, and surgery. Central diagnostic pathology review and molecular testing for fusion genes were carried out on prospectively collected specimens. Event-free (EFS) and overall survival (OS) at 5 years were correlated with histological subtype and PAX-FOXO1 status. RESULTS: Of 616 eligible D9803 enrollees, 434 cases had adequate clinical, molecular, and pathology data for definitive classification as ERMS, ARMS P3F+ or P7F+, or ARMSn (without detectable fusion). EFS was worse for those with ARMS P3F+ (54%) and P7F+ (65%) than those with ERMS (77%; P < 0.001). EFS for ARMSn and ERMS were not statistically different (90% vs. 77%, P = 0.15). ARMS P3F+ had poorer OS (64%) than ARMS P7F+ (87%), ARMSn (89%), and ERMS (82%; P = 0.006). CONCLUSIONS: ARMSn has an outcome similar to ERMS and superior EFS compared to ARMS with either P3F or P7F, when given therapy designed for children with intermediate risk RMS. This prospective analysis supports incorporation of PAX-FOXO1 fusion status into risk stratification and treatment allocation.
URLPMID:22454413 [本文引用: 1]
Abstract PURPOSE: To improve the risk stratification of patients with rhabdomyosarcoma (RMS) through the use of clinical and molecular biologic data. PATIENTS AND METHODS: Two independent data sets of gene-expression profiling for 124 and 101 patients with RMS were used to derive prognostic gene signatures by using a meta-analysis. These and a previously published metagene signature were evaluated by using cross validation analyses. A combined clinical and molecular risk-stratification scheme that incorporated the PAX3/FOXO1 fusion gene status was derived from 287 patients with RMS and evaluated. RESULTS: We showed that our prognostic gene-expression signature and the one previously published performed well with reproducible and significant effects. However, their effect was reduced when cross validated or tested in independent data and did not add new prognostic information over the fusion gene status, which is simpler to assay. Among nonmetastatic patients, patients who were PAX3/FOXO1 positive had a significantly poorer outcome compared with both alveolar-negative and PAX7/FOXO1-positive patients. Furthermore, a new clinicomolecular risk score that incorporated fusion gene status (negative and PAX3/FOXO1 and PAX7/FOXO1 positive), Intergroup Rhabdomyosarcoma Study TNM stage, and age showed a significant increase in performance over the current risk-stratification scheme. CONCLUSION: Gene signatures can improve current stratification of patients with RMS but will require complex assays to be developed and extensive validation before clinical application. A significant majority of their prognostic value was encapsulated by the fusion gene status. A continuous risk score derived from the combination of clinical parameters with the presence or absence of PAX3/FOXO1 represents a robust approach to improving current risk-adapted therapy for RMS.
URLPMID:31 [本文引用: 1]
Abstract Alveolar rhabdomyosarcoma comprises a rare highly malignant tumor presumed to be associated with skeletal muscle lineage in children. The hallmark of the majority of alveolar rhabdomyosarcoma is a chromosomal translocation that generates the PAX3-FOXO1 fusion protein, which is an oncogenic transcription factor responsible for the development of the malignant phenotype of this tumor. Alveolar rhabdomyosarcoma cells are dependent on the oncogenic activity of PAX3-FOXO1 , and its expression status in alveolar rhabdomyosarcoma tumors correlates with worst patient outcome, suggesting that blocking this activity of PAX3-FOXO1 may be an attractive therapeutic strategy against this fusion-positive disease. In this study, we screened small molecule chemical libraries for inhibitors of PAX3-FOXO1 transcriptional activity using a cell-based readout system. We identified the Sarco/endoplasmic reticulum Ca(2+)-ATPases (SERCA) inhibitor thapsigargin as an effective inhibitor of PAX3-FOXO1 . Subsequent experiments in alveolar rhabdomyosarcoma cells showed that activation of AKT by thapsigargin inhibited PAX3-FOXO1 activity via phosphorylation. Moreover, this AKT activation appears to be associated with the effects of thapsigargin on intracellular calcium levels. Furthermore, thapsigargin inhibited the binding of PAX3-FOXO1 to target genes and subsequently promoted its proteasomal degradation. In addition, thapsigargin treatment decreases the growth and invasive capacity of alveolar rhabdomyosarcoma cells while inducing apoptosis in vitro. Finally, thapsigargin can suppress the growth of an alveolar rhabdomyosarcoma xenograft tumor in vivo. These data reveal that thapsigargin-induced activation of AKT is an effective mechanism to inhibit PAX3-FOXO1 and a potential agent for targeted therapy against alveolar rhabdomyosarcoma . 漏2013 AACR.
URLPMID:25821947 [本文引用: 1]
Patients with translocation-positive alveolar rhabdomyosarcoma (ARMS), an aggressive childhood tumor primarily characterized by the PAX3-FOXO1 oncogenic fusion protein, have a poor prognosis because of lack of therapies that specifically target ARMS tumors. This fact highlights the need for novel pharmaceutical interventions. Posttranslational modifications such as phosphorylation are becoming attractive biological targets for the development of such interventions. Along these lines, we demonstrated that PAX3-FOXO1 is phosphorylated at three specific sites and that its pattern of phosphorylation is altered relative to wild-type Pax3 throughout early myogenesis and in ARMS tumor cells. However, little work has been performed examining the effect of directly inhibiting phosphorylation at these sites on ARMS development. To address this gap in knowledge, we used small molecule inhibitors or mutational analysis to specifically inhibit phosphorylation of PAX3-FOXO1 to investigate how altering phosphorylation of the oncogenic fusion protein affects ARMS phenotypes. We found that inhibiting the phosphorylation of PAX3-FOXO1 at Ser201 significantly reduced migration, invasion and proliferation in two independent ARMS tumor cell lines. Further, we found that inhibition of phosphorylation at Ser205 also decreased proliferation and anchorage-independent growth. Consistent with these in vitro results, we demonstrate for the first time that PAX3-FOXO1 is phosphorylated at Ser201 and Ser205 in a primary tumor sample and in tumor cells actively invading the surrounding normal tissue. This report is the first to demonstrate that the direct inhibition of PAX3-FOXO1 phosphorylation reduces ARMS tumor phenotypes in vitro and that these phosphorylation events are present in primary human ARMS tumors and invading tumor cells. These results identify phosphorylation of PAX3-FOXO1, especially at Ser201, as a novel biological target that can be explored as a promising avenue for ARMS therapies.
URLPMID:20351326 [本文引用: 1]
Abstract PURPOSE: To determine whether the clinical and molecular biologic characteristics of the alveolar rhabdomyosarcoma (ARMS) and embryonal rhabdomyosarcoma (ERMS) subtypes have relevance independent of the presence or absence of the PAX/FOXO1 fusion gene. PATIENTS AND METHODS: The fusion gene status of 210 histopathologically reviewed, clinically annotated rhabdomyosarcoma samples was determined by reverse transcriptase polymerase chain reaction. Kaplan-Meier analysis was used to assess event-free survival and overall survival in fusion gene-negative ARMS (ARMSn; n = 39), fusion gene-positive ARMS (ARMSp; n = 94), and ERMS (n = 77). A total of 101 RMS samples were also profiled for whole-genome expression, and 128 were profiled for genomic copy number imbalances. Profiling data were analyzed by supervised and unsupervised methods to compare features related to histopathology and fusion gene status. Results were also projected by meta-analysis techniques across three separate publically available data sets. RESULTS: Overall and event-free survival, frequency of metastases, and distribution of site at initial presentation were not significantly different between ARMSn and ERMS. Consistent with this, analysis of gene expression signatures could not reproducibly distinguish ARMSn from ERMS whereas fusion gene-positive cases were distinct. ARMSn and ERMS frequently show whole-chromosome copy number changes, notably gain of chromosome 8 with associated high levels of expression of genes from this chromosome. CONCLUSION: The clinical behavior and molecular characteristics of alveolar cases without a fusion gene are indistinguishable from embryonal cases and significantly different from fusion-positive alveolar cases. This implies that fusion gene status irrespective of histology is a critical factor in risk stratification of RMS.
URLPMID:12450792 [本文引用: 1]
We report that human secretory breast carcinoma (SBC), a rare subtype of infiltrating ductal carcinoma, expresses the ETV6-NTRK3 gene fusion previously cloned in pediatric mesenchymal cancers. This gene fusion encodes a chimeric tyrosine kinase with potent transforming activity in fibroblasts. ETV6-NTRK3 expression was confirmed in 12 (92%) of 13 SBC cases, but not in other ductal carcinomas. Retroviral transfer of ETV6-NTRK3 (EN) into murine mammary epithelial cells resulted in transformed cells that readily formed tumors in nude mice. Phenotypically, tumors produced glands and expressed epithelial antigens, confirming that EN transformation is compatible with epithelial differentiation. This represents a recurrent chromosomal rearrangement and expression of a dominantly acting oncogene as a primary event in human breast carcinoma.
[本文引用: 1]
[本文引用: 1]
URLPMID:23341890 [本文引用: 1]
The EML4-ALK fusion gene has been recently identified in a small subset of non-small cell lung cancer (NSCLC) patients who respond positively to ALK inhibitors. The characteristics of the EML4-ALK fusion gene in Chinese patients with NSCLC are poorly understood. Here, we report on the prevalence of EML4-ALK, EGFR status and KRAS mutations in 208 Chinese patients with NSCLC. EGFR mutations were found in 24.5% (51/208) of patients. In concordance with previous reports, these mutations were identified at high frequencies in females (47.5% vs 15.0% in males;P<0.05); never-smokers (42.3% vs 13.9% in smokers;P<0.05), and adenocarcinoma patients (44.2% vs 8.0% in non-adenocarcinoma patients;P<0.05). There were only 2.88% (6/208) patients with KRAS mutations in our study group. We identified 7 patients who harbored the EML4-ALK fusion gene (3.37%, 7/208), including 4 cases with variant 3 (57.1%), 2 with variant 1, and 1 with variant 2. All positive cases corresponded to female patients (11.5%, 7/61). Six of the positive cases were non-smokers (7.69%, 6/78). The incidence of EML4-ALK translocation in female, non-smoking adenocarcinoma patients was as high as 15.2% (5/33). No EGFR/KRAS mutations were detected among the EML4-ALK positive patients. Pathological analysis showed no difference between solid signet-ring cell pattern (4/7) and mucinous cribriform pattern (3/7) in ALK-positive patients. Immunostaining showed intratumor heterogeneity of ALK rearrangement in primary carcinomas and 50% (3/6) of metastatic tumors with ALK-negative staining. Meta-analysis demonstrated that EML4-ALK translocation occurred in 4.84% (125/2580) of unselected patients with NSCLC, and was also predominant in non-smoking patients with adenocarcinoma. Taken together, EML4-ALK translocations were infrequent in the entire NSCLC patient population, but were frequent in the NSCLC subgroup of female, non-smoker, adenocarcinoma patients. There was intratumor heterogeneity of ALK rearrangement in primary carcinomas and at metastatic sites.
[本文引用: 1]
[本文引用: 1]
URLPMID:2711688 [本文引用: 1]
The discovery of recurrent gene fusions in a majority of prostate cancers has important clinical and biological implications in the study of common epithelial tumours. Gene fusion and chromosomal rearrangements were previously thought to be primarily the oncogenic mechanism of haematological malignancies and sarcomas. The prostate cancer gene fusions that have been identified thus far are characterized by 5' genomic regulatory elements, most commonly controlled by androgen, fused to members of the Ets family of transcription factors, leading to the overexpression of oncogenic transcription factors. Ets gene fusions probably define a distinct class of prostate cancer, and this might have a bearing on diagnosis, prognosis and rational therapeutic targeting.
URLPMID:4824033 [本文引用: 1]
Abstract Pre-RNA splicing is an essential step in generating mature mRNA. RNA trans-splicing combines two separate pre-mRNA molecules to form a chimeric non-co-linear RNA, which may exert a function distinct from its original molecules. Trans-spliced RNAs may encode novel proteins or serve as noncoding or regulatory RNAs. These novel RNAs not only increase the complexity of the proteome but also provide new regulatory mechanisms for gene expression. An increasing amount of evidence indicates that trans-splicing occurs frequently in both physiological and pathological processes. In addition, mRNA reprogramming based on trans-splicing has been successfully applied in RNA-based therapies for human genetic diseases. Nevertheless, clarifying the extent and evolution of trans-splicing in vertebrates and developing detection methods for trans-splicing remain challenging. In this review, we summarize previous research, highlight recent advances in trans-splicing, and discuss possible splicing mechanisms and functions from an evolutionary viewpoint. 脗漏 The Author(s) 2016. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
URLPMID:16442775 [本文引用: 2]
NM23-H1 and NM23-H2 are neighboring genes on chromosome 17q. They encode nucleoside diphosphate kinases that have additional roles in signal transduction, transcription, and apoptosis. NM23-H1 expression is a strong marker for prognosis and metastatic behavior in many tumor types. A new bioinformatic tool, TranscriptView, identified read-through transcripts that start in the NM23-H1 gene and continue in the neighboring NM23-H2 gene. Splicing results in a transcript containing exons 1 to 4 of NM23-H1 and exons 2 to 5 of NM23-H2. The resulting mRNA encodes a novel and long variant of the NM23 protein family, NM23-LV, which contains part of NM23-H1 and the complete NM23-H2 protein. The transcript was amplified and sequenced from two neuroblastoma cell lines, confirming the presence of the predicted NM23-LV mRNA in vivo. Tissue analysis showed that NM23-LV is ubiquitously expressed, with the exception of the kidney. Neuroblastoma tumors show high-level expression of NM23-H1 and-H2 as well as NM23-LV mRNA. In neuroblastoma cells, the NM23-LV protein has mainly a cytoplasmic localization, but some nuclear staining was observed as well.
URLPMID:4422297 [本文引用: 8]
Chimeric read-through RNAs are transcripts originating from two directly adjacent genes (<10 kb) on the same DNA strand. Although they are found in next-generation whole transcriptome sequencing (RNA-Seq) data on a regular basis, investigating them further has usually been refrained from. Therefore, their expression patterns or functions in general, and in oncogenesis in particular, are poorly understood. We used paired-end RNA-Seq and a specifically designed computational data analysis pipeline (FusionSeq) to nominate read-through events in a small discovery set of renal cell carcinomas (RCC) and confirmed them in a larger validation cohort. 324 read-through events were called overall; 22/27 (81%) selected nominees passed validation with conventional PCR and were sequenced at the junction region. We frequently identified various isoforms of a given read-through event. 2/22 read-throughs were up-regulated: BC039389-GATM was higher expressed in RCC compared to benign adjacent kidney; KLK4-KRSP1 was expressed in 46/169 (27%) RCCs, but rarely in normal tissue. KLK4-KRSP1 expression was associated with worse clinical outcome in the patient cohort. In cell lines, both read-throughs influenced molecular mechanisms (i.e. target gene expression or migration/invasion) in a way that counteracted the effect of the respective parent transcript GATM or KLK4. Our data suggests that the up-regulation of read-through RNA chimeras in tumors is not random but causes regulatory effects on cellular mechanisms and may impact patient survival.
URLPMID:28030848 [本文引用: 5]
Recent findings on the existence of oncogenic fusion genes in a wide array of solid tumors, including head and neck squamous cell carcinoma (HNSCC), suggests that fusion genes have become attractive targets for cancer diagnosis and treatment. In this study, we showed for the first time that a read-through fusion gene JMJD7-PLA2G4B is presented in HNSCC, splicing neighboring jumonji domain containing 7 (JMJD7) and phospholipase A2, group IVB (PLA2G4B) genes together. Ablation of JMJD7-PLA2G4B significantly inhibited proliferation of HNSCC cells by promoting G1 cell cycle arrest and increased starvation-induced cell death compared to JMJD7-only knockdown HNSCC cells. Mechanistically, we found that JMJD7-PLA2G4B modulates phosphorylation of Protein Kinase B (AKT) to promote HNSCC cell survival. Moreover, JMJD7-PLA2G4B also regulated an E3 ligase S-phase kinase-associated protein 2 (SKP2) to control the cell cycle progression from G1 phase to S phase by inhibiting Cyclin-dependent kinase inhibitor 1 (p21) and 1B (p27) expression. Our study provides novel insights into the oncogenic control of JMJD7-PLA2G4B in HNSCC cell proliferation and survival, and suggests that JMJD7-PLA2G4B may serve as an important therapeutic target and prognostic marker for HNSCC development and progression.
URLPMID:19158498 [本文引用: 1]
Abstract Chimeric gene products, most often resulting from chromosome translocations, have been considered unique features of cancer, or at least of cells at high risk for becoming cancerous. Chimeric JAZF1-JJAZ1 mRNA transcribed from DNA spanning the site of recombination in the (7;17)(p15;q21) chromosomal translocation found in half of endometrial stromal sarcomas and most cases of benign stromal nodules is one such example. The recent finding that chimeric JAZF1-JJAZ1 mRNA can also be detected in normal endometrial stromal cells suggests that chimeric gene products are not limited to cancer or pre-cancerous cells. The JAZF1-JJAZ1 mRNA and the protein encoded by it appear to be identical to that synthesized from the gene fusion in neoplastic cells. In cultured cells, the chimeric protein has anti-apoptotic properties and is pro-proliferative when unrearranged JJAZ1 alleles are silenced, as they are in endometrial stromal sarcomas but not in the stromal nodules. These observations are consistent with the conclusion that chromosomal rearrangements and gene fusions in neoplastic cells may represent mechanisms for the deregulated expression of chimeric gene products that are generated at specific stages in cell development and have physiologic functions in normal cells. Furthermore, it may be possible that other means for abnormal production of chimeric gene products, such as hyperactive transsplicing of RNA, may be another mechanism underlying the neoplastic properties of tumor cells.
URLPMID:16344562 [本文引用: 8]
An international, peer-reviewed genome sciences journal featuring outstanding original research that offers novel insights into the biology of all organisms
URLPMID:2953495 [本文引用: 9]
From the ENCODE project, it is realized that almost every base of the entire human genome is transcribed. One class of transcripts resulting from this arises from the conjoined gene, which is formed by combining the exons of two or more distinct (parent) genes lying on the same strand of a chromosome. Only a very limited number of such genes are known, and the definition and terminologies used for them are highly variable in the public databases. In this work, we have computationally identified and manually curated 751 conjoined genes (CGs) in the human genome that are supported by at least one mRNA or EST sequence available in the NCBI database. 353 representative CGs, of which 291 (82%) could be confirmed, were subjected to experimental validation using RT-PCR and sequencing methods. We speculate that these genes are arising out of novel functional requirements and are not merely artifacts of transcription, since more than 70% of them are conserved in other vertebrate genomes. The unique splicing patterns exhibited by CGs reveal their possible roles in protein evolution or gene regulation. Novel CGs, for which no transcript is available, could be identified in 80% of randomly selected potential CG forming regions, indicating that their formation is a routine process. Formation of CGs is not only limited to human, as we have also identified 270 CGs in mouse and 227 in drosophila using our approach. Additionally, we propose a novel mechanism for the formation of CGs. Finally, we developed a database, ConjoinG, which contains detailed information about all the CGs (800 in total) identified in the human genome. In summary, our findings reveal new insights about the functionality of CGs in terms of another possible mechanism for gene regulation and genomic evolution and the mechanism leading to their formation.
URLPMID:21261984 [本文引用: 2]
Readthrough fusions across adjacent genes in the genome, or transcription-induced chimeras (TICs), have been estimated using expressed sequence tag (EST) libraries to involve 4-6% of all genes. Deep t
[本文引用: 3]
URLPMID:24929677 [本文引用: 4]
Read-through fusion transcripts that result from the splicing of two adjacent genes in the same coding orientation are a recently discovered type of chimeric RNA. We sought to determine if read-through fusion transcripts exist in breast cancer. We performed paired-end RNA-seq of 168 breast samples, including 28 breast cancer cell lines, 42 triple negative breast cancer primary tumors, 42 estrogen receptor positive (ER+) breast cancer primary tumors, and 56 non-malignant breast tissue samples. We analyzed the sequencing data to identify breast cancer associated read-through fusion transcripts. We discovered two recurrent read-through fusion transcripts that were identified in breast cancer cell lines, confirmed across breast cancer primary tumors, and were not detected in normal tissues ( SCNN1A - TNFRSF1A and CTSD - IFITM10 ). Both fusion transcripts use canonical splice sites to join the last splice donor of the 5鈥 gene to the first splice acceptor of the 3鈥 gene, creating an in-frame fusion transcript. Western blots indicated that the fusion transcripts are translated into fusion proteins in breast cancer cells. Custom small interfering RNAs targeting the CTSD - IFITM10 fusion junction reduced expression of the fusion transcript and reduced breast cancer cell proliferation. Read-through fusion transcripts between adjacent genes with different biochemical functions represent a new type of recurrent molecular defect in breast cancer that warrant further investigation as potential biomarkers and therapeutic targets. Both breast cancer associated fusion transcripts identified in this study involve membrane proteins ( SCNN1A - TNFRSF1A and CTSD - IFITM10 ), which raises the possibility that they could be breast cancer-specific cell surface markers.
URLPMID:26718406 [本文引用: 1]
The expression of transcription-induced chimeras (TICs) was underestimated due to strategic and logical reasons. In order to thoroughly examine TICs, systematic survey of TIC events was conducted in mammalian genomes using ESTs, followed by experimental validation using RT-PCR and real-time quantitative PCR (qPCR). The expression of 6598% TIC events in at least one tissue or cell line from both mouse and human was verified. Besides, 6540% TICs were broadly expressed, and 6533% of TICs showed expression levels comparable to or higher than their upstream parental genes. We further identified putative chimeric proteins in public databases and validated two using Western blotting. GO analysis revealed that proteins resided in one multi-protein complex or functioning in metabolic or signaling pathway tended to produce fused products. Taken together, we have shown substantial evidence for the underestimated TIC events; and TICs could be a novel regulated way to further increases the proteome complexity in mammalian genomes. Possible regulation mechanisms and evolution of TICs were also discussed.
URLPMID:22719019 [本文引用: 5]
Abstract Gene fusion is a common event in cancer. The fusion RNA and protein products often play causal roles in tumorigenesis and therefore represent ideal diagnostic and therapeutic targets. Formerly, fusion chimeric products in cancer were thought to be produced solely by chromosomal translocation. Here, we show that a chimeric SLC45A3-ELK4 RNA is generated in the absence of chromosomal rearrangement. We showed that it is not a product of RNA trans-splicing, but formed by cis-splicing of adjacent genes/read-through. The binding of CCCTC-binding factor (CTCF) to the insulator sequences inversely correlates with the expression of the chimera transcript. The SLC45A3-ELK4 fusion, but not wild-type, ELK4 plays important roles in regulating cell growth in both androgen-dependent and -independent prostate cancer cells. The level of the chimeric transcript correlates with disease progression, with the highest levels in prostate cancer metastases. Our results suggest that gene fusions can arise from cis-splicing of adjacent genes without corresponding DNA changes. SIGNIFICANCE: With the absence of corresponding DNA rearrangement, chimeric fusion SLC45A3-ELK4 transcript in prostate cancer cells is generated by cis-splicing of adjacent genes/gene read-through instead of trans-splicing. SLC45A3-ELK4 controls prostate cancer cell proliferation, and the chimera level correlates with prostate cancer disease progression.
URLPMID:4136775 [本文引用: 1]
Abstract Chimeric RNAs originating from two or more different genes are known to exist not only in cancer, but also in normal tissues, where they can play a role in human evolution. However, the exact mechanism of their formation is unknown. Here, we use RNA sequencing data from 462 healthy individuals representing 5 human populations to systematically identify and in depth characterize 81 RNA tandem chimeric transcripts, 13 of which are novel. We observe that 6 out of these 81 chimeras have been regarded as cancer-specific. Moreover, we show that a prevalence of long introns at the fusion breakpoint is associated with the chimeric transcripts formation. We also find that tandem RNA chimeras have lower abundances as compared to their partner genes. Finally, by combining our results with genomic data from the same individuals we uncover intronic genetic variants associated with the chimeric RNA formation. Taken together our findings provide an important insight into the chimeric transcripts formation and open new avenues of research into the role of intronic genetic variants in post-transcriptional processing events.
URLPMID:4667522 [本文引用: 1]
Fusion transcripts are found in many tissues and have the potential to create novel functional products. Here, we investigate the genomic sequences around fusion junctions to better understand the transcriptional mechanisms mediating fusion transcription/splicing. We analyzed data from prostate (cancer) cells as02previous studies have02shown02extensively that these cells readily undergo fusion transcription.We used the FusionMap program to identify high-confidence fusion transcripts from RNAseq data. The RNAseq datasets were from our (N65=658) and other (N65=6514) clinical prostate tumors with adjacent non-cancer cells, and from the LNCaP prostate cancer cell line that were mock-, androgen- (DHT), and anti-androgen- (bicalutamide, enzalutamide) treated. In total, 185 fusion transcripts were identified from all RNAseq datasets. The majority (7602%) of these fusion transcripts were 'read-through chimeras' derived from adjacent genes in the genome. Characterization of sequences at fusion loci were carried out using a combination of the FusionMap program, custom Perl scripts, and the RNAfold program. Our computational analysis indicated that most fusion junctions (7602%) use the consensus GT-AG intron donor-acceptor splice site, and most fusion transcripts (8502%) maintained the open reading frame. We assessed whether parental genes of fusion transcripts have the potential to form complementary base pairing between parental genes which might bring them into physical proximity. Our computational analysis of sequences flanking fusion junctions at parental loci indicate that these loci have a similar propensity as non-fusion loci to hybridize. The abundance of repetitive sequences at fusion and non-fusion loci was also investigated given that SINE repeats are involved in aberrant gene transcription. We found few instances of repetitive sequences at both fusion and non-fusion junctions. Finally, RT-qPCR was performed on RNA from both clinical prostate tumors and adjacent non-cancer cells (N65=657), and LNCaP cells treated as above to validate the expression of seven fusion transcripts and their respective parental genes. We reveal that fusion transcript expression is similar to the expression of parental genes.Fusion transcripts maintain the open reading frame, and likely use the same transcriptional machinery as non-fusion transcripts as they share many genomic features at splice/fusion junctions.
URLPMID:22231539 [本文引用: 9]
Recently, conjoined genes (CGs) have emerged as important genetic factors necessary for understanding the human genome. However, their formation mechanism and precise structures have remained mysterious. Based on a detailed structural analysis of 57 human CG transcript variants (CGTVs, discovered in this study) and all (833) known CGs in the human genome, we discovered that the poly(A) signal site from the upstream parent gene region is completely removed via the skipping or truncation of the final exon; consequently, CG transcription is terminated at the poly(A) signal site of the downstream parent gene. This result led us to propose a novel mechanism of CG formation: the complete removal of the poly(A) signal site from the upstream parent gene is a prerequisite for the CG transcriptional machinery to continue transcribing uninterrupted into the intergenic region and downstream parent gene. The removal of the poly(A) signal sequence from the upstream gene region appears to be caused by a deletion or truncation mutation in the human genome rather than post-transcriptional trans-splicing events. With respect to the characteristics of CG sequence structures, we found that intergenic regions are hot spots for novel exon creation during CGTV formation and that exons farther from the intergenic regions are more highly conserved in the CGTVs. Interestingly, many novel exons newly created within the intergenic and intragenic regions originated from transposable element sequences. Additionally, the CGTVs showed tumor tissue-biased expression. In conclusion, our study provides novel insights into the CG formation mechanism and expands the present concepts of the genetic structural landscape, gene regulation, and gene formation mechanisms in the human genome.
[本文引用: 1]
URLPMID:11753382 [本文引用: 1]
Nat Genet. 2002 Jan;30(1):13-9. Research Support, U.S. Gov't, Non-P.H.S.
URLPMID:18978789 [本文引用: 1]
Abstract We carried out the first analysis of alternative splicing complexity in human tissues using mRNA-Seq data. New splice junctions were detected in approximately 20% of multiexon genes, many of which are tissue specific. By combining mRNA-Seq and EST-cDNA sequence data, we estimate that transcripts from approximately 95% of multiexon genes undergo alternative splicing and that there are approximately 100,000 intermediate- to high-abundance alternative splicing events in major human tissues. From a comparison with quantitative alternative splicing microarray profiling data, we also show that mRNA-Seq data provide reliable measurements for exon inclusion levels.
URLPMID:2593745 [本文引用: 1]
Abstract Through alternative processing of pre-messenger RNAs, individual mammalian genes often produce multiple mRNA and protein isoforms that may have related, distinct or even opposing functions. Here we report an in-depth analysis of 15 diverse human tissue and cell line transcriptomes on the basis of deep sequencing of complementary DNA fragments, yielding a digital inventory of gene and mRNA isoform expression. Analyses in which sequence reads are mapped to exon-exon junctions indicated that 92-94% of human genes undergo alternative splicing, 86% with a minor isoform frequency of 15% or more. Differences in isoform-specific read densities indicated that most alternative splicing and alternative cleavage and polyadenylation events vary between tissues, whereas variation between individuals was approximately twofold to threefold less common. Extreme or 'switch-like' regulation of splicing between tissues was associated with increased sequence conservation in regulatory regions and with generation of full-length open reading frames. Patterns of alternative splicing and alternative cleavage and polyadenylation were strongly correlated across tissues, suggesting coordinated regulation of these processes, and sequence conservation of a subset of known regulatory motifs in both alternative introns and 3' untranslated regions suggested common involvement of specific factors in tissue-level regulation of both splicing and polyadenylation.
[本文引用: 2]
[本文引用: 2]
URLPMID:14759258 [本文引用: 1]
Background Nonsense-mediated mRNA decay (NMD) is a eukaryotic mRNA surveillance mechanism that detects and degrades mRNAs with premature termination codons (PTC + mRNAs). In mammals, a termination codon is recognized as premature if it lies more than about 50 nucleotides upstream of the final intron position. More than a third of reliably inferred alternative splicing events in humans have been shown to result in PTC + mRNA isoforms. As the mechanistic details of NMD have only recently been elucidated, we hypothesized that many PTC + isoforms may have been cloned, characterized and deposited in the public databases, even though they would be targeted for degradation in vivo . Results We analyzed the human alternative protein isoforms described in the SWISS-PROT database and found that 144 (5.8% of 2,483) isoform sequences amenable to analysis, from 107 (7.9% of 1,363) SWISS-PROT entries, derive from PTC + mRNA. Conclusions For several of the PTC + isoforms we identified, existing experimental evidence can be reinterpreted and is consistent with the action of NMD to degrade the transcripts. Several genes with mRNA isoforms that we identified as PTC + - calpain-10, the CDC-like kinases (CLKs) and LARD - show how previous experimental results may be understood in light of NMD.
URLPMID:12824192 [本文引用: 2]
Classic Hodgkin's lymphoma (HL) tissue contains a small population of morphologically distinct malignant cells called Hodgkin and Reed-Sternberg (HRS) cells, associated with the development of HL. Using 3'-rapid amplification of cDNA ends (RACE) we identified an alternative mRNA for the DEC-205 multilectin receptor in the HRS cell line L428. Sequence analysis revealed that the mRNA encodes a fusion protein between DEC-205 and a novel C-type lectin DCL-1. Although the 7.5-kb DEC-205 and 4.2-kb DCL-1 mRNA were expressed independently in myeloid and B lymphoid cell lines, the DEC-205/DCL-1 fusion mRNA (9.5 kb) predominated in the HRS cell lines (L428, KM-H2, and HDLM-2). The DEC-205 and DCL-1 genes comprising 35 and 6 exons, respectively, are juxtaposed on chromosome band 2q24 and separated by only 5.4 kb. We determined the DCL-1 transcription initiation site within the intervening sequence by 5'-RACE, confirming that DCL-1 is an independent gene. Two DEC-205/DCL-1 fusion mRNA variants may result from cotranscription of DEC-205 and DCL-1, followed by splicing DEC-205 exon 35 or 34-35 along with DCL-1 exon 1. The resulting reading frames encode the DEC-205 ectodomain plus the DCL-1 ectodomain, the transmembrane, and the cytoplasmic domain. Using DCL-1 cytoplasmic domain-specific polyclonal and DEC-205 monoclonal antibodies for immunoprecipitation/Western blot analysis, we showed that the fusion mRNA is translated into a DEC-205/DCL-1 fusion protein, expressed in the HRS cell lines. These results imply an unusual transcriptional control mechanism in HRS cells, which cotranscribe an mRNA containing DEC-205 and DCL-1 prior to generating the intergenically spliced mRNA to produce a DEC-205/DCL-1 fusion protein.
URLPMID:5458218 [本文引用: 2]
Traditional gene fusions are involved in the development of various neoplasia.DUS4L-BCAP29, a chimeric fusion RNA, has been reported to be a cancer-fusion in prostate and gastric cancer, in addition to playing a tumorigenic role. Here, we showed that theDUS4L-BCAP29fusion transcript exists in a variety of normal tissues. It is also present in non-cancer epithelial, as well as in fibroblast cell lines. Quantitatively, the fusion transcript has a comparable expression in non-cancerous, gastric and prostate cell lines and tissues as in the cancer cell lines and tissues. The loss-of-function approach as previously reported is not sufficient to prove the functionality of the fusion. On the other hand, the gain-of-function approach showed that overexpression ofDUS4L-BCAP29promotes cell growth and motility, even in non-cancer cells. Finally, we provide further evidence that the fusion transcript is a product of cis-splicing between adjacent genes. In summary, we believe that in contrast to traditional gene fusions,DUS4L-BCAP29cannot be used as a cancer biomarker. Instead, it is a fusion transcript that exists in normal physiology and that its pro-growth effect is not unique to cancer cells.
URLPMID:28716526 [本文引用: 2]
Abstract Gene fusions in cancer typically lead to the expression of a fusion protein or disrupt the expression of one of the parental genes. Here we report a new phenomenon whereby a fusion transcript functions as a long non-coding chimeric RNA (lnccRNA). This fusion RNA, SLC45A3-ELK4, generated by cis-splicing between neighboring genes, was found in prostate cancer. The fusion RNA encodes the same protein as ELK4. Intriguingly, we found that the fusion RNA level is less than 1% of wild type ELK4, unlikely to perturb the general pool of ELK4 protein. Nonetheless, when the fusion RNA, but not ELK4 is silenced, cell proliferation is inhibited in both androgen-dependent and castration-resistant prostate cancer cells. This growth arrest can be rescued by exogenous expression of the fusion and a mutant designed to prevent translation of the ELK4 protein. In the same setting, the mutant could also suppress CDKN1A and several other targets of SLC45A3-ELK4. In addition, similar to many long non-coding RNAs, the fusion RNA is enriched in the nuclear fraction. Altogether, these results indicate that SLC45A3-ELK4 regulates cancer cell proliferation by its transcript, not translated protein. Copyright 脗漏 2017 Elsevier B.V. All rights reserved.
URLPMID:15289480 [本文引用: 1]
Abstract It is estimated that between 35% and 74% of all human genes can undergo alternative splicing. Currently, the most efficient methods for large-scale detection of alternative splicing use expressed sequence tags (ESTs) or microarray analysis. As these methods merely sample the transcriptome, splice variants that do not appear in deeply sampled tissues have a low probability of being detected. We present a new method by which we can predict that an internal exon is skipped (namely whether it is a cassette-exon) merely based on its naked genomic sequence and on the sequence of its mouse ortholog. No other data, such as ESTs, are required for the prediction. Using our method, which was experimentally validated, we detected hundreds of novel splice variants that were not detectable using ESTs. We show that a substantial fraction of the splice variants in the human genome could not be identified through current human EST or cDNA data. Copyright 2004 Cold Spring Harbor Laboratory Press ISSN
URLPMID:16344564 [本文引用: 1]
An international, peer-reviewed genome sciences journal featuring outstanding original research that offers novel insights into the biology of all organisms
URLPMID:21571633 [本文引用: 1]
Transcription-induced chimeric RNAs, possessing sequences from different genes, are expected to increase the proteomic diversity through chimeric proteins or altered regulation. Despite their importance, few studies have focused on chimeric RNAs especially regarding their presence/roles in human cancers. By deep sequencing the transcriptome of 20 human prostate cancer and 10 matched benign prostate tissues, we obtained 1.3 billion sequence reads, which led to the identification of 2,369 chimeric RNA candidates. Chimeric RNAs occurred in significantly higher frequency in cancer than in matched benign samples. Experimental investigation of a selected 46 set led to the confirmation of 32 chimeric RNAs, of which 27 were highly recurrent and previously undescribed in prostate cancer. Importantly, a subset of these chimeras was present in prostate cancer cell lines, but not detectable in primary human prostate epithelium cells, implying their associations with cancer. These chimeras contain discernable 5' and 3' splice sites at the RNA junction, indicating that their formation is mediated by splicing. Their presence is also largely independent of the expression of parental genes, suggesting that other factors are involved in their production and regulation. One chimera, TMEM79-SMG5, is highly differentially expressed in human cancer samples and therefore a potential biomarker. The prevalence of chimeric RNAs may allow the limited number of human genes to encode a substantially larger number of RNAs and proteins, forming an additional layer of cellular complexity. Together, our results suggest that chimeric RNAs are widespread, and increased chimeric RNA events could represent a unique class of molecular alteration in cancer.
[本文引用: 2]
URLPMID:26898937 [本文引用: 1]
Urothelial carcinoma (UC) is the most common type of bladder cancer and is the second most frequently diagnosed genitourinary tumor. The identification of fusion genes in bladder cancer might provide new perspectives for its classification and significance. In this study, we present a thorough search on three UC samples for novel fusion transcripts in bladder cancer using high-throughput RNA sequencing. We used stringent requirements for 819 fusion candidates and nominated 10 candidate fusion transcripts. Among them four novel fusion genesSEPT9/CYHR,IGF1R/TTC23, SYT8/TNNI2andCASZ1/DFFAwere validated and characterized in 48 formalin-fixed paraffin-embedded (FFPE) specimens of bladder cancer.Chromosomal rearrangements of regions 17q25, 15q26.3 and 1p36.22 resulting in the fusion transcriptsSEPT9/CYHR, IGF1R/TTC23andCASZ1/DFFA,appeared to be rare or unique events because they were not detected in the 48 UC samples. In contrast, theSYT8/TNNI2fusion transcript resulting from transcription-induced chimerism by read-through mechanisms was a rather common and tumor-specific event occurring in 37.5% (18/48) of the UC specimens. Further investigation of functional and clinical relevance of novel fusion genes remains to be elucidated to reveal their role in bladder carcinogenesis.
URL [本文引用: 1]
URLPMID:21376230 [本文引用: 1]
Cell. 2011 Mar 4;144(5):646-74. doi: 10.1016/j.cell.2011.02.013. Research Support, N.I.H., Extramural; Review
URLPMID:25239642 [本文引用: 2]
Abstract Gene fusion is among the primary processes that generate new genes and has been well characterized as potent pathway of oncogenesis. Here, by high-throughput RNA sequencing in nine paired human endometrial carcinoma (EC) and matched non-cancerous tissues, we obtained that chimeric translin-associated factor X-disrupted-in-schizophrenia 1 (TSNAX-DISC1) occurred significantly upregulated in multiple EC samples. Experimental investigation showed that TSNAX-DISC1 appears to be formed by splicing without chromosomal rearrangement. The chimera expression inversely correlated with the binding of CCCTC-binding factor (CTCF) to the insulators. Subsequent investigations indicate that long intergenic non-coding RNA lincRNA-NR_034037, separating TSNAX from DISC1, regulates TSNAX -DISC1 production and TSNAX/DISC1 expression levels by extricating CTCF from insulators. Dysregulation of TSNAX influences steroidogenic factor-1-stimulated transcription on the StAR promoter, altering progesterone actions, implying the association with cancer. Together, these results advance our understanding of the mechanism in which lincRNA-NR_034037 regulates TSNAX-DISC1 formation programs that tightly regulate EC development. 漏 The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
URLPMID:24240688 [本文引用: 1]
Gene fusion is involved in the development of various types of malignancies. Recent advances in sequencing technology have facilitated identification of gene fusions and have stimulated the research of this field in cancer. In the present study, we performed next-generation transcriptome sequencing in order to discover novel gene fusions in gastric cancer. A total of 282 fusion transcript candidates were detected from 12 gastric cancer cell lines by bioinformatic filtering. Among the candidates, we have validated 19 fusion transcripts, which are 7 inter-chromosomal and 12 intra-chromosomal fusions. A novel DUS4L-BCAP29 fusion transcript was found in 2 out of 12 cell lines and 10 out of 13 gastric cancer tissues. Knockdown of DUS4L-BCAP29 transcript using siRNA inhibited cell proliferation. Soft agar assay further confirmed that this novel fusion transcript has tumorigenic potential. We also identified that microRNA-coding gene PVT1, which is amplified in double minute chromosomes in SNU-16 cells, is recurrently involved in gene fusion. PVT1 produced six different fusion transcripts involving four different genes as fusion partners. Our findings provide better insight into transcriptional and genetic alterations of gastric cancer: namely, the tumorigenic effects of transcriptional read-through and a candidate region for genetic instability.
URLPMID:17442748 [本文引用: 1]
Conserved noncoding elements (CNEs) constitute the majority of sequences under purifying selection in the human genome, yet their function remains largely unknown. Experimental evidence suggests that many of these elements play regulatory roles, but little is known about regulatory motifs contained within them. Here we describe a systematic approach to discover and characterize regulatory motifs within mammalian CNEs by searching for long motifs (12-22 nt) with significant enrichment in CNEs and studying their biochemical and genomic properties. Our analysis identifies 233 long motifs (LMs), matching a total of approximately 60,000 conserved instances across the human genome. These motifs include 16 previously known regulatory elements, such as the histone 3'-UTR motif and the neuron-restrictive silencer element, as well as striking examples of novel functional elements. The most highly enriched motif (LM1) corresponds to the X-box motif known from yeast and nematode. We show that it is bound by the RFX1 protein and identify thousands of conserved motif instances, suggesting a broad role for the RFX family in gene regulation. A second group of motifs (LM2*) does not match any previously known motif. We demonstrate by biochemical and computational methods that it defines a binding site for the CTCF protein, which is involved in insulator function to limit the spread of gene activation. We identify nearly 15,000 conserved sites that likely serve as insulators, and we show that nearby genes separated by predicted CTCF sites show markedly reduced correlation in gene expression. These sites may thus partition the human genome into domains of expression.
URLPMID:16117655 [本文引用: 1]
Felsenfeld G(1), Burgess-Beusse B, Farrell C, Gaszner M, Ghirlando R, Huang S, Jin C, Litt M, Magdinier F, Mutskov V, Nakatani Y, Tagami H, West A, Yusufzai T.
URLPMID:17512414 [本文引用: 1]
Abstract Histone modifications are implicated in influencing gene expression. We have generated high-resolution maps for the genome-wide distribution of 20 histone lysine and arginine methylations as well as histone variant H2A.Z, RNA polymerase II, and the insulator binding protein CTCF across the human genome using the Solexa 1G sequencing technology. Typical patterns of histone methylations exhibited at promoters, insulators, enhancers, and transcribed regions are identified. The monomethylations of H3K27, H3K9, H4K20, H3K79, and H2BK5 are all linked to gene activation, whereas trimethylations of H3K27, H3K9, and H3K79 are linked to repression. H2A.Z associates with functional regulatory elements, and CTCF marks boundaries of histone methylation domains. Chromosome banding patterns are correlated with unique patterns of histone modifications. Chromosome breakpoints detected in T cell cancers frequently reside in chromatin regions associated with H3K4 methylations. Our data provide new insights into the function of histone methylation and chromatin organization in genome function.
[本文引用: 2]
URLPMID:11909521 [本文引用: 1]
Abstract The messenger RNA processing reactions of capping, splicing, and polyadenylation occur cotranscriptionally. They not only influence one another's efficiency and specificity, but are also coordinated by transcription. The phosphorylated CTD of RNA polymerase II provides key molecular contacts with these mRNA processing reactions throughout transcriptional elongation and termination.
URLPMID:15647503 [本文引用: 1]
mRNA polyadenylation is a critical cellular process in eukaryotes. It involves 3' end cleavage of nascent mRNAs and addition of the poly(A) tail, which plays important roles in many aspects of the cellular metabolism of mRNA. The process is controlled by various cis-acting elements surrounding the cleavage site, and their binding factors. In this study, we surveyed genome regions containing cleavage sites [herein called poly(A) sites], for 13,942 human and 11,155 mouse genes. We found that a great proportion of human and mouse genes have alternative polyadenylation ( approximately 54 and 32%, respectively). The conservation of alternative polyadenylation type or polyadenylation configuration between human and mouse orthologs is statistically significant, indicating that alternative polyadenylation is widely employed by these two species to produce alternative gene transcripts. Genes belonging to several functional groups, indicated by their Gene Ontology annotations, are biased with respect to polyadenylation configuration. Many poly(A) sites harbor multiple cleavage sites (51.25% human and 46.97% mouse sites), leading to heterogeneous 3' end formation for transcripts. This implies that the cleavage process of polyadenylation is largely imprecise. Different types of poly(A) sites, with regard to their relative locations in a gene, are found to have distinct nucleotide composition in surrounding genomic regions. This large-scale study provides important insights into the mechanism of polyadenylation in mammalian species and represents a genomic view of the regulation of gene expression by alternative polyadenylation.
URLPMID:15741508 [本文引用: 1]
Alternative initiation, splicing, and polyadenylation are key mechanisms used by many organisms to generate diversity among mature mRNA transcripts originating from the same transcription unit. While previous computational analyses of alternative polyadenylation have focused on polyadenylation activities within or downstream of the normal 3'-terminal exons, we present the results of the first genome-wide analysis of patterns of alternative polyadenylation in the human, mouse, and rat genomes occurring over the entire transcribed regions of mRNAs using 3'-ESTs with poly(A) tails aligned to genomic sequences. Four distinct classes of patterns of alternative polyadenylation result from this analysis: tandem poly(A) sites, composite exons, hidden exons, and truncated exons. We estimate that at least 49% (human), 31% (mouse), and 28% (rat) of polyadenylated transcription units have alternative polyadenylation. A portion of these alternative polyadenylation events result in new protein isoforms.
URLPMID:18817380 [本文引用: 1]
Regulation of gene expression by RNA processing mechanisms is now understood to be an important level of control in mammalian cells. Regulation at the level of RNA transcription, splicing, polyadenylation, nucleo-cytoplasmic transport, and translation into polypeptides has been well-studied. Alternative RNA processing events, such as alternative splicing, also have been recognized as key contributors to the complexity of mammalian gene expression. Pre-messenger RNAs (pre-mRNAs) may be polyadenylated in several different ways due to more than one polyadenylation signal, allowing a single gene to encode multiple mRNA transcripts. However, alternative polyadenylation has only recently taken the field as a major player in gene regulation. This review summarizes what is currently known about alternative polyadenylation. It covers results from bioinformatics, as well as those from investigations of viral and tissue-specific studies and, importantly, will set the stage for what is yet to come.
URLPMID:1414089 [本文引用: 1]
Bioinformatic analyses of the occurrence and mechanism of alternative polyadenylation in different human tissues reveals systematic differences among tissues and suggests the involvement of bothtrans- andcis-regulatory elements. Alternative polyadenylation is one of the mechanisms in human cells that give rise to a variety of transcripts from a single gene. More than half of the human genes have multiple polyadenylation sites (poly(A) sites), leading to variable mRNA and protein products. Previous studies of individual genes have indicated that alternative polyadenylation could occur in a tissue-specific manner. We set out to systematically investigate the occurrence and mechanism of alternative polyadenylation in different human tissues using bioinformatic approaches. Using expressed sequence tag (EST) data, we investigated 42 distinct tissue types. We found that several tissues tend to use poly(A) sites that are biased toward certain locations of a gene, such as sites located in introns or internal exons, and various sites in the exon located closest to the 3' end. We also identified several tissues, including eye, retina and placenta, that tend to use poly(A) sites not frequently used in other tissues. By exploring microarray expression data, we analyzed over 20 genes whose protein products are involved in the process or regulation of mRNA polyadenylation. Several brain tissues showed high concordance of gene expression of these genes with each other, but low concordance with other tissue types. By comparing genomic regions surrounding poly(A) sites preferentially used in brain tissues with those in other tissues, we identified severalcis-regulatory elements that were significantly associated with brain-specific poly(A) sites. Our results indicate that there are systematic differences in poly(A) site usage among human tissues, and bothtrans-acting factors andcis-regulatory elements may be involved in regulating alternative polyadenylation in different tissues.
URLPMID:26190259 [本文引用: 1]
Vilborg et al. describe stress-induced transcripts generated by readthrough downstream of protein-coding genes (DoGs). DoGs are regulated by IP3 receptor signaling and remain chromatin-bound. Being long (often >45 kb) and diverse (>2,000 species), DoGs contribute significantly to the human transcriptome.
[本文引用: 1]
URLPMID:11389087 [本文引用: 1]
Microsatellite (MS) instability occurs in tumors with DNA mismatch repair (MMR) deficiencies but is typically absent in adjacent normal tissue. However, MS mutations have been observed in normal tissues from rare individuals with congenital MMR deficiencies. Autopsy tissues from a 4-year-old with congenital MMR deficiency (MLH1-/-) were examined for MS mutations. Insertions and deletions were observed in CA-repeat MS loci. Approximately 0.26 to 1.4 mutations per MS locus per cell were estimated to be present in normal heart, lymph node, kidney, and bladder epithelium. These findings illustrate that phenotypically normal MMR-deficient cells commonly accumulate MS mutations. Loss of MMR and the accumulation of some MS mutations may occur early in MMR-deficient tumor progression, even before a gatekeeper mutation.
[本文引用: 1]
URLPMID:2725402 [本文引用: 1]
Abstract Recurrent gene fusions, typically associated with haematological malignancies and rare bone and soft-tissue tumours, have recently been described in common solid tumours. Here we use an integrative analysis of high-throughput long- and short-read transcriptome sequencing of cancer cells to discover novel gene fusions. As a proof of concept, we successfully used integrative transcriptome sequencing to 're-discover' the BCR-ABL1 (ref. 10) gene fusion in a chronic myelogenous leukaemia cell line and the TMPRSS2-ERG gene fusion in a prostate cancer cell line and tissues. Additionally, we nominated, and experimentally validated, novel gene fusions resulting in chimaeric transcripts in cancer cell lines and tumours. Taken together, this study establishes a robust pipeline for the discovery of novel gene chimaeras using high-throughput sequencing, opening up an important class of cancer-related mutations for comprehensive characterization.

{kind=link}
{kind=link}