 ,
, ,1,2,*
,1,2,*Techniques and methods of microbiomics and their applications
Gui-Feng GAO1, Hai-Yan CHU,,1,2,*通讯作者: * 褚海燕: ORCID: 0000-0001-9004-8750,hychu@issas.ac.cn
编委: 温学发
责任编辑: 赵航
收稿日期:2019-08-19接受日期:2019-10-23网络出版日期:2020-04-20
| 基金资助: |
Corresponding authors: * ORCID: 0000-0001-9004-8750,hychu@issas.ac.cn
Received:2019-08-19Accepted:2019-10-23Online:2020-04-20
| Fund supported: |

摘要
微生物组是指一个特定环境或生态系统中全部微生物及其遗传信息的集合, 其蕴藏着极为丰富的微生物资源。全面系统地解析微生物组的结构和功能, 将为解决人类面临的能源、生态环境、工农业生产和人体健康等重大问题带来新思路。然而, 微生物组学研究在很大程度上取决于其技术与方法的发展。在高通量测序技术出现以前, 微生物研究主要基于分离培养和指纹图谱等技术, 然而, 由于这些技术存在的缺陷, 人们对于微生物的认识十分有限。自21世纪初以来, 尽管高通量测序和质谱技术的革命性突破极大地促进了人们对于微生物的认识, 微生物组学技术在微生物组研究中的应用仍面临着诸多挑战。此外, 目前微生物组的结构和多样性等描述性研究已臻成熟, 微生物组学研究正处于从数量到质量、从结构到功能的关键转变时期。因此, 该文首先介绍了微生物组学的基本概念及其发展简史, 其次简述了微生物组学研究的相关技术和方法及其发展历程, 并进一步阐述了微生物组学的技术和方法在生态学研究中的应用及存在的主要问题, 最后从技术、理论和应用层面阐述了未来微生物组学技术和方法发展的前沿方向, 并提出了今后微生物组学研究的优先发展领域。
关键词:
Abstract
Microbiome is the combination of all microorganisms and their genetic information in a specific environment or ecosystem, which contains abundant microbial resources. A comprehensive and systematic analysis of the structure and function of microbiome will provide new ideas in solving the core issues in the fields of energy, ecological environment, industrial and agricultural production and human health. However, the study of microbiome largely depends on the development of relevant technologies and methods. Before to the advent of high-throughput sequencing technology, microbial research was mainly based on techniques such as isolation, pure-culture and fingerprint. However, due to the technical restrictions, scientists could only get limited knowledge of microorganisms. Since the beginning of 21st century, the revolutionary advances in the technology of high-throughput sequencing and mass spectrometry have greatly improved our understanding on the structure and ecological functions of environmental microbiome. However, the application of microbiomics technology in microbial research still faces many challenges. In addition, the descriptive studies focusing on the structure and diversity of microbiome have already matured, and the study of microbiomics is facing a critical transition period from quantity to quality and from structure to function. Hence, this paper will firstly introduce the basic concepts of microbiomics and a brief development history. Secondly, this paper introduces the related technologies and methods of microbiomics with their development process, and further expounds the applications and main problems of microbiomics technologies and methods in ecological study. Finally, this paper expounds the frontier direction of the development of microbiomics technology and methods from the technical, theoretical and application levels, and proposes the priority development areas of microbiome research in the future.
Keywords:
PDF (1065KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
引用本文
高贵锋, 褚海燕. 微生物组学的技术和方法及其应用. 植物生态学报, 2020, 44(4): 395-408. DOI: 10.17521/cjpe.2019.0222
GAO Gui-Feng, CHU Hai-Yan.
微生物数量庞大、种类繁多, 是地球生物化学循环过程的驱动者, 是工农业生产、医药卫生和环境保护等关键领域的核心资源, 因此微生物组研究已成为新一轮科技革命的战略高地(朱永官等, 2017)。然而, 微生物并不是孤立存在的, 而是与其所处环境紧密联系, 其活性和生长在很大程度上受周围环境的影响, 包括生物环境和非生物环境(贺纪正等, 2014)。微生物与其所处环境共同构成了复杂的微生物组, 其在支撑生态系统过程和功能中发挥着不可替代的作用。为了充分发挥微生物组在各前沿领域的关键作用和巨大潜力, 深入理解微生物组的结构和功能至关重要。然而, 微生物组学研究的发展强烈依赖于微生物组学的技术和方法。
在20世纪80年代以前, 微生物研究主要基于微生物的分离培养, 但绝大多数微生物无法被分离培养, 因此, 这阶段人们对于微生物的认识基本停留在形态观察、分类及生理学研究。20世纪末期, DNA指纹图谱等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接在DNA水平对环境整体微生物群落进行分析, 开创了微生物分子生态学研究的新时代。然而, DNA指纹图谱只能靶标优势类群, 不能真实反映微生物的多样性及物种组成。自21世纪初以来, 高通量测序和质谱等技术的突破, 使得我们可以从DNA、RNA、蛋白质和代谢物等不同水平解析微生物组, 以获得更为全面的微生物组信息。然而, 目前各组学技术仍存在一定局限性, 比如, 现有测序技术均基于使用PCR的DNA扩增, 这将会导致有些序列可能被测了多次, 而有些量少的序列则无法被大量扩增, 同时PCR过程中可能会引入错配碱基, 从而造成信息的丢失。在宏代谢组分析中, 气相色谱-质谱联用(GC-MS)只能对其中的挥发性物质实现直接分析, 而对那些难挥发的物质分析效果不佳。在理论层面上, 现代生态学经过20世纪的发展已经累积了大量成熟的理论和模型, 而这些理论大都建立在宏观生态学的基础上, 这些理论和模型是否也适用于微生物领域, 目前还没有明确的结论, 仍需要更多的微生物组数据支撑。在技术应用上, 仅利用单一组学技术并不能完全揭示微生物组信息, 近年来多组学技术结合的研究策略更有利于全面解析微生物组(Deng et al., 2019)。因此, 如何解决各组学技术的缺陷, 充分发挥各组学技术的优势, 注重多组学技术的并行应用和多学科的交叉融合, 将是未来微生物组研究的必然趋势。不仅如此, 由于高通量测序较低的测序成本, 目前微生物组研究已积累了海量的测序数据, 微生物组研究正面临着从数量到质量、从结构到功能研究的关键转变过程。
本文将首先介绍微生物组学的基本概念及其发展简史; 其次, 简要阐述微生物组学技术的概念和内涵, 进一步从宏基因组、宏转录组、宏蛋白质组和宏代谢组四部分分别概述微生物组学的技术和方法及其发展历程; 此外, 本文将着重阐述微生物组学相关技术和方法在生态学研究中的应用及目前存在的主要问题; 最后, 本文将从技术和理论层面简要阐述未来微生物组学技术和方法的发展方向, 强调多组学结合与多学科交叉在微生物组学研究中的重要意义, 并进一步提出未来微生物组学研究的优先发展领域, 即微生物生物地理、土壤-植物-微生物互作和微生物组功能定向调控。
1 微生物组学的基本概念及其发展简史
微生物与其所处环境构成了复杂的生态系统, 是地球上生物多样性的重要组成部分。微生物组(microbiome)是指一个特定环境或生态系统中全部微生物及其遗传信息的集合, 其内涵包括了微生物与其环境和宿主的相互作用(刘双江等, 2017; 吴庆龙和江和龙, 2017)。微生物组蕴藏着极为丰富的微生物资源, 是工农业生产、医药卫生和环境保护等领域的核心资源(朱永官等, 2017)。微生物组学(microbiomics)是以微生物组为研究对象, 探究其内部群体间的相互关系、结构、功能及其与环境或宿主间相互关系的学科(刘双江等, 2017)。微生物学研究大致可分为三个阶段。第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段。第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代。值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(Sanger et al., 1977)。1998年, 威斯康辛大学的Handelsman等(1998)首次提出宏基因组的概念, 其研究对象为特定环境中的总DNA。宏转录组则兴起于宏基因组之后, 它以特定环境中的全部RNA为研究对象, 探究全部基因组的转录水平, Leininger等(2006)首次使用454焦磷酸测序技术对复杂微生物群落进行宏转录组研究。第三阶段: 从2006年开始, 高通量测序(第二代测序技术)和质谱技术的革命性突破以及生物信息学的快速发展极大推动了微生物组研究(Falkowski et al., 2008; Herbst et al., 2013; Zhong et al., 2017; Bahram et al., 2018; Kleiner et al., 2018)。根据Web of Science核心文集的检索结果, 出现关键词“microbiome”的文章数量正以指数型增长(图1A)。微生物之间以及微生物与其所处环境间的相互作用极为复杂, 通过宏基因组学结合宏转录组学以及新一代质谱技术催生下的宏蛋白质组学和宏代谢组学, 人们可以更全面、系统地解析微生物组的结构和功能。随着微生物组研究的发展, Egert等(2006)最早提出了“microbiomics”一词, 基于微生物组学相关技术和方法(目前主要为宏基因组学和宏代谢组学, 相比之下, 宏转录组学和宏蛋白组学仍处于起步阶段), 微生物组学研究开始进入了空前的发展时期(图1B)。目前, 各个国家、地区和组织正积极推进“微生物组计划” (表1), 研究对象涵括人体、土壤、海洋、大气等, 力求为解决21世纪人类面临的农业、能源、环境、海洋和气候等重大问题提供新的思路和途径。
图1
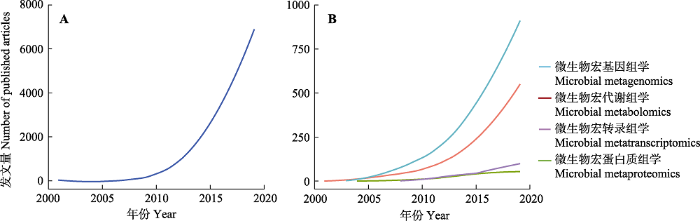 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1历年来发表的微生物组学相关文章数量趋势图。A, 包含关键词“微生物组”的文章数。B, 包含关键词“微生物”且分别包含“宏代谢组学”“宏蛋白质组学”“宏基因组学”“宏转录组学”的文章数量。数据来源于Web of Science核心文集(时间: 1900-2019年)。
Fig. 1Tendency of the published articles in microbiome over years. A, Number of articles with the keyword “microbiome”. B, Number of articles with the keyword “(‘microorganism*’ or ‘microbe*’ or ‘bacter*’ or ‘archae*’ or ‘fung*’)” and “metagenomics”, “metatranscriptomics”, “metaproteomics” and “metabolomics”, respectively. Data were collected from Web of Science Core Collection (Time: 1900-2019).
Table 1
表1
表1全球重大“微生物组计划”一览
Table 1
| 主导部门 Department | 启动时间 Starting year | 计划名称 Project name |
|---|---|---|
| 美国国立卫生研究院 US National institutes of Health | 2007 | 人体微生物组计划 The Human Microbiome Project |
| 美国阿贡国家实验室 Argonne National Laboratory | 2010 | 地球微生物组计划 Earth Microbiome Project |
| 巴西国家科学技术研究院 Instituto Nacional de Pesquisas Espaciais | 2013 | 巴西微生物组计划 The Brazilian Microbiome Project |
| 美国白宫科学和技术政策办公室与联邦机构、私营基金管理机构 Office of Science and Technology Policy, Federal Agency, Private Fund Management Agency | 2016 | 国家微生物组计划 National Microbiome Initiative |
| 南非和美国国际开发署 South Africa and United States Agency for International Development | 2016 | 非洲土壤微生物组计划 African Soil Microbiology Project |
| 中国科学院 Chinese Academy of Sciences | 2017 | 中国微生物组计划 China Microbiome Initiative |
新窗口打开|下载CSV
2 微生物组学的技术和方法及其发展
微生物组学技术是指不依赖于微生物培养, 而利用高通量测序和质谱鉴定等技术来研究微生物组的手段, 目前已被广泛应用于环境微生物组研究, 其研究对象包括土壤、水体、大气和人体等。目前, 微生物组学技术主要包括以下4种:(1)微生物宏基因组学(microbial metagenomics): 通过提取环境微生物的全部DNA, 研究其群落组成、遗传信息及其与所处环境的协同进化关系(Turnbaugh & Gordon, 2008)。
(2)微生物宏转录组学(microbial metatranscriptomics): 研究环境中全部微生物的转录组信息, 揭示相关基因在时空尺度上的表达水平, 从而对微生物群落的相关功能进行研究(Aguiar-Pulido et al., 2016)。
(3)微生物宏蛋白质组学(microbial metaproteomics): 定性和定量地分析环境微生物在特定环境条件和特定时间下的全部蛋白质组分(Wang et al., 2016)。
(4)微生物宏代谢组学(microbial metabolomics): 对微生物在特定生理时期内所有低分子量代谢物(包括代谢中间产物、激素、信号分子和次生代谢产物等)进行定性和定量分析, 并研究其与环境之间的相互作用(Tang, 2011)。
微生物组学技术在微生物组研究中的广泛应用主要得益于以下几点。
(1)测序技术的突破
在20世纪80年代以前, 人们对于微生物的认识主要基于微生物的分离培养, 但绝大多数微生物不可被分离培养。为了克服这个技术缺陷, 研究人员开展了大量工作, 比如延长培养时间和优化培养条件等, 以提高微生物培养效率, 但效果仍不理想。从20世纪80年代开始, 为了克服纯培养的缺陷, 研究人员开始利用微生物细胞内的DNA来评估微生物的种类和结构多样性, 这种在遗传分子水平来研究微生物群落的方法即为现代分子生物学方法, 主要包括DNA指纹图谱、基因芯片和稳定同位素探针等(刘国华等, 2012)。值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术(Sanger法)。Sanger法始于1977年, 其原理是基于核酸模板在复制或转录时双脱氧碱基会终止PCR的原理, 进一步利用凝胶电泳分离长度仅差一个碱基的核酸片段。使用该技术完成的噬菌体X174全长5 375个碱基的基因测序标志着微生物研究进入了基因组学时代(Sanger et al., 1977)。Sanger法测序具有读长长(1 000-1 500 bp)、准确度高等优点, 2001年完成的首个人类基因组图谱也是以Sanger法为基础, 目前该技术仍被用于获取高准确度的测序数据。然而, Sanger法每次只能测一条单独的序列, 导致其测序成本高, 无法大规模应用。
二代测序的原理是边合成边测序, 通过捕捉新合成的末端的标记来确定DNA的序列, 该技术在保持测序准确度的同时, 主要解决了Sanger法测序通量低的问题, 二代测序可同时对几万到几百万条DNA序列进行测序, 因此也被称为高通量测序。现有平台主要包括Illumina Hiseq、MiSeq、NovaSeq 6000等。二代测序的进步极大推动了微生物组学的发展, 2006年, Leininger等(2006)首次使用454焦磷酸测序技术对农田土壤氨氧化微生物群落进行了研究。迄今为止, 大规模的微生物组测序项目仍主要依赖于二代测序技术, 其被广泛应用于土壤、水体和沙漠等复杂环境样品中的土壤微生物群落分析(Bailly et al., 2007; Shi et al., 2009; Damon et al., 2012; Zhang et al., 2019)。然而, 二代测序也存在不足, 其测序片段被限制在了250-300 bp, 由于存在系统偏好性且建库过程中使用了PCR扩增, 将会导致有些序列可能被测了多次, 有些量少的序列则无法被大量扩增, 同时PCR过程中可能会引入错配碱基, 从而造成信息的丢失(Shendure & Ji, 2008)。
三代测序(单分子测序)的原理是通过现代光学、高分子和纳米技术等手段来区分碱基信号的差异, 直接读取DNA的序列信息, 从而解决了二代测序信息丢失和碱基错配等问题(Schadt et al., 2010)。该技术是一个新的里程碑, 它整合了一代和二代测序的优点, 即测序过程无需PCR扩增, 且具有高通量、长读长(10-15 kb)的特点, 但三代测序依赖于DNA聚合酶的活性, 错误率偏高(15%-40%), 幸运的是, 三代测序的错误是完全随机发生的, 因此可通过多次重复测序来纠正, 但这将增加测序成本。目前市场上以PacBio SMRT和Oxford Nanopore技术为主, 主要应用于复杂环境样品的基因组测序(Schloss et al., 2016)和单细胞基因组测序(Rice et al., 2016)。
(2)质谱技术的发展
微生物数量巨大、种类繁多, 尽管高通量测序技术的突破大大增加了人们对于微生物的认识, 但随着研究的深入, 人们意识到单从基因和转录水平并不能完全揭示微生物的奥秘, 并逐渐意识到蛋白质和代谢产物在微生物功能研究中的重要性。对复杂环境中的微生物群落功能研究一直是生物学研究的重点和难点, 长期以来的研究大多依赖于代谢产物或底物浓度的变化, 遗漏了绝大多数微生物, 难以真实反映微生物在复杂环境中的生理代谢信息(贺纪正等, 2014), 因此微生物多样性及其功能也常被认为是“灰箱”。质谱技术是宏蛋白质组学和宏代谢组学的核心技术, 通常结合色谱技术对复杂样品中的蛋白质或代谢物进行分析(White et al., 2016)。质谱分析是先将物质离子化, 然后按离子的质荷比(m/e)进行分 离, 最后测量各离子谱峰的强度而实现物质定性和定量分析, 是研究、分析和鉴定生物大分子的前沿方法(王桂友等, 2009; Herbst et al., 2013; Zhong et al., 2017; Kleiner et al., 2018)。传统的蛋白分析方法有凝集素亲和层析和Western蛋白印迹分析, 但一次实验仅能分析一到几个蛋白, 无法满足大规模分析的需求。20世纪70年代, 双向电泳的出现, 使得细胞系或细胞器的大量蛋白质能展示在双向凝胶上, 形成了狭义上的蛋白质组学概念, 该技术能对复杂蛋白质混合样品进行定性和定量分析, 但该技术对部分低丰度蛋白、极端等电点的蛋白以及一些疏水性蛋白分离效果不佳。近年来, 随着多重色谱分离与质谱联用技术的发展, 分离法在蛋白质组学分析中的应用日益广泛, 与传统双向凝胶电泳相比, 其分离效果要更优, 可以得到更为精确的蛋白质信息(White et al., 2016)。在过去十年间, 宏蛋白质组学由传统的双向凝胶电泳、蛋白质从头测序等发展为“鸟枪法”, 可同时进行蛋白质分离、鉴定和定量。与双向凝胶法相比, “鸟枪法”大大增加了蛋白质的覆盖率, 允许短时间内对上千个蛋白质进行高通量鉴定, 还可鉴定不溶性膜蛋白。
应用代谢组学对大量代谢物进行全面分析严重依赖于分离技术的发展。代谢组学常用的分析方法有气相色谱-质谱联用(GC-MS)、液相色谱-质谱联用(LC-MS)、核磁共振(NMR)和傅里叶转换红外光谱(FTIR)。其中, GC-MS、LC-MS和NMR是最常用的手段, 能在一次运行中检测多种代谢物。GC-MS是一种高分辨率的色谱分离方法, 广泛用于挥发性和半挥发性有机物的分析, 适用于检测初级代谢产物脂肪酸、碳水化合物等, 大多数基于质谱的宏代谢组学研究均使用GC-MS进行, 但GC-MS对那些难挥发的物质分析效果不佳。LC-MS则是检测非挥发性化合物最常用的方法, 样品无需衍生化, 可直接检测代谢物, 比如黄酮类化合物、生物碱等次级代谢物, 以及氨基酸、碳水化合物等初级代谢物, 是分离分析复杂有机混合物的有效手段。除了质谱技术, NMR无需大量的样品制备和分馏工作, 可迅速检测样品中最丰富的代谢物, 也可以检测出质谱难以电离的化合物, 但与质谱技术相比, 其灵敏性较差、覆盖率低(王琳和田璐, 2019)。迄今为止, 仍没有一种单一的分析方法能够全面覆盖样品的代谢组分, 每种方法各有优劣, 因此可结合使用, 相互补充。
近年来, 从单细胞水平上分析微生物的生理代谢的单细胞技术迅速发展, 其中单细胞成像技术可将检测结果可视化, 能较好反映环境微生物类群、丰度及其功能活性(Musat et al., 2012)。将超高分辨率显微成像技术和同位素示踪技术相结合的纳米二次离子质谱(NanoSIMS)在微生物生态学研究领域中显现出了巨大潜力, 该技术具有较高的灵敏度和准确度, 使得研究者不仅能够检测环境中低丰度但发挥重要作用的微生物类群, 还能从单细胞水平上提供微生物的生理生态特征, 这对于认识复杂环境中微生物介导的功能至关重要。目前, NanoSIMS技术在环境微生物研究中主要应用于碳氮硫元素生物地球化学循环的微生物驱动机制研究(Tourna et al., 2011; Huang et al., 2019)和土壤、微生物和植物互作关系研究(Gorka et al., 2019)。
(3)生物信息学的发展
生物信息学是伴随着人类基因组计划发展而来的学科, 包含了生物信息的获取、加工、存储、分配、分析、解释等在内的所有方面, 它综合运用数学、计算机科学和生物学的各种工具来阐明和理解大量数据所包含的生物学意义(赵屹等, 2012)。微生物组学研究依赖于生物信息学的发展, 使得后者成为微生物组学技术应用的一个主要的技术瓶颈, 可以说, 面对海量的数据, 没有生物信息学的参与, 研究工作将寸步难行(杨云锋, 2013)。而伴随着大规模测序的发展及数据积累程度的增加, 其重要性和难度也将随之增大(贺纪正等, 2014)。在发展过程中, 各微生物组学技术都已初步建立了各自的数据库、算法和软件(Breitwieser et al., 2019)。以扩增子测序为例, QIIME、Mothur和QIIME2是最常用的分析平台。2009年, 美国密歇根大学的Patrick D. Schloss教授团队发布了首个扩增子分析流程Mothur, 它整合了之前发表的操作分类单元(OTU)定义软件DOTUR、OTU差异比较工具SONS等, 从而实现了第一套较完整的分析流程(刘永鑫等, 2019)。2010年, 现美国加州大学圣地亚哥分校的Rob Knight团队发布了QIIME分析流程。QIIME的优势主要在于它整合了200多款相关软件和包, 使用者可以有更多软件和方法选择, 提供了150多个脚本, 可实现多种个性化分析, 增强统计和可视化, 实现多样性、物种组成、差异比较和网络等方法和出版级图表绘制, 因此QIIME大受研究者的青睐(刘永鑫等, 2019)。为了满足日益增长的测序数据和可重复分析的要求, Gregory J. Caporaso教授于2016年发起了基于Python 3从头编写的QIIME2, 该平台实现了分析流程的可追溯, 同时整合了一系列新算法, 如物种分类新方法2-feature-classifier和序列质量控制DADA2等, 极大提高了分析流程的适用范围和易用性。近年来, 也不断涌现出一些在线的数据分析平台, 如SILVAngs、MicrobiomeAnalyst、Qiita、MetaPhlAn2和gcMeta等。不仅如此, 相比扩增子测序, 宏基因组测序成本相对较高, 为此, 研究人员开发了一些可基于扩增子序列进行微生物组功能和表型预测的软件, 如PICRUSt、FAPROTAX和BugBase等。可以说, 高通量测序的进步推动了生物信息学的发展, 反之, 生物信息学的发展使得海量测序数据的序列比对、分析、拼接、功能注释、统计学检验和可视化等各种分析成为 可能。
3 微生物组学技术在生态学研究中的应用及存在问题
3.1 微生物组学技术在生态学研究中的应用
3.1.1 微生物组学技术在微生物群落研究中的应用微生物组学技术被广泛应用于揭示复杂环境中的微生物群落(包括多样性和群落组成)及其与周围环境或宿主之间的关系, 其研究对象涵盖土壤(Tedersoo et al., 2014; Thompson et al., 2017)、海洋(Sogin et al., 2006; Moran, 2015)和冰川(Rime et al., 2015; Anesio et al., 2017)等, 主要包括以下内容:
(1)微生物生物地理。生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(Martiny et al., 2006)。之前普遍认为微生物的分布不同于动植物, 不具有明显的地带性和区域分布特征, 而是呈全球性随机分布(O’ Malley, 2007)。事实上, 通过构建距离-衰减关系和物种面积曲线, 发现微生物物种并非随机分布(Horner-Devine et al., 2004b), 不同生境类型的微生物群落存在显著差异。因此, 明确微生物的分布规律, 对生态学和生物地理学的发展以及微生物资源的充分保护和利用具有重要意义。目前, 生物地理学家和生态学家将影响微生物物种丰富度格局的因素定义为两大类(即历史因素和当代环境因子), 通常这两类因子之间是相互影响的, 并与时间和空间尺度密切相关。以土壤为例, 通过整合全球189个样地的土壤宏基因组数据, Bahram等(2018)发现, 土壤细菌的物种和功能多样性在温带地区最高, 环境因子对微生物基因组成的影响大于地理距离。研究表明, 影响土壤微生物群落的环境因子主要包括土壤理化性质(Tripathi et al., 2018; Lupatini et al., 2019; Zhang et al., 2019), 植物生产力和系统发育(Yang et al., 2017, 2019)及气候条件(Zhang et al., 2016; Guo et al., 2018)等。
(2)微生物对全球变化的响应。微生物对环境变化的响应较为敏感, 是全球气候变化的调节器, 因此, 研究微生物组对于环境变化的响应及反馈具有重要意义, 其结果可为预测全球变化背景下生态系统结构和功能的演变提供理论依据。利用微生物组学技术, 可探究土壤微生物群落对全球气候变化(Guo et al., 2018; Raut et al., 2018), 土地利用方式改变(Li et al., 2019; Moreno et al., 2019)和农业管理措施转变(Xiang et al., 2017; Feng et al., 2018)等的响应。例如, 研究发现从亚马孙雨林到牛牧场的土地利用方式的转变对微生物多样性具有显著影响, 土壤细菌的局部分类和系统发育多样性在转化后增加, 但群落在空间上变得更加相似, 这种均质化是由有限范围的森林土壤细菌的丧失引起的, 并导致了多样性的净丧失(Rodrigues et al., 2013)。其次, 通过分析当前细菌分布与历史气候之间的联系, Ladau等(2018)的研究预测了未来在区域和大陆尺度上土壤微生物的分布, 如果细菌群落与现有气候条件相平衡, 则青藏高原和北美大部分(约75%)地区土壤中的细菌多样性将增加。
3.1.2 微生物组学技术在微生物功能研究中的应用
微生物参与了所有已知的生物地球化学循环过程, 其与植物生长、污染物降解和其他生态系统功能密切相关(Falkowski et al., 2008), 利用微生物组学技术, 已开展了以下研究:
(1)微生物生物地球化学循环。采用微生物宏转录组学可以探究微生物的相关功能, 发现大量的微生物资源及新的基因, 为探索微生物群落的生态功能奠定基础, 有助于生态系统功能和服务的调控(Gilbert et al., 2009)。例如, Poretsky等(2009)利用宏转录组学分析成功地从淡水和海洋浮游细菌中获得编码碳氮循环中重要功能蛋白的信使RNA。此外, Damon等(2012)通过对欧洲山毛榉(Fagus sylvatica)和挪威云杉(Picea abies)林下的土壤进行宏转录组学分析, 发现了129条代谢通路, 包括氮循环、糖代谢、磷酸盐代谢和有机质降解等。不仅如此, 宏蛋白质组学也被用于测定微生物群落的碳源及同化途径(Kleiner et al., 2018), 以及鉴定环境中参与元素生物地球化学循环及有机物氧化的潜力蛋白质(Bastida et al., 2014)。
(2)微生物与植物生长和健康。宏基因组学和宏代谢组学也被广泛用于探究微生物与宿主、环境之间的相互关系, 进而了解生物间相互作用的机制(Han et al., 2010; Wagner et al., 2016; Yang et al., 2017; ?apek et al., 2018)。例如, Shi等(2019a)通过扩增子高通量测序, 研究发现发病轻的马铃薯表土病原菌和细菌丰度较低、细菌多样性较高, 且菌群共存网络较复杂, 为进一步解析微生物群落功能与疮痂病发生的联系, 研究人员进一步对薯表土进行了宏基因组测序, 结果表明参与ABC transporters、bacterial secretion system、quorum sensing、nitrogen metabolism和一些cytochrome P450相关代谢途径的基因显著富集在重病薯表土中, 而一些抗生素合成相关代谢途径显著富集在轻病薯表土中, 该研究首次系统研究了土壤微生物组成和功能与疮痂病的关系, 对揭示疮痂病的发生机制和防治疮痂病具有重要意义。此外, 也有研究通过宏代谢组学分析, 发现丛枝菌根真菌的定殖改变了新疆千里光(Senecio jacobaea)的多种根际化合物组分(如吡咯啶生物碱), 从而起到关键的生物防御作用(Hill et al., 2018)。
(3)环境污染微生物修复。微生物组学技术在环境污染治理方面也表现出了巨大潜力, 包括有机污染物降解、重金属转化和生物污染等领域。例如, 微生物组学技术可用于研究环境中多环芳烃(PAHs)的微生物降解机制。首先, 可以先通过对受到PAHs污染的土壤进行宏基因组学研究, 鉴定能降解PAHs的微生物及其关键基因(Gutierrez et al., 2015)。其次, 通过比较宏蛋白质组学和宏代谢组学来挖掘相关蛋白和代谢物, 并对其进行鉴定和功能分析, 从而解析微生物降解PAHs的代谢通路和内在机理(Herbst et al., 2013; Zhong et al., 2017)。
3.2 微生物组学技术在生态学研究中存在的主要问题
3.2.1 样本制备从复杂环境中获得高保真性和高质量的样本是开展微生物组学研究的第一步。然而, 样品获取方法上的偏差仍是阻碍微生物组学研究的巨大挑战(Nesme et al., 2016)。环境微生物组学研究最常见的样本类型就是土壤, 其种类繁多、组分复杂、存在大量的抑制剂, 此时就需要根据不同样品的来源及特征采取不同的提取方法, 以获得高多样性和高质量的环境基因组(Jones et al., 2015; Poussin et al., 2018)。例如, 对于腐殖酸含量特别高的样品, 可以在DNA提取过程中增加使用异硫氰酸胍(Guanidine thiocyanate)洗涤硅珠的步骤(Antony-Babu et al., 2013)。针对低丰度的微生物、蛋白或代谢物等, 目前仍缺乏有效的富集和纯化技术, 这也是微生物组学研究面临的又一重大挑战。在蛋白质提取过程中, 直接对样品进行原位裂解可以获得较为完整的蛋白, 包括细胞蛋白和胞外蛋白, 但该方法会造成分类学上的困难, 同时由于土壤干扰物的存在, 该方法用于提取土壤蛋白还有较大技术难度(于仁涛等, 2009)。2007年, 研究人员报道了一种从土壤中进行蛋白分离的方法, 先使用0.1 mol·L-1 NaOH溶液从土壤中提取蛋白质、微生物和腐殖酸等, 然后进一步经过酚提取, 将蛋白质分离(Benndorf et al., 2007)。此外, 在样本分析前, 对获得的样本进行质量评估以确保达到分析要求尤为重要, 然而, 目前还没有统一的样本评估标准。
3.2.2 样本检测
基于标记基因的扩增子是大部分微生物组研究的基础, 但其样本检测步骤较多, 高通量测序读长较短、存在系统偏好性等问题, 且建库过程中使用了PCR扩增, 会导致某些序列可能被测了多次, 而有些量少的序列则无法被大量扩增, 不仅如此, PCR过程中可能会引入错配碱基, 从而造成信息的丢失。针对这些不足, 研究人员比较了不同文库制备方法, 利用标准样品详细分析扩增过程, 揭示了PCR扩增中造成错误和偏差的来源, 并提出了条件优化后的基于两步法的PCR扩增方法, 改进后的新方法能检测到更多现有方法中经常无法检测的类群, 同时提高微生物组研究的准确性(Gohl et al., 2016)。近年来, 三代测序(单分子测序)整合了一代和二代测序的优点, 测序过程无需进行PCR扩增方法, 从而解决了二代测序信息丢失和碱基错配等问题(Schadt et al., 2010), 在微生物组研究中显现出了巨大潜力, 但其错误率较高。尽管高通量测序在鉴定物种及丰度上很有用, 但它并不适合于准确地确定群落中物种的绝对丰度。近年来, 通过向样品中加入合成的嵌合体DNA作为内参, 可定量计算环境样品中微生物的绝对丰度。但利用该方法, 研究人员发现18S和ITS扩增子可能分别高估和低估了土壤中的霉菌数量(Tkacz et al., 2018)。因此, 实验前最好通过预实验来选择内参的加入量。
3.2.3 数据分析
参考数据库的发展是限制微生物组数据分析的重要因素。据估计, 因缺乏参考基因组数据, 测序所得的宏基因组序列, 有7%-60%无法被准确分类(Vilanova & Porcar, 2016; Thompson et al., 2017)。同样, 蛋白质的鉴定在很大程度上也取决于数据库, 其可信度受到数据库中存在的物种的限制(Wang et al., 2016)。目前各数据库管理标准不统一, 仍缺乏跨领域的数据整合(马俊才等, 2017)。
在数据处理上, 高通量测序数据的质量控制不是单一指标或操作即可完成的, 但目前还未建立统一、规范化的数据质量控制标准。目前比较成熟的序列拼接均基于一个或少数几个基因组为前提, 在面对海量数据和复杂的基因组时, 现有算法基本都无法满足需求。在微生物组学研究中, 大部分扩增子研究主要利用UCLUST或UPARSE等算法对97%相似性的序列进行OTU聚类, 然而不同的OTU聚类方法会对微生物组数据产生较大影响(Horner-Devine et al., 2004a)。近年来, 也出现了一些新的算法, 如DADA2和unoise3, 相当于基于100%的序列相似度进行聚类, 该方法大大提高了准确度和物种多样性。此外, 被广泛应用的OTU分类只能注释到属水平, 极少能鉴定到种水平, 这使得在某些特定微生物的功能研究上略显乏力, 因为较粗略的物种划分标准会导致可观测到的微生物空间分布格局减弱甚至消失(Horner-Devine et al., 2004a)。对于宏代谢组学来说, 其数据具有高维、小样本、高噪声、相互作用关系复杂和冗余性等特征, 如何从复杂的宏代谢组学数据中提取出有价值的信息, 筛选出潜在的生物标志物成为近年来研究的热点和难点。
4 研究展望
4.1 开发、整合新技术与新方法, 建设微生物组学大数据平台
微生物组学研究在很大程度上取决于微生物组学的技术和方法水平的高低, 随着微生物组学的发展, 新算法和计算平台层出不穷。然而, 不同组学技术各有优劣, 因此, 除了要发展新技术和新方法以应对不断产生的微生物数据, 更要注重各方法间的整合和互补, 在技术和方法的选择上应结合研究目的, 在条件允许的情况下, 对不同方法进行比较, 提高准确度和互补性。例如, 单细胞技术是21世纪最重要的新技术之一, 土壤中有90%以上的微生物功能尚未可知, 单细胞技术将能有效突破这一瓶颈, 为系统评价土壤微生物资源和定向挖掘微生物功能提供关键技术支撑。在数据分析方面, 微生物组学研究涉及海量数据的获取、统计分析和建模等, 因此, 生物信息学的发展显得尤为重要。只有进一步提升分析技术, 开发相适应的算法和软件, 将分析技术、数据处理、多元统计分析及可视化有机结合起来, 才能更好地推动微生物组学研究。Xu等(2017)指出, 在未来十年, 微生物组学数据分析将转变为微生物组数据科学。微生物组学大数据的收集、存储、功能挖掘和开发利用是制约微生物组学发展的核心问题(马俊才等, 2017)。在数据收集上, 应制定并执行标准化分析流程, 确保数据具有再现性、稳健性、可复制性和普遍性(Poussin et al., 2018; Schloss, 2018)。在数据分析过程中, 要进一步丰富相关参考数据库, 目前, 地球微生物组计划已经呼吁并鼓励世界各地的科学家按照协议共享数据(Gilbert et al., 2018; Langille et al., 2018)。近年来, 随着测序成本的降低, 微生物组学数据急剧增加, 但绝大多数数据都是一次性利用, 因此, 如何充分挖掘、利用现有的海量测序数据是微生物组学研究面临的另一个重要挑战。在微生物组学研究发展过程中, 应大力支持微生物组数据科学的发展, 进行有效的经费支持、制定有效的数据标准和实现互通, 建立对数据进行比较分析、整合并实现数据标准化的微生物组数据中心(Becher et al., 2013; Kyrpides et al., 2016)。目前, 我国也开发了部分微生物组学大数据平台, 如gcMeta, 其建立了一个微生物宏基因组和宏转录组的管理、在线分析、可视化及数据发布的一站式系统, 目前已整合了来自国际相关平台(NCBI、EBI等)重要项目(HMP、Tara等)超过12万个样本数据(Shi et al., 2019b)。未来还可进一步整合其他组学数据, 包括宏蛋白组学、宏代谢组学和单细胞测序等数据, 从而能更全面、系统地解析微生物组, 为我国微生物组学研究发展奠定坚实的基础。
4.2 多学科交叉与多组学结合
微生物组学的发展需要多学科交叉, 这种交叉是新技术、新方法的活水之源。微生物组学的信息分析最终目标是要阐明微生物群落组成、功能、结构, 以及群落与环境的相互作用, 所以如何有效地利用和挖掘微生物组学数据来建立分子生态的理论, 是微生物组信息分析过程中的重中之重。在这点上, 微生物组学研究需要与生态学相结合, 可以先借鉴宏观生态学已经建立起来的生态理论和模型, 运用于微观的微生物群落上, 然后通过改进这些理论和模型来理解和改造微生物群落, 从而为环境变化的预测提供依据。此外, 微生物不能独立于周围环境而存在, 因此, 微生物学和土壤物理、土壤化学等学科的交叉, 将更有利于系统解析微生物组结构、功能及其环境影响机制, 其所呈现出的系统生物学研究模式, 将有望成为未来生命科学领域令人激动的新前沿。微生物组学的发展也离不开多组学结合的研究策略, 包括微生物分离培养、生态学鉴定和代谢产物分析等。尽管每种组学方法都对微生物组学研究提供了有价值的信息, 但仅仅基于DNA测序的方法分析微生物群落的多样性和群落组成, 并不能很好地研究微生物组的功能特征(Emerson et al., 2017)。倘若将多组学结合起来, 一方面可描绘出更为全面的微生物组信息(Aguiar-Pulido et al., 2016; Jansson & Baker, 2016), 另一方面也能丰富现有数据库(Wang et al., 2016)。因此, 多组学结合是未来微生物组学研究发展的必然趋势, 现阶段的研究除了要简化复杂的多组学数据, 还应注重微生物培养组学的发展和应用, 以便更好地解析微生物群落结构及其功能(Bashiardes et al., 2016; Vilanova & Porcar, 2016)。
4.3 建议优先研究领域
4.3.1 微生物的时空分布我国幅员辽阔, 生境复杂多样, 不同地区气候、植被和土壤类型差异较大, 且受人类活动干扰程度不同, 借助微生物组学技术研究微生物的时空分布有助于深入挖掘土壤中的未知生物资源(包括细菌、古菌、真核生物和病毒等)和微生物组的代谢产物(如氨基酸、抗生素和激素等), 深刻理解土壤中微生物多样性的产生和维持机制, 阐明影响微生物群落的主要环境因子。目前, 大多数微生物生物地理学研究在不同空间尺度上进行, 但在较大时间尺度上的研究仍较少, 已有研究表明微生物和动植物一样, 都具有昼夜节律和季节变化规律, 因此, 未来微生物组在较大时空尺度下的动态变化值得关注, 特别是在一些特殊生境, 如气候变化敏感区、干旱半干旱区和海陆交错带等。此外, 不局限于单个营养级, 也可从食物网角度, 解析微生物地理分布格局及其与驱动因子、植物区系等直接的关系。在此基础上, 研究环境微生物组对全球变化的响应, 包括全球气候变化、农业管理措施和土地利用方式改变等, 并可进一步预测全球变化背景下微生物群落及其功能的演变方向。以往研究大多仅关注某一个全球变化驱动因子, 缺乏微生物组对多个环境因子综合作用的响应研究, 未来这方面的工作将对评估全球变化背景下微生物组的演变提供重要依据。
4.3.2 植物-微生物互作
在自然界中, 正常生长的植物(包括地上和地下部分)表面和内部都富集了数量庞大且种类繁多的微生物, 这些微生物为植物微生物组(Müller et al., 2016)。这些微生物编码了比宿主植物更多的基因, 通过协作和竞争形成稳定的群落结构, 对植物生长发育、抗病、抗逆等至关重要。然而, 目前对这些微生物的认识还比较片面, 大多数研究主要集中在豆科植物-根瘤菌、共生菌根真菌、某些病原菌和非共生植物根际促生菌上, 这些微生物仅是植物微生物组中比例很小的一部分。人们对于绝大部分植物微生物组还并不了解, 因此在继续开展这方面功能微生物组研究的基础上, 未来还应扩展到植物整体微生物组, 以更全面系统地解析植物微生物组的结构和功能。此外, 由于技术的限制, 传统的研究主要依赖于在实验室条件下开展植物与单一微生物的互作关系研究, 很少在自然状态下研究微生物组与宿主植物共存的机制, 因此, 未来应注重从植物整体微生物组层面上, 对土壤-植物-微生物系统的营养物质, 生长调节化合物, 信号分子(酚类化合物、黄酮类化合物、独脚金内酯等)的生物合成、生物活性及调控机制等物质流和信息流进行深入分析, 深刻理解生物互作过程中的分子生物学和生物化学基础, 从而为微生物合成生物学的进一步开展提供重要的科学依据。
4.3.3 微生物组功能调控
微生物作为地球上生命的先驱, 与人类生产、生活和生存息息相关。目前, 人类正面临着粮食安全、环境污染、全球气候变化和能源等多方面威胁, 基于微生物组结构和功能对生物圈的广泛影响, 深入理解、发掘并利用微生物组资源, 定向调控微生物组的生态调控作用和功能, 将为解决以上问题提供革命性的新思路和新方法。基于微生物组工程的常规原则和最佳方法, 研究人员提出了一种重复的设计-构建-检测-学习(DBTL)循环, 以推进微生物组工程研究和技术发展(Lawson et al., 2019)。在农业生产上, 结合作物根际微生物组和叶片微生物组等, 研究微生物组对作物抗病、抗逆、产量、品质等的调控机制, 研究微生物组对作物连作障碍的影响和克服手段。在此基础上, 广泛筛选促进农作物生长的微生物, 开发微生物肥料和菌剂, 了解其作用机理, 研究其对土壤肥力保持、植物抗病、克服连作障碍和提高作物品质等方面的作用。此外, 进一步构建作物的简易微生物组, 以增强抗逆、增加固氮, 减轻现代农业对化肥、农药和除草剂等的严重依赖, 同时大幅度提高作物的产量和品质, 促进农业可持续发展。在污染治理上, 随着现代工农业的飞速发展, 一些难降解的新兴污染物不断出现, 如抗生素、内分泌干扰物和阻燃剂等。尽管微生物具有强大的环境修复能力, 但其进化速度远不及新兴污染物出现的速度, 因此亟需应用合成生物学来解决这一难题。通过研究微生物的代谢通路, 不断挖掘微生物代谢通路中与代谢产物相关的重要元件, 包括降解元件、转运元件、趋化元件和抗逆元件等。在此基础上, 运用合成生物学手段, 定向设计和改造现有降解菌株, 一方面有选择性地开发和利用这些生物元件, 构建具备全新代谢网络的工程菌, 使其能够降解一种或多种污染物; 另一方面提高现有降解菌株的降解效率, 增强菌株的环境适应性, 使其能够在高盐、酸碱和高渗透压等极端条件下保持降解活性, 为环境污染的微生物修复提供技术支持。展望未来, 微生物组学技术将继续在环境生态领域发挥巨大作用, 相关研究成果将为解决农业可持续发展、环境污染修复、应对气候变化等关键问题提供崭新思路。

参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
URLPMID:28649411 [本文引用: 1]
URLPMID:23581622 [本文引用: 1]
URLPMID:30069051 [本文引用: 2]
DOI:10.1038/ismej.2007.68URLPMID:18043670 [本文引用: 1]

To appreciate the functional diversity of communities of soil eukaryotic micro-organisms we evaluated an experimental approach based on the construction and screening of a cDNA library using polyadenylated mRNA extracted from a forest soil. Such a library contains genes that are expressed by each of the different organisms forming the community and represents its metatranscriptome. The diversity of the organisms that contributed to this library was evaluated by sequencing a portion of the 18S rDNA gene amplified from either soil DNA or reverse-transcribed RNA. More than 70% of the sequences were from fungi and unicellular eukaryotes (protists) while the other most represented group was the metazoa. Calculation of richness estimators suggested that more than 180 species could be present in the soil samples studied. Sequencing of 119 cDNA identified genes with no homologues in databases (32%) and genes coding proteins involved in different biochemical and cellular processes. Surprisingly, the taxonomic distribution of the cDNA and of the 18S rDNA genes did not coincide, with a marked under-representation of the protists among the cDNA. Specific genes from such an environmental cDNA library could be isolated by expression in a heterologous microbial host, Saccharomyces cerevisiae. This is illustrated by the functional complementation of a histidine auxotrophic yeast mutant by two cDNA originating possibly from an ascomycete and a basidiomycete fungal species. Study of the metatranscriptome has the potential to uncover adaptations of whole microbial communities to local environmental conditions. It also gives access to an abundant source of genes of biotechnological interest.
URLPMID:27127406 [本文引用: 1]
URLPMID:24530626 [本文引用: 1]
URLPMID:23894095 [本文引用: 1]
DOI:10.1038/ismej.2007.39URLPMID:18043633 [本文引用: 1]
Using proteins from soil or groundwater as functional biomarkers requires efficient extraction. We developed an extraction method in which the separation of proteins from the inorganic and organic constituents of the soil matrix was achieved by a combination of 0.1 M NaOH treatment and phenol extraction. Incubation with NaOH released humic acids and proteins from soil minerals, and simultaneously, disrupted microorganisms. The subsequent phenol extraction separated the proteins from the humic organic matter. Protein extracts were applied to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and 2D-electrophoresis (2-DE). Spots and bands were excised and individual proteins identified by liquid chromatography online linked to mass spectrometry (MS) via electrospray ionization source (LC-ESI-MS). To assess the suitability of the method for the functional analysis of environmental metaproteomes, it was applied to soil that had been enriched in chlorophenoxy acid-degrading bacteria by incubation with 2,4-dichlorophenoxy acetic acid (2,4-D) for 22 days. The method was also used to analyze groundwater from the aquifer of a chlorobenzene-contaminated site. The identification of enzymes such as chlorocatechol dioxygenases was consistent with bacterial metabolic pathways expected to be expressed in these samples. The protocol enabled thus the analysis of the metaproteome of soil and groundwater samples. It thereby provides a means to study the diversity of environmental microbial communities while addressing functional aspects more directly than metagenome or even metatranscriptome analysis.
URLPMID:29028872 [本文引用: 1]
URLPMID:30201963 [本文引用: 1]
URLPMID:22238585 [本文引用: 2]
DOI:10.1016/j.envint.2019.105085URLPMID:31415965 [本文引用: 1]
Suspended floc and fixed biofilm are two commonly applied strategies for heterotrophic denitrification in wastewater treatment. These two strategies use different carbon sources and reside within different ecological niches for microbial aggregation, which were hypothesized to show distinct microbial structures and metabolic fitness. We surveyed three floc reactors and three biofilm reactors for denitrification and determined if there were distinct microbial aggregations. Multiple molecular omics approaches were used to determine the microbial community composition, co-occurrence network and metabolic pathways. Proteobacteria was the dominating and most active phylum among all samples. Carbon source played an important role in shaping the microbial community composition while the distribution of functional protein was largely influenced by salinity. We found that the topological network features had different ecological patterns and that the microorganisms in the biofilm reactors had more nodes but less interactions than those in floc reactors. The large niche differences in the biofilm reactors explained the observed high microbial diversity, functional redundancy and resulting high system stability. We also observed a lower proportion of denitrifiers and higher resistance to oxygen and salinity perturbation in the biofilm reactors than the floc reactors. Our findings support our hypothesis that niche differences caused a distinct microbial structure and increased microbial ecology distribution, which has the potential to improve system efficiency and stability.
DOI:10.1016/j.tim.2005.12.007URLPMID:16406528 [本文引用: 1]
Molecular tools have revealed wide microbial diversity in the human alimentary tract. Most intestinal microorganisms have not been cultured and the in situ functions of distinct groups of the intestinal microbiota are largely unknown but pivotal to understanding the role of these microorganisms in health and disease. Promising strategies to gain more insight into the functionality of the complex microbial communities in the human alimentary tract, including fermentation processes in the colon, are discussed. These research approaches could provide a basis for the definition of a healthy gut based on key properties of microbial functionality. This will also enable the development of direct nutritional strategies for intestinal disease prevention and health promotion.
URLPMID:28810907 [本文引用: 1]
URLPMID:18497287 [本文引用: 2]
[本文引用: 1]
URLPMID:29657969 [本文引用: 1]
DOI:10.1111/j.1462-2920.2008.01745.xURLPMID:18783384 [本文引用: 1]
Phosphonates are organic compounds that contain a C-P bond and are a poorly characterized component of the marine phosphorus cycle. They may represent a potential source of bioavailable phosphorus, particularly in oligotrophic conditions. This study has investigated the distribution of the phnA gene which encodes phosphonoacetate hydrolase, the enzyme that mineralizes phosphonoacetate. Using newly designed degenerate primers targeting the phnA gene we analysed the potential for phosphonoacetate utilization in DNA and cDNA libraries constructed from a phytoplankton bloom in the Western English Channel during July 2006. Total RNA was isolated and reverse transcribed and phosphonoacetate hydrolase (phnA) transcripts were PCR amplified from the cDNA with the degenerate primers, cloned and sequenced. Phylogenetic analysis demonstrated considerable diversity with 14 sequence types yielding five unique phnA protein groups. We also identified 28 phnA homologues in a 454-pyrosequencing metagenomic and metatranscriptomic study from a coastal marine mesocosm, indicating that > 3% of marine bacteria in this study contained phnA. phnA homologues were also present in a metagenomic fosmid library from this experiment. Finally, cultures of four isolates of potential coral pathogens belonging to the Vibrionaceae contained the phnA gene. In the laboratory, these isolates were able to grow with phosphonoacetate as sole P and C source. The fact that the capacity to utilize phosphonoacetate was evident in each of the three coastal environments suggests the potential for widespread utilization of this bioavailable P source.
DOI:10.1038/nbt.3601URLPMID:27454739 [本文引用: 1]
Amplicon-based marker gene surveys form the basis of most microbiome and other microbial community studies. Such PCR-based methods have multiple steps, each of which is susceptible to error and bias. Variance in results has also arisen through the use of multiple methods of next-generation sequencing (NGS) amplicon library preparation. Here we formally characterized errors and biases by comparing different methods of amplicon-based NGS library preparation. Using mock community standards, we analyzed the amplification process to reveal insights into sources of experimental error and bias in amplicon-based microbial community and microbiome experiments. We present a method that improves on the current best practices and enables the detection of taxonomic groups that often go undetected with existing methods.
URLPMID:30863368 [本文引用: 1]
[本文引用: 2]
DOI:10.3389/fmicb.2015.00695URLPMID:26217326 [本文引用: 1]
Marine hydrocarbon-degrading bacteria perform a fundamental role in the biodegradation of crude oil and its petrochemical derivatives in coastal and open ocean environments. However, there is a paucity of knowledge on the diversity and function of these organisms in deep-sea sediment. Here we used stable-isotope probing (SIP), a valuable tool to link the phylogeny and function of targeted microbial groups, to investigate polycyclic aromatic hydrocarbon (PAH)-degrading bacteria under aerobic conditions in sediments from Guaymas Basin with uniformly labeled [(13)C]-phenanthrene (PHE). The dominant sequences in clone libraries constructed from (13)C-enriched bacterial DNA (from PHE enrichments) were identified to belong to the genus Cycloclasticus. We used quantitative PCR primers targeting the 16S rRNA gene of the SIP-identified Cycloclasticus to determine their abundance in sediment incubations amended with unlabeled PHE and showed substantial increases in gene abundance during the experiments. We also isolated a strain, BG-2, representing the SIP-identified Cycloclasticus sequence (99.9% 16S rRNA gene sequence identity), and used this strain to provide direct evidence of PHE degradation and mineralization. In addition, we isolated Halomonas, Thalassospira, and Lutibacterium sp. with demonstrable PHE-degrading capacity from Guaymas Basin sediment. This study demonstrates the value of coupling SIP with cultivation methods to identify and expand on the known diversity of PAH-degrading bacteria in the deep-sea.
URLPMID:20143941 [本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 3]
URLPMID:23603340 [本文引用: 3]
DOI:10.1007/s10886-017-0921-1URLPMID:29392532 [本文引用: 1]
Arbuscular mycorrhizal fungal (AMF) colonisation of plant roots is one of the most ancient and widespread interactions in ecology, yet the systemic consequences for plant secondary chemistry remain unclear. We performed the first metabolomic investigation into the impact of AMF colonisation by Rhizophagus irregularis on the chemical defences, spanning above- and below-ground tissues, in its host-plant ragwort (Senecio jacobaea). We used a non-targeted metabolomics approach to profile, and where possible identify, compounds induced by AMF colonisation in both roots and shoots. Metabolomics analyses revealed that 33 compounds were significantly increased in the root tissue of AMF colonised plants, including seven blumenols, plant-derived compounds known to be associated with AMF colonisation. One of these was a novel structure conjugated with a malonyl-sugar and uronic acid moiety, hitherto an unreported combination. Such structural modifications of blumenols could be significant for their previously reported functional roles associated with the establishment and maintenance of AM colonisation. Pyrrolizidine alkaloids (PAs), key anti-herbivore defence compounds in ragwort, dominated the metabolomic profiles of root and shoot extracts. Analyses of the metabolomic profiles revealed an increase in four PAs in roots (but not shoots) of AMF colonised plants, with the potential to protect colonised plants from below-ground organisms.
DOI:10.1098/rspb.2003.2549URLPMID:15058386 [本文引用: 2]
Bacteria may be one of the most abundant and species-rich groups of organisms, and they mediate many critical ecosystem processes. Despite the ecological importance of bacteria, past practical and theoretical constraints have limited our ability to document patterns of bacterial diversity and to understand the processes that determine these patterns. However, recent advances in molecular techniques that allow more thorough detection of bacteria in nature have made it possible to examine such patterns and processes. Here, we review recent studies of the distribution of free-living bacterial diversity and compare our current understanding with what is known about patterns in plant and animal diversity. From these recent studies a preliminary picture is emerging: bacterial diversity may exhibit regular patterns, and in some cases these patterns may be qualitatively similar to those observed for plants and animals.
URLPMID:15592412 [本文引用: 1]
URLPMID:31177774 [本文引用: 1]
DOI:10.1038/nmicrobiol.2016.49URLPMID:27572648 [本文引用: 1]
URLPMID:26512100 [本文引用: 1]
DOI:10.1073/pnas.1717590115URLPMID:29735704 [本文引用: 3]
Recently identified Parkinson's disease (PD) genes involved in synaptic vesicle endocytosis, such as DNAJC6 (auxilin), have further implicated synaptic dysfunction in PD pathogenesis. However, how synaptic dysfunction contributes to the vulnerability of human dopaminergic neurons has not been previously explored. Here, we demonstrate that commonly mutated, PD-linked leucine-rich repeat kinase 2 (LRRK2) mediates the phosphorylation of auxilin in its clathrin-binding domain at Ser627. Kinase activity-dependent LRRK2 phosphorylation of auxilin led to differential clathrin binding, resulting in disrupted synaptic vesicle endocytosis and decreased synaptic vesicle density in LRRK2 patient-derived dopaminergic neurons. Moreover, impaired synaptic vesicle endocytosis contributed to the accumulation of oxidized dopamine that in turn mediated pathogenic effects such as decreased glucocerebrosidase activity and increased alpha-synuclein in mutant LRRK2 neurons. Importantly, these pathogenic phenotypes were partially attenuated by restoring auxilin function in mutant LRRK2 dopaminergic neurons. Together, this work suggests that mutant LRRK2 disrupts synaptic vesicle endocytosis, leading to altered dopamine metabolism and dopamine-mediated toxic effects in patient-derived dopaminergic neurons.
DOI:10.1016/j.tim.2016.02.011URLPMID:27197692 [本文引用: 1]
Microbiology is experiencing a revolution brought on by recent developments in sequencing technology. The unprecedented volume of microbiome data being generated poses significant challenges that are currently hindering progress in the field. Here, we outline the major bottlenecks and propose a vision to advance microbiome research as a data-driven science.
URLPMID:29657967 [本文引用: 1]
DOI:10.1186/s40168-017-0394-zURLPMID:29321060 [本文引用: 1]
URLPMID:31548653 [本文引用: 1]
DOI:10.1038/nature04983URLPMID:16915287 [本文引用: 2]
Ammonia oxidation is the first step in nitrification, a key process in the global nitrogen cycle that results in the formation of nitrate through microbial activity. The increase in nitrate availability in soils is important for plant nutrition, but it also has considerable impact on groundwater pollution owing to leaching. Here we show that archaeal ammonia oxidizers are more abundant in soils than their well-known bacterial counterparts. We investigated the abundance of the gene encoding a subunit of the key enzyme ammonia monooxygenase (amoA) in 12 pristine and agricultural soils of three climatic zones. amoA gene copies of Crenarchaeota (Archaea) were up to 3,000-fold more abundant than bacterial amoA genes. High amounts of crenarchaeota-specific lipids, including crenarchaeol, correlated with the abundance of archaeal amoA gene copies. Furthermore, reverse transcription quantitative PCR studies and complementary DNA analysis using novel cloning-independent pyrosequencing technology demonstrated the activity of the archaea in situ and supported the numerical dominance of archaeal over bacterial ammonia oxidizers. Our results indicate that crenarchaeota may be the most abundant ammonia-oxidizing organisms in soil ecosystems on Earth.
URLPMID:30368524 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.16288/j.yczz.19-222URLPMID:31549683 [本文引用: 2]
Development of high-throughput sequencing stimulates a series of microbiome technologies, such as amplicon sequencing, metagenome, metatranscriptome, which have rapidly promoted microbiome research. Microbiome data analysis involves a lot of basic knowledge, software and databases, and it is difficult for peers to learn and select proper methods. This review systematically outlines the basic ideas of microbiome data analysis and the basic knowledge required to conduct analysis. In addition, it summarizes the advantages and disadvantages of commonly used software and databases used in the comparison, visualization, network, evolution, machine learning and association analysis. This review aims to provide a convenient and flexible guide for selecting analytical tools and suitable databases for mining the biological significance of microbiome data.
DOI:10.16288/j.yczz.19-222URLPMID:31549683 [本文引用: 2]
Development of high-throughput sequencing stimulates a series of microbiome technologies, such as amplicon sequencing, metagenome, metatranscriptome, which have rapidly promoted microbiome research. Microbiome data analysis involves a lot of basic knowledge, software and databases, and it is difficult for peers to learn and select proper methods. This review systematically outlines the basic ideas of microbiome data analysis and the basic knowledge required to conduct analysis. In addition, it summarizes the advantages and disadvantages of commonly used software and databases used in the comparison, visualization, network, evolution, machine learning and association analysis. This review aims to provide a convenient and flexible guide for selecting analytical tools and suitable databases for mining the biological significance of microbiome data.
DOI:10.1007/s00248-018-1310-1URLPMID:30607437 [本文引用: 1]
Moisture and temperature play important roles in the assembly and functioning of prokaryotic communities in soil. However, how moisture and temperature regulate the function of niche- versus neutral-based processes during the assembly of these communities has not been examined considering both the total microbial community and the sole active portion with potential for growth in native subtropical grassland. We set up a well-controlled microcosm-based experiment to investigate the individual and combined effects of moisture and temperature on soil prokaryotic communities by simulating subtropical seasons in grassland. The prokaryotic populations with potential for growth and the total prokaryotic community were assessed by 16S rRNA transcript and 16S rRNA gene analyses, respectively. Moisture was the major factor influencing community diversity and structure, with a considerable effect of this factor on the total community. The prokaryotic populations with potential for growth and the total communities were influenced by the same assembly rules, with the niche-based mechanism being more influential in communities under dry condition. Our results provide new information regarding moisture and temperature in microbial communities of soil and elucidate how coexisting prokaryotic populations, under different physiological statuses, are shaped in native subtropical grassland soil.
[本文引用: 2]
[本文引用: 2]
DOI:10.1038/nrmicro1341URLPMID:16415926 [本文引用: 1]
We review the biogeography of microorganisms in light of the biogeography of macroorganisms. A large body of research supports the idea that free-living microbial taxa exhibit biogeographic patterns. Current evidence confirms that, as proposed by the Baas-Becking hypothesis, 'the environment selects' and is, in part, responsible for spatial variation in microbial diversity. However, recent studies also dispute the idea that 'everything is everywhere'. We also consider how the processes that generate and maintain biogeographic patterns in macroorganisms could operate in the microbial world.
URLPMID:26659059 [本文引用: 1]
DOI:10.1016/j.scitotenv.2018.08.214URLPMID:30340250 [本文引用: 1]
The aim of this study was to understand the responses of the microbial community of soil under different land uses to drought in a semi-arid Mediterranean area. In a laboratory incubation, soil samples from different land uses (natural forest, drip-irrigated orchard, rain-fed almond tree cultivation and abandoned area) were maintained at 20% and 60% of the WHC. The microbial biomass and potential enzyme activities were determined after four and fifty days of soil incubation. The diversity and composition of the microbial community were studied after 50days of incubation. The total mineralisation of soil organic C (SOC), as well as, the mineralisation of fresh organic matter (FOM) and the
DOI:10.1146/annurev-genet-120215-034952URLPMID:27648643 [本文引用: 1]
Plants do not grow as axenic organisms in nature, but host a diverse community of microorganisms, termed the plant microbiota. There is an increasing awareness that the plant microbiota plays a role in plant growth and can provide protection from invading pathogens. Apart from intense research on crop plants, Arabidopsis is emerging as a valuable model system to investigate the drivers shaping stable bacterial communities on leaves and roots and as a tool to decipher the intricate relationship among the host and its colonizing microorganisms. Gnotobiotic experimental systems help establish causal relationships between plant and microbiota genotypes and phenotypes and test hypotheses on biotic and abiotic perturbations in a systematic way. We highlight major recent findings in plant microbiota research using comparative community profiling and omics analyses, and discuss these approaches in light of community establishment and beneficial traits like nutrient acquisition and plant health.
URLPMID:22092433 [本文引用: 1]
DOI:10.3389/fmicb.2016.00073URLPMID:26903960 [本文引用: 1]
DOI:10.1038/nrmicro1711URLPMID:17603517 [本文引用: 1]
The identification of geographical patterns in microbial distributions has begun to challenge purely ecological explanations of biogeography and the underlying principle of
URLPMID:19207571 [本文引用: 1]
URLPMID:29890228 [本文引用: 2]
DOI:10.1111/gcb.14453URLPMID:30230661 [本文引用: 1]
Rising atmospheric CO2 concentration directly stimulates plant productivity and affects nutrient dynamics in the soil. However, the influence of CO2 enrichment on soil bacterial communities remains elusive, likely due to their complex interactions with a wide range of plant and soil properties. Here, we investigated the bacterial community response to a decade long preindustrial-to-future CO2 gradient (250-500 ppm) among three contrasting soil types using 16S rRNA gene amplicon sequencing. In addition, we examined the effect of seasonal variation and plant species composition on bacterial communities. We found that Shannon index (H') and Faith's phylogenetic diversity (PD) did not change in response to the CO2 gradient (R(2) = 0.01, p > 0.05). CO2 gradient and season also had a negligible effect on overall community structure, although silty clay soil communities were better structured on a CO2 gradient (p < 0.001) among three soils. Similarly, CO2 gradient had no significant effect on the relative abundance of different phyla. However, we observed soil-specific variation of CO2 effects in a few individual families. For example, the abundance of Pirellulaceae family decreased linearly with CO2 gradient, but only in sandy loam soils. Conversely, the abundance of Micromonosporaceae and Gaillaceae families increased with CO2 gradient in clay soils. Soil water content (SWC) and nutrient properties were the key environmental constraints shaping bacterial community structure, one manifestation of which was a decline in bacterial diversity with increasing SWC. Furthermore, the impact of plant species composition on community structure was secondary to the strong influence of soil properties. Taken together, our findings indicate that bacterial communities may be largely unresponsive to indirect effects of CO2 enrichment through plants. Instead, bacterial communities are strongly regulated by edaphic conditions, presumably because soil differences create distinct environmental niches for bacteria.
URLPMID:27471578 [本文引用: 1]
URLPMID:25533315 [本文引用: 1]
URLPMID:23271810 [本文引用: 1]
DOI:10.1038/265687a0URLPMID:870828 [本文引用: 2]
A DNA sequence for the genome of bacteriophage phi X174 of approximately 5,375 nucleotides has been determined using the rapid and simple 'plus and minus' method. The sequence identifies many of the features responsible for the production of the proteins of the nine known genes of the organism, including initiation and termination sites for the proteins and RNAs. Two pairs of genes are coded by the same region of DNA using different reading frames.
[本文引用: 2]
URLPMID:29871915 [本文引用: 1]
DOI:10.7717/peerj.1869URLPMID:27069806 [本文引用: 1]
Over the past 10 years, microbial ecologists have largely abandoned sequencing 16S rRNA genes by the Sanger sequencing method and have instead adopted highly parallelized sequencing platforms. These new platforms, such as 454 and Illumina's MiSeq, have allowed researchers to obtain millions of high quality but short sequences. The result of the added sequencing depth has been significant improvements in experimental design. The tradeoff has been the decline in the number of full-length reference sequences that are deposited into databases. To overcome this problem, we tested the ability of the PacBio Single Molecule, Real-Time (SMRT) DNA sequencing platform to generate sequence reads from the 16S rRNA gene. We generated sequencing data from the V4, V3-V5, V1-V3, V1-V5, V1-V6, and V1-V9 variable regions from within the 16S rRNA gene using DNA from a synthetic mock community and natural samples collected from human feces, mouse feces, and soil. The mock community allowed us to assess the actual sequencing error rate and how that error rate changed when different curation methods were applied. We developed a simple method based on sequence characteristics and quality scores to reduce the observed error rate for the V1-V9 region from 0.69 to 0.027%. This error rate is comparable to what has been observed for the shorter reads generated by 454 and Illumina's MiSeq sequencing platforms. Although the per base sequencing cost is still significantly more than that of MiSeq, the prospect of supplementing reference databases with full-length sequences from organisms below the limit of detection from the Sanger approach is exciting.
URLPMID:18846087 [本文引用: 1]
URLPMID:30709420 [本文引用: 1]
[本文引用: 1]
DOI:10.1038/nature08055URLPMID:19444216 [本文引用: 1]
Microbial gene expression in the environment has recently been assessed via pyrosequencing of total RNA extracted directly from natural microbial assemblages. Several such 'metatranscriptomic' studies have reported that many complementary DNA sequences shared no significant homology with known peptide sequences, and so might represent transcripts from uncharacterized proteins. Here we report that a large fraction of cDNA sequences detected in microbial metatranscriptomic data sets are comprised of well-known small RNAs (sRNAs), as well as new groups of previously unrecognized putative sRNAs (psRNAs). These psRNAs mapped specifically to intergenic regions of microbial genomes recovered from similar habitats, displayed characteristic conserved secondary structures and were frequently flanked by genes that indicated potential regulatory functions. Depth-dependent variation of psRNAs generally reflected known depth distributions of broad taxonomic groups, but fine-scale differences in the psRNAs within closely related populations indicated potential roles in niche adaptation. Genome-specific mapping of a subset of psRNAs derived from predominant planktonic species such as Pelagibacter revealed recently discovered as well as potentially new regulatory elements. Our analyses show that metatranscriptomic data sets can reveal new information about the diversity, taxonomic distribution and abundance of sRNAs in naturally occurring microbial communities, and indicate their involvement in environmentally relevant processes including carbon metabolism and nutrient acquisition.
URLPMID:16880384 [本文引用: 1]
URLPMID:22379393 [本文引用: 1]
URLPMID:25430773 [本文引用: 1]
DOI:10.1038/nature24621URLPMID:29088705 [本文引用: 2]
Our growing awareness of the microbial world's importance and diversity contrasts starkly with our limited understanding of its fundamental structure. Despite recent advances in DNA sequencing, a lack of standardized protocols and common analytical frameworks impedes comparisons among studies, hindering the development of global inferences about microbial life on Earth. Here we present a meta-analysis of microbial community samples collected by hundreds of researchers for the Earth Microbiome Project. Coordinated protocols and new analytical methods, particularly the use of exact sequences instead of clustered operational taxonomic units, enable bacterial and archaeal ribosomal RNA gene sequences to be followed across multiple studies and allow us to explore patterns of diversity at an unprecedented scale. The result is both a reference database giving global context to DNA sequence data and a framework for incorporating data from future studies, fostering increasingly complete characterization of Earth's microbial diversity.
DOI:10.1186/s40168-018-0491-7URLPMID:29921326 [本文引用: 1]
BACKGROUND: Microbial communities (microbiota) influence human and animal disease and immunity, geochemical nutrient cycling and plant productivity. Specific groups, including bacteria, archaea, eukaryotes or fungi, are amplified by PCR to assess the relative abundance of sub-groups (e.g. genera). However, neither the absolute abundance of sub-groups is revealed, nor can different amplicon families (i.e. OTUs derived from a specific pair of PCR primers such as bacterial 16S, eukaryotic 18S or fungi ITS) be compared. This prevents determination of the absolute abundance of a particular group and domain-level shifts in microbiota abundance can remain undetected. RESULTS: We have developed absolute quantitation of amplicon families using synthetic chimeric DNA spikes. Synthetic spikes were added directly to environmental samples, co-isolated and PCR-amplified, allowing calculation of the absolute abundance of amplicon families (e.g. prokaryotic 16S, eukaryotic 18S and fungal ITS per unit mass of sample). CONCLUSIONS: Spikes can be adapted to any amplicon-specific group including rhizobia from soils, Firmicutes and Bifidobacteria from human gut or Enterobacteriaceae from food samples. Crucially, using highly complex soil samples, we show that the absolute abundance of specific groups can remain steady or increase, even when their relative abundance decreases. Thus, without absolute quantitation, the underlying pathology, physiology and ecology of microbial groups may be masked by their relative abundance.
DOI:10.1073/pnas.1013488108URLPMID:21525411 [本文引用: 1]
Genes of archaea encoding homologues of ammonia monooxygenases have been found on a widespread basis and in large amounts in almost all terrestrial and marine environments, indicating that ammonia oxidizing archaea (AOA) might play a major role in nitrification on Earth. However, only one pure isolate of this group from a marine environment has so far been obtained, demonstrating archaeal ammonia oxidation coupled with autotrophic growth similar to the bacterial counterparts. Here we describe the cultivation and isolation of an AOA from soil. It grows on ammonia or urea as an energy source and is capable of using higher ammonia concentrations than the marine isolate, Nitrosopumilus maritimus. Surprisingly, although it is able to grow chemolithoautotrophically, considerable growth rates of this strain are obtained only upon addition of low amounts of pyruvate or when grown in coculture with bacteria. Our findings expand the recognized metabolic spectrum of AOA and help explain controversial results obtained in the past on the activity and carbon assimilation of these globally distributed organisms.
DOI:10.1038/s41396-018-0082-4URLPMID:29515169 [本文引用: 1]
Little is known about the factors affecting the relative influences of stochastic and deterministic processes that govern the assembly of microbial communities in successional soils. Here, we conducted a meta-analysis of bacterial communities using six different successional soil datasets distributed across different regions. Different relationships between pH and successional age across these datasets allowed us to separate the influences of successional age (i.e., time) from soil pH. We found that extreme acidic or alkaline pH conditions lead to assembly of phylogenetically more clustered bacterial communities through deterministic processes, whereas pH conditions close to neutral lead to phylogenetically less clustered bacterial communities with more stochasticity. We suggest that the influence of pH, rather than successional age, is the main driving force in producing trends in phylogenetic assembly of bacteria, and that pH also influences the relative balance of stochastic and deterministic processes along successional soils. Given that pH had a much stronger association with community assembly than did successional age, we evaluated whether the inferred influence of pH was maintained when studying globally distributed samples collected without regard for successional age. This dataset confirmed the strong influence of pH, suggesting that the influence of soil pH on community assembly processes occurs globally. Extreme pH conditions likely exert more stringent limits on survival and fitness, imposing strong selective pressures through ecological and evolutionary time. Taken together, these findings suggest that the degree to which stochastic vs. deterministic processes shape soil bacterial community assembly is a consequence of soil pH rather than successional age.
DOI:10.1016/j.cell.2008.08.025URLPMID:18775300 [本文引用: 1]
Metagenomics seeks to characterize the composition of microbial communities, their operations, and their dynamically coevolving relationships with the habitats they occupy without having to culture community members. Uniting metagenomics with analyses of the products of microbial community metabolism (metabolomics) will shed light on how microbial communities function in a variety of environments, including the human body.
DOI:10.1038/nmicrobiol.2016.101URLPMID:27573112 [本文引用: 2]
DOI:10.1038/ncomms12151URLPMID:27402057 [本文引用: 1]
Bacteria living on and in leaves and roots influence many aspects of plant health, so the extent of a plant's genetic control over its microbiota is of great interest to crop breeders and evolutionary biologists. Laboratory-based studies, because they poorly simulate true environmental heterogeneity, may misestimate or totally miss the influence of certain host genes on the microbiome. Here we report a large-scale field experiment to disentangle the effects of genotype, environment, age and year of harvest on bacterial communities associated with leaves and roots of Boechera stricta (Brassicaceae), a perennial wild mustard. Host genetic control of the microbiome is evident in leaves but not roots, and varies substantially among sites. Microbiome composition also shifts as plants age. Furthermore, a large proportion of leaf bacterial groups are shared with roots, suggesting inoculation from soil. Our results demonstrate how genotype-by-environment interactions contribute to the complexity of microbiome assembly in natural environments.
[本文引用: 3]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nprot.2016.148URL [本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/nph.14606URLPMID:28542845 [本文引用: 2]
Previous studies have revealed inconsistent correlations between fungal diversity and plant diversity from local to global scales, and there is a lack of information about the diversity-diversity and productivity-diversity relationships for fungi in alpine regions. Here we investigated the internal relationships between soil fungal diversity, plant diversity and productivity across 60 grassland sites on the Tibetan Plateau, using Illumina sequencing of the internal transcribed spacer 2 (ITS2) region for fungal identification. Fungal alpha and beta diversities were best explained by plant alpha and beta diversities, respectively, when accounting for environmental drivers and geographic distance. The best ordinary least squares (OLS) multiple regression models, partial least squares regression (PLSR) and variation partitioning analysis (VPA) indicated that plant richness was positively correlated with fungal richness. However, no correlation between plant richness and fungal richness was evident for fungal functional guilds when analyzed individually. Plant productivity showed a weaker relationship to fungal diversity which was intercorrelated with other factors such as plant diversity, and was thus excluded as a main driver. Our study points to a predominant effect of plant diversity, along with other factors such as carbon : nitrogen (C : N) ratio, soil phosphorus and dissolved organic carbon, on soil fungal richness.
DOI:10.1038/s41396-018-0303-xURLPMID:30353037 [本文引用: 1]
Recent studies have detected strong phylogenetic signals in tree-fungus associations for diseased leaves and mycorrhizal symbioses. However, the extent of plant phylogenetic constraints on the free-living soil mycobiome remains unknown, especially at broad geographic scales. Here, 343 soil samples were collected adjacent to individual tree trunks, representing 58 woody plant species located in five mountain forests of eastern China. Integrating plant species identity and phylogenetic information, we aimed to unravel the relative contributions of phylogenetic relationships among tree species, abiotic environmental filtering, and geographic isolation to the geographic distribution of soil mycobiome. We found that the community dissimilarities of total fungi and each dominant guild (viz. saprotrophs, plant pathogens, and ectomycorrhizal fungi) significantly increased with increasing plant phylogenetic distance. Plant phylogenetic eigenvectors explained 11.4% of the variation in community composition, whereas environmental and spatial factors explained 24.1% and 7.2% of the variation, respectively. The communities of ectomycorrhizal fungi and plant pathogens were relatively more strongly affected by plant phylogeny than those of saprotrophs (13.7% and 10.4% vs. 8.5%). Overall, our results demonstrate how plant phylogeny, environment, and geographic space contribute to forest soil fungal distributions and suggest that the influence of plant phylogeny on fungal association may differ by guilds.
[本文引用: 1]
[本文引用: 1]
URLPMID:19835134 [本文引用: 1]
Metaproteomics is an emerging proteomics technology to analyze large scale protein expression in environmental microbial ecosystem. It is termed as the large-scale characterization of the entire protein complement of environmental microbial community at a given point in time. This review focuses on the research strategies and the recent applications in this field based on the published reports and in combination with our own research experiences.
URLPMID:19835134 [本文引用: 1]
Metaproteomics is an emerging proteomics technology to analyze large scale protein expression in environmental microbial ecosystem. It is termed as the large-scale characterization of the entire protein complement of environmental microbial community at a given point in time. This review focuses on the research strategies and the recent applications in this field based on the published reports and in combination with our own research experiences.
DOI:10.1128/mSystems.00225-18URLPMID:30801023 [本文引用: 2]
Soil salinization is a growing environmental problem caused by both natural and human activities. Excessive salinity in soil suppresses growth, decreases species diversity, and alters the community composition of plants; however, the effect of salinity on soil microbial communities is poorly understood. Here, we characterize the soil microbial community along a natural salinity gradient in Gurbantunggut Desert, Northwestern China. Microbial diversity linearly decreased with increases in salinity, and community dissimilarity significantly increased with salinity differences. Soil salinity showed a strong effect on microbial community dissimilarity, even after controlling for the effects of spatial distance and other environmental variables. Microbial phylotypes (n = 270) belonging to Halobacteria, Nitriliruptoria, [Rhodothermi], Gammaproteobacteria, and Alphaproteobacteria showed a high-salinity niche preference. Out of nine potential phenotypes predicted by BugBase, oxygen-related phenotypes showed a significant relationship with salinity content. To explore the community assembly processes, we used null models of within-community (nearest-taxon index [NTI]) and between-community (betaNTI) phylogenetic composition. NTI showed a significantly negative relationship with salinity, suggesting that the microbial community was less phylogenetically clustered in more-saline soils. betaNTI, the between-community analogue of NTI, showed that deterministic processes have overtaken stochastic processes across all sites, suggesting the importance of environmental filtering in microbial community assembly. Taken together, these results suggest the importance of salinity in soil microbial community composition and assembly processes in a desert ecosystem. IMPORTANCE Belowground microorganisms are indispensable components for nutrient cycling in desert ecosystems, and understanding how they respond to increased salinity is essential for managing and ameliorating salinization. Our sequence-based data revealed that microbial diversity decreased with increasing salinity, and certain salt-tolerant phylotypes and phenotypes showed a positive relationship with salinity. Using a null modeling approach to estimate microbial community assembly processes along a salinity gradient, we found that salinity imposed a strong selection pressure on the microbial community, which resulted in a dominance of deterministic processes. Studying microbial diversity and community assembly processes along salinity gradients is essential in understanding the fundamental ecological processes in desert ecosystems affected by salinization.
DOI:10.3389/fmicb.2016.01032URLPMID:27446064 [本文引用: 1]
Soil microbial communities are influenced by climate change drivers such as warming and altered precipitation. These changes create abiotic stresses, including desiccation and nutrient limitation, which act on microbes. However, our understanding of the responses of microbial communities to co-occurring climate change drivers is limited. We surveyed soil bacterial and fungal diversity and composition after a 1-year warming and altered precipitation manipulation in the Tibetan plateau alpine grassland. In isolation, warming and decreased precipitation treatments each had no significant effects on soil bacterial community structure; however, in combination of both treatments altered bacterial community structure (p = 0.03). The main effect of altered precipitation specifically impacted the relative abundances of Bacteroidetes and Gammaproteobacteria compared to the control, while the main effect of warming impacted the relative abundance of Betaproteobacteria. In contrast, the fungal community had no significant response to the treatments after 1-year. Using structural equation modeling (SEM), we found bacterial community composition was positively related to soil moisture. Our results indicate that short-term climate change could cause changes in soil bacterial community through taxonomic shifts. Our work provides new insights into immediate soil microbial responses to short-term stressors acting on an ecosystem that is particularly sensitive to global climate change.
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.marpolbul.2016.11.020URLPMID:27865521 [本文引用: 3]
Alkylated polycyclic aromatic hydrocarbons (PAHs) are abundant in petroleum, and alkylated phenanthrenes are considered as the primary PAHs during some oil spill events. Bacterial strain of Sphingobium sp. MP9-4, isolated from petroleum-contaminated soil, was efficient to degrade 1-methylphenanthrene (1-MP). A detailed metabolism map of 1-MP in this strain was delineated based on analysis of metabolites with gas chromatograph-mass spectrometer (GC-MS). 1-MP was initially oxidized via two different biochemical strategies, including benzene ring and methyl-group attacks. Benzene ring attack was initiated with dioxygenation of the non-methylated aromatic ring via similar degradation pathways of phenanthrene (PHE) by bacteria. For methyl-group attack, mono oxygenase system was involved and more diverse enzymes were needed than that of PHE degradation. This study enhances the understanding of the metabolic pathways of alkylated PAHs and shows the significant potential of Sphingobium sp. MP9-4 for the bioremediation of alkylated PAHs contaminated environments.
[本文引用: 2]
[本文引用: 2]
Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis
2
2016
... (2)微生物宏转录组学(microbial metatranscriptomics): 研究环境中全部微生物的转录组信息, 揭示相关基因在时空尺度上的表达水平, 从而对微生物群落的相关功能进行研究(
... 微生物组学的发展也离不开多组学结合的研究策略, 包括微生物分离培养、生态学鉴定和代谢产物分析等.尽管每种组学方法都对微生物组学研究提供了有价值的信息, 但仅仅基于DNA测序的方法分析微生物群落的多样性和群落组成, 并不能很好地研究微生物组的功能特征(
The microbiome of glaciers and ice sheets
1
2017
... 微生物组学技术被广泛应用于揭示复杂环境中的微生物群落(包括多样性和群落组成)及其与周围环境或宿主之间的关系, 其研究对象涵盖土壤(
An improved method compatible with metagenomic analyses to extract genomic DNA from soils in Tuber melanosporum orchards
1
2013
... 从复杂环境中获得高保真性和高质量的样本是开展微生物组学研究的第一步.然而, 样品获取方法上的偏差仍是阻碍微生物组学研究的巨大挑战(
Structure and function of the global topsoil microbiome
2
2018
... 微生物学研究大致可分为三个阶段.第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段.第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代.值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
Soil eukaryotic functional diversity, a metatranscriptomic approach
1
2007
... 二代测序的原理是边合成边测序, 通过捕捉新合成的末端的标记来确定DNA的序列, 该技术在保持测序准确度的同时, 主要解决了Sanger法测序通量低的问题, 二代测序可同时对几万到几百万条DNA序列进行测序, 因此也被称为高通量测序.现有平台主要包括Illumina Hiseq、MiSeq、NovaSeq 6000等.二代测序的进步极大推动了微生物组学的发展, 2006年,
Use of metatranscriptomics in microbiome research
1
2016
... 微生物组学的发展也离不开多组学结合的研究策略, 包括微生物分离培养、生态学鉴定和代谢产物分析等.尽管每种组学方法都对微生物组学研究提供了有价值的信息, 但仅仅基于DNA测序的方法分析微生物群落的多样性和群落组成, 并不能很好地研究微生物组的功能特征(
Metaproteomics of soils from semiarid environment: functional and phylogenetic information obtained with different protein extraction methods
1
2014
... (1)微生物生物地球化学循环.采用微生物宏转录组学可以探究微生物的相关功能, 发现大量的微生物资源及新的基因, 为探索微生物群落的生态功能奠定基础, 有助于生态系统功能和服务的调控(
Metaproteomics to unravel major microbial players in leaf litter and soil environments: challenges and perspectives
1
2013
...
Functional metaproteome analysis of protein extracts from contaminated soil and groundwater
1
2007
... 从复杂环境中获得高保真性和高质量的样本是开展微生物组学研究的第一步.然而, 样品获取方法上的偏差仍是阻碍微生物组学研究的巨大挑战(
A review of methods and databases for metagenomic classification and assembly
1
2019
... 在发展过程中, 各微生物组学技术都已初步建立了各自的数据库、算法和软件(
A plantmicrobe interaction framework explaining nutrient effects on primary production
1
2018
... (2)微生物与植物生长和健康.宏基因组学和宏代谢组学也被广泛用于探究微生物与宿主、环境之间的相互关系, 进而了解生物间相互作用的机制(
Metatranscriptomics reveals the diversity of genes expressed by eukaryotes in forest soils
2
2012
... 二代测序的原理是边合成边测序, 通过捕捉新合成的末端的标记来确定DNA的序列, 该技术在保持测序准确度的同时, 主要解决了Sanger法测序通量低的问题, 二代测序可同时对几万到几百万条DNA序列进行测序, 因此也被称为高通量测序.现有平台主要包括Illumina Hiseq、MiSeq、NovaSeq 6000等.二代测序的进步极大推动了微生物组学的发展, 2006年,
... (1)微生物生物地球化学循环.采用微生物宏转录组学可以探究微生物的相关功能, 发现大量的微生物资源及新的基因, 为探索微生物群落的生态功能奠定基础, 有助于生态系统功能和服务的调控(
Multiomics analysis reveals niche and fitness differences in typical denitrification microbial aggregations
1
2019
... 在20世纪80年代以前, 微生物研究主要基于微生物的分离培养, 但绝大多数微生物无法被分离培养, 因此, 这阶段人们对于微生物的认识基本停留在形态观察、分类及生理学研究.20世纪末期, DNA指纹图谱等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接在DNA水平对环境整体微生物群落进行分析, 开创了微生物分子生态学研究的新时代.然而, DNA指纹图谱只能靶标优势类群, 不能真实反映微生物的多样性及物种组成.自21世纪初以来, 高通量测序和质谱等技术的突破, 使得我们可以从DNA、RNA、蛋白质和代谢物等不同水平解析微生物组, 以获得更为全面的微生物组信息.然而, 目前各组学技术仍存在一定局限性, 比如, 现有测序技术均基于使用PCR的DNA扩增, 这将会导致有些序列可能被测了多次, 而有些量少的序列则无法被大量扩增, 同时PCR过程中可能会引入错配碱基, 从而造成信息的丢失.在宏代谢组分析中, 气相色谱-质谱联用(GC-MS)只能对其中的挥发性物质实现直接分析, 而对那些难挥发的物质分析效果不佳.在理论层面上, 现代生态学经过20世纪的发展已经累积了大量成熟的理论和模型, 而这些理论大都建立在宏观生态学的基础上, 这些理论和模型是否也适用于微生物领域, 目前还没有明确的结论, 仍需要更多的微生物组数据支撑.在技术应用上, 仅利用单一组学技术并不能完全揭示微生物组信息, 近年来多组学技术结合的研究策略更有利于全面解析微生物组(
Beyond diversity: functional microbiomics of the human colon
1
2006
... 微生物学研究大致可分为三个阶段.第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段.第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代.值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(
Schr?dinger’s microbes: tools for distinguishing the living from the dead in microbial ecosystems
1
2017
... 微生物组学的发展也离不开多组学结合的研究策略, 包括微生物分离培养、生态学鉴定和代谢产物分析等.尽管每种组学方法都对微生物组学研究提供了有价值的信息, 但仅仅基于DNA测序的方法分析微生物群落的多样性和群落组成, 并不能很好地研究微生物组的功能特征(
The microbial engines that drive Earth’s biogeochemical cycles
2
2008
... 微生物学研究大致可分为三个阶段.第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段.第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代.值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(
... 微生物参与了所有已知的生物地球化学循环过程, 其与植物生长、污染物降解和其他生态系统功能密切相关(
Long-term fertilization influences community assembly processes of soil diazotrophs
1
2018
... (2)微生物对全球变化的响应.微生物对环境变化的响应较为敏感, 是全球气候变化的调节器, 因此, 研究微生物组对于环境变化的响应及反馈具有重要意义, 其结果可为预测全球变化背景下生态系统结构和功能的演变提供理论依据.利用微生物组学技术, 可探究土壤微生物群落对全球气候变化(
Earth microbiome project and global systems biology
1
2018
...
Potential for phosphonoacetate utilization by marine bacteria in temperate coastal waters
1
2009
... (1)微生物生物地球化学循环.采用微生物宏转录组学可以探究微生物的相关功能, 发现大量的微生物资源及新的基因, 为探索微生物群落的生态功能奠定基础, 有助于生态系统功能和服务的调控(
Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies
1
2016
... 基于标记基因的扩增子是大部分微生物组研究的基础, 但其样本检测步骤较多, 高通量测序读长较短、存在系统偏好性等问题, 且建库过程中使用了PCR扩增, 会导致某些序列可能被测了多次, 而有些量少的序列则无法被大量扩增, 不仅如此, PCR过程中可能会引入错配碱基, 从而造成信息的丢失.针对这些不足, 研究人员比较了不同文库制备方法, 利用标准样品详细分析扩增过程, 揭示了PCR扩增中造成错误和偏差的来源, 并提出了条件优化后的基于两步法的PCR扩增方法, 改进后的新方法能检测到更多现有方法中经常无法检测的类群, 同时提高微生物组研究的准确性(
Rapid transfer of plant photosynthates to soil bacteria via ectomycorrhizal hyphae and its interaction with nitrogen availability
1
2019
... 近年来, 从单细胞水平上分析微生物的生理代谢的单细胞技术迅速发展, 其中单细胞成像技术可将检测结果可视化, 能较好反映环境微生物类群、丰度及其功能活性(
Climate warming leads to divergent succession of grassland microbial communities
2
2018
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
... (2)微生物对全球变化的响应.微生物对环境变化的响应较为敏感, 是全球气候变化的调节器, 因此, 研究微生物组对于环境变化的响应及反馈具有重要意义, 其结果可为预测全球变化背景下生态系统结构和功能的演变提供理论依据.利用微生物组学技术, 可探究土壤微生物群落对全球气候变化(
Cultivation- dependent and cultivation-independent characterization of hydrocarbon-degrading bacteria in Guaymas Basin sediments
1
2015
... (3)环境污染微生物修复.微生物组学技术在环境污染治理方面也表现出了巨大潜力, 包括有机污染物降解、重金属转化和生物污染等领域.例如, 微生物组学技术可用于研究环境中多环芳烃(PAHs)的微生物降解机制.首先, 可以先通过对受到PAHs污染的土壤进行宏基因组学研究, 鉴定能降解PAHs的微生物及其关键基因(
Metabolomics: towards understanding host-microbe interactions
1
2010
... (2)微生物与植物生长和健康.宏基因组学和宏代谢组学也被广泛用于探究微生物与宿主、环境之间的相互关系, 进而了解生物间相互作用的机制(
Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products
1
1998
... 微生物学研究大致可分为三个阶段.第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段.第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代.值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(
3
2014
... 微生物数量庞大、种类繁多, 是地球生物化学循环过程的驱动者, 是工农业生产、医药卫生和环境保护等关键领域的核心资源, 因此微生物组研究已成为新一轮科技革命的战略高地(
... 微生物数量巨大、种类繁多, 尽管高通量测序技术的突破大大增加了人们对于微生物的认识, 但随着研究的深入, 人们意识到单从基因和转录水平并不能完全揭示微生物的奥秘, 并逐渐意识到蛋白质和代谢产物在微生物功能研究中的重要性.对复杂环境中的微生物群落功能研究一直是生物学研究的重点和难点, 长期以来的研究大多依赖于代谢产物或底物浓度的变化, 遗漏了绝大多数微生物, 难以真实反映微生物在复杂环境中的生理代谢信息(
... 生物信息学是伴随着人类基因组计划发展而来的学科, 包含了生物信息的获取、加工、存储、分配、分析、解释等在内的所有方面, 它综合运用数学、计算机科学和生物学的各种工具来阐明和理解大量数据所包含的生物学意义(
3
2014
... 微生物数量庞大、种类繁多, 是地球生物化学循环过程的驱动者, 是工农业生产、医药卫生和环境保护等关键领域的核心资源, 因此微生物组研究已成为新一轮科技革命的战略高地(
... 微生物数量巨大、种类繁多, 尽管高通量测序技术的突破大大增加了人们对于微生物的认识, 但随着研究的深入, 人们意识到单从基因和转录水平并不能完全揭示微生物的奥秘, 并逐渐意识到蛋白质和代谢产物在微生物功能研究中的重要性.对复杂环境中的微生物群落功能研究一直是生物学研究的重点和难点, 长期以来的研究大多依赖于代谢产物或底物浓度的变化, 遗漏了绝大多数微生物, 难以真实反映微生物在复杂环境中的生理代谢信息(
... 生物信息学是伴随着人类基因组计划发展而来的学科, 包含了生物信息的获取、加工、存储、分配、分析、解释等在内的所有方面, 它综合运用数学、计算机科学和生物学的各种工具来阐明和理解大量数据所包含的生物学意义(
Sulfur-34S stable isotope labeling of amino acids for quantification (SULAQ34) of proteomic changes in Pseudomonas fluorescens during naphthalene degradation
3
2013
... 微生物学研究大致可分为三个阶段.第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段.第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代.值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(
... 质谱技术是宏蛋白质组学和宏代谢组学的核心技术, 通常结合色谱技术对复杂样品中的蛋白质或代谢物进行分析(
... (3)环境污染微生物修复.微生物组学技术在环境污染治理方面也表现出了巨大潜力, 包括有机污染物降解、重金属转化和生物污染等领域.例如, 微生物组学技术可用于研究环境中多环芳烃(PAHs)的微生物降解机制.首先, 可以先通过对受到PAHs污染的土壤进行宏基因组学研究, 鉴定能降解PAHs的微生物及其关键基因(
Arbuscular mycorrhizal fungi and plant chemical defence: effects of colonisation on aboveground and belowground metabolomes
1
2018
... (2)微生物与植物生长和健康.宏基因组学和宏代谢组学也被广泛用于探究微生物与宿主、环境之间的相互关系, 进而了解生物间相互作用的机制(
An ecological perspective on bacterial biodiversity
2
2004a
... 在数据处理上, 高通量测序数据的质量控制不是单一指标或操作即可完成的, 但目前还未建立统一、规范化的数据质量控制标准.目前比较成熟的序列拼接均基于一个或少数几个基因组为前提, 在面对海量数据和复杂的基因组时, 现有算法基本都无法满足需求.在微生物组学研究中, 大部分扩增子研究主要利用UCLUST或UPARSE等算法对97%相似性的序列进行OTU聚类, 然而不同的OTU聚类方法会对微生物组数据产生较大影响(
... ).近年来, 也出现了一些新的算法, 如DADA2和unoise3, 相当于基于100%的序列相似度进行聚类, 该方法大大提高了准确度和物种多样性.此外, 被广泛应用的OTU分类只能注释到属水平, 极少能鉴定到种水平, 这使得在某些特定微生物的功能研究上略显乏力, 因为较粗略的物种划分标准会导致可观测到的微生物空间分布格局减弱甚至消失(
A taxa-area relationship for bacteria
1
2004b
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
Enrichment of lignin-derived carbon in mineral-associated soil organic matter
1
2019
... 近年来, 从单细胞水平上分析微生物的生理代谢的单细胞技术迅速发展, 其中单细胞成像技术可将检测结果可视化, 能较好反映环境微生物类群、丰度及其功能活性(
A multi-omic future for microbiome studies
1
2016
... 微生物组学的发展也离不开多组学结合的研究策略, 包括微生物分离培养、生态学鉴定和代谢产物分析等.尽管每种组学方法都对微生物组学研究提供了有价值的信息, 但仅仅基于DNA测序的方法分析微生物群落的多样性和群落组成, 并不能很好地研究微生物组的功能特征(
Library preparation methodology can influence genomic and functional predictions in human microbiome research
1
2015
... 从复杂环境中获得高保真性和高质量的样本是开展微生物组学研究的第一步.然而, 样品获取方法上的偏差仍是阻碍微生物组学研究的巨大挑战(
Metaproteomics method to determine carbon sources and assimilation pathways of species in microbial communities
3
2018
... 微生物学研究大致可分为三个阶段.第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段.第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代.值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(
... 质谱技术是宏蛋白质组学和宏代谢组学的核心技术, 通常结合色谱技术对复杂样品中的蛋白质或代谢物进行分析(
... (1)微生物生物地球化学循环.采用微生物宏转录组学可以探究微生物的相关功能, 发现大量的微生物资源及新的基因, 为探索微生物群落的生态功能奠定基础, 有助于生态系统功能和服务的调控(
Microbiome data science: understanding our microbial planet
1
2016
...
Existing climate change will lead to pronounced shifts in the diversity of soil prokaryotes
1
2018
... (2)微生物对全球变化的响应.微生物对环境变化的响应较为敏感, 是全球气候变化的调节器, 因此, 研究微生物组对于环境变化的响应及反馈具有重要意义, 其结果可为预测全球变化背景下生态系统结构和功能的演变提供理论依据.利用微生物组学技术, 可探究土壤微生物群落对全球气候变化(
“Available upon request”: not good enough for microbiome data!
1
2018
...
Common principles and best practices for engineering microbiomes
1
2019
... 微生物作为地球上生命的先驱, 与人类生产、生活和生存息息相关.目前, 人类正面临着粮食安全、环境污染、全球气候变化和能源等多方面威胁, 基于微生物组结构和功能对生物圈的广泛影响, 深入理解、发掘并利用微生物组资源, 定向调控微生物组的生态调控作用和功能, 将为解决以上问题提供革命性的新思路和新方法.基于微生物组工程的常规原则和最佳方法, 研究人员提出了一种重复的设计-构建-检测-学习(DBTL)循环, 以推进微生物组工程研究和技术发展(
Archaea predominate among ammonia-oxidizing prokaryotes in soils
2
2006
... 微生物学研究大致可分为三个阶段.第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段.第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代.值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(
... 二代测序的原理是边合成边测序, 通过捕捉新合成的末端的标记来确定DNA的序列, 该技术在保持测序准确度的同时, 主要解决了Sanger法测序通量低的问题, 二代测序可同时对几万到几百万条DNA序列进行测序, 因此也被称为高通量测序.现有平台主要包括Illumina Hiseq、MiSeq、NovaSeq 6000等.二代测序的进步极大推动了微生物组学的发展, 2006年,
Legacy of land use history determines reprogramming of plant physiology by soil microbiome
1
2019
... (2)微生物对全球变化的响应.微生物对环境变化的响应较为敏感, 是全球气候变化的调节器, 因此, 研究微生物组对于环境变化的响应及反馈具有重要意义, 其结果可为预测全球变化背景下生态系统结构和功能的演变提供理论依据.利用微生物组学技术, 可探究土壤微生物群落对全球气候变化(
土壤微生物群落多样性解析法: 从培养到非培养
1
2012
... 从20世纪80年代开始, 为了克服纯培养的缺陷, 研究人员开始利用微生物细胞内的DNA来评估微生物的种类和结构多样性, 这种在遗传分子水平来研究微生物群落的方法即为现代分子生物学方法, 主要包括DNA指纹图谱、基因芯片和稳定同位素探针等(
土壤微生物群落多样性解析法: 从培养到非培养
1
2012
... 从20世纪80年代开始, 为了克服纯培养的缺陷, 研究人员开始利用微生物细胞内的DNA来评估微生物的种类和结构多样性, 这种在遗传分子水平来研究微生物群落的方法即为现代分子生物学方法, 主要包括DNA指纹图谱、基因芯片和稳定同位素探针等(
中国微生物组计划: 机遇与挑战
2
2017
... 微生物与其所处环境构成了复杂的生态系统, 是地球上生物多样性的重要组成部分.微生物组(microbiome)是指一个特定环境或生态系统中全部微生物及其遗传信息的集合, 其内涵包括了微生物与其环境和宿主的相互作用(
... ).微生物组学(microbiomics)是以微生物组为研究对象, 探究其内部群体间的相互关系、结构、功能及其与环境或宿主间相互关系的学科(
中国微生物组计划: 机遇与挑战
2
2017
... 微生物与其所处环境构成了复杂的生态系统, 是地球上生物多样性的重要组成部分.微生物组(microbiome)是指一个特定环境或生态系统中全部微生物及其遗传信息的集合, 其内涵包括了微生物与其环境和宿主的相互作用(
... ).微生物组学(microbiomics)是以微生物组为研究对象, 探究其内部群体间的相互关系、结构、功能及其与环境或宿主间相互关系的学科(
微生物组数据分析方法与应用
2
2019
... 在发展过程中, 各微生物组学技术都已初步建立了各自的数据库、算法和软件(
... ).2010年, 现美国加州大学圣地亚哥分校的Rob Knight团队发布了QIIME分析流程.QIIME的优势主要在于它整合了200多款相关软件和包, 使用者可以有更多软件和方法选择, 提供了150多个脚本, 可实现多种个性化分析, 增强统计和可视化, 实现多样性、物种组成、差异比较和网络等方法和出版级图表绘制, 因此QIIME大受研究者的青睐(
微生物组数据分析方法与应用
2
2019
... 在发展过程中, 各微生物组学技术都已初步建立了各自的数据库、算法和软件(
... ).2010年, 现美国加州大学圣地亚哥分校的Rob Knight团队发布了QIIME分析流程.QIIME的优势主要在于它整合了200多款相关软件和包, 使用者可以有更多软件和方法选择, 提供了150多个脚本, 可实现多种个性化分析, 增强统计和可视化, 实现多样性、物种组成、差异比较和网络等方法和出版级图表绘制, 因此QIIME大受研究者的青睐(
Moisture is more important than temperature for assembly of both potentially active and whole prokaryotic communities in subtropical grassland
1
2019
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
关于中国微生物组数据中心建设的思考
2
2017
... 参考数据库的发展是限制微生物组数据分析的重要因素.据估计, 因缺乏参考基因组数据, 测序所得的宏基因组序列, 有7%-60%无法被准确分类(
...
关于中国微生物组数据中心建设的思考
2
2017
... 参考数据库的发展是限制微生物组数据分析的重要因素.据估计, 因缺乏参考基因组数据, 测序所得的宏基因组序列, 有7%-60%无法被准确分类(
...
Microbial biogeography: putting microorganisms on the map
1
2006
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
The global ocean microbiome
1
2015
... 微生物组学技术被广泛应用于揭示复杂环境中的微生物群落(包括多样性和群落组成)及其与周围环境或宿主之间的关系, 其研究对象涵盖土壤(
Land use shapes the resistance of the soil microbial community and the C cycling response to drought in a semi-arid area
1
2019
... (2)微生物对全球变化的响应.微生物对环境变化的响应较为敏感, 是全球气候变化的调节器, 因此, 研究微生物组对于环境变化的响应及反馈具有重要意义, 其结果可为预测全球变化背景下生态系统结构和功能的演变提供理论依据.利用微生物组学技术, 可探究土壤微生物群落对全球气候变化(
The plant microbiota: systems-level insights and perspectives
1
2016
... 在自然界中, 正常生长的植物(包括地上和地下部分)表面和内部都富集了数量庞大且种类繁多的微生物, 这些微生物为植物微生物组(
Detecting metabolic activities in single cells, with emphasis on nano-SIMS
1
2012
... 近年来, 从单细胞水平上分析微生物的生理代谢的单细胞技术迅速发展, 其中单细胞成像技术可将检测结果可视化, 能较好反映环境微生物类群、丰度及其功能活性(
Back to the future of soil metagenomics
1
2016
... 从复杂环境中获得高保真性和高质量的样本是开展微生物组学研究的第一步.然而, 样品获取方法上的偏差仍是阻碍微生物组学研究的巨大挑战(
The nineteenth century roots of “everything is everywhere”
1
2007
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific subtropical gyre
1
2009
... (1)微生物生物地球化学循环.采用微生物宏转录组学可以探究微生物的相关功能, 发现大量的微生物资源及新的基因, 为探索微生物群落的生态功能奠定基础, 有助于生态系统功能和服务的调控(
Interrogating the microbiome: experimental and computational considerations in support of study reproducibility
2
2018
... 从复杂环境中获得高保真性和高质量的样本是开展微生物组学研究的第一步.然而, 样品获取方法上的偏差仍是阻碍微生物组学研究的巨大挑战(
...
Bacterial community response to a preindustrial-to-future CO2 gradient is limited and soil specific in Texas Prairie grassland
1
2018
... (2)微生物对全球变化的响应.微生物对环境变化的响应较为敏感, 是全球气候变化的调节器, 因此, 研究微生物组对于环境变化的响应及反馈具有重要意义, 其结果可为预测全球变化背景下生态系统结构和功能的演变提供理论依据.利用微生物组学技术, 可探究土壤微生物群落对全球气候变化(
Complete genome of Nitrosospira briensis C-128, an ammonia-oxidizing bacterium from agricultural soil
1
2016
... 三代测序(单分子测序)的原理是通过现代光学、高分子和纳米技术等手段来区分碱基信号的差异, 直接读取DNA的序列信息, 从而解决了二代测序信息丢失和碱基错配等问题(
Vertical distribution of the soil microbiota along a successional gradient in a glacier forefield
1
2015
... 微生物组学技术被广泛应用于揭示复杂环境中的微生物群落(包括多样性和群落组成)及其与周围环境或宿主之间的关系, 其研究对象涵盖土壤(
Conversion of the Amazon rainforest to agriculture results in biotic homogenization of soil bacterial communities
1
2013
... (2)微生物对全球变化的响应.微生物对环境变化的响应较为敏感, 是全球气候变化的调节器, 因此, 研究微生物组对于环境变化的响应及反馈具有重要意义, 其结果可为预测全球变化背景下生态系统结构和功能的演变提供理论依据.利用微生物组学技术, 可探究土壤微生物群落对全球气候变化(
Nucleotide sequence of bacteriophage phi X174 DNA
2
1977
... 微生物学研究大致可分为三个阶段.第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段.第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代.值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(
... 从20世纪80年代开始, 为了克服纯培养的缺陷, 研究人员开始利用微生物细胞内的DNA来评估微生物的种类和结构多样性, 这种在遗传分子水平来研究微生物群落的方法即为现代分子生物学方法, 主要包括DNA指纹图谱、基因芯片和稳定同位素探针等(
A window into third-generation sequencing
2
2010
... 三代测序(单分子测序)的原理是通过现代光学、高分子和纳米技术等手段来区分碱基信号的差异, 直接读取DNA的序列信息, 从而解决了二代测序信息丢失和碱基错配等问题(
... 基于标记基因的扩增子是大部分微生物组研究的基础, 但其样本检测步骤较多, 高通量测序读长较短、存在系统偏好性等问题, 且建库过程中使用了PCR扩增, 会导致某些序列可能被测了多次, 而有些量少的序列则无法被大量扩增, 不仅如此, PCR过程中可能会引入错配碱基, 从而造成信息的丢失.针对这些不足, 研究人员比较了不同文库制备方法, 利用标准样品详细分析扩增过程, 揭示了PCR扩增中造成错误和偏差的来源, 并提出了条件优化后的基于两步法的PCR扩增方法, 改进后的新方法能检测到更多现有方法中经常无法检测的类群, 同时提高微生物组研究的准确性(
Identifying and overcoming threats to reproducibility, replicability, robustness, and generalizability in microbiome research
1
2018
...
Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system
1
2016
... 三代测序(单分子测序)的原理是通过现代光学、高分子和纳米技术等手段来区分碱基信号的差异, 直接读取DNA的序列信息, 从而解决了二代测序信息丢失和碱基错配等问题(
Next-generation DNA sequencing
1
2008
... 二代测序的原理是边合成边测序, 通过捕捉新合成的末端的标记来确定DNA的序列, 该技术在保持测序准确度的同时, 主要解决了Sanger法测序通量低的问题, 二代测序可同时对几万到几百万条DNA序列进行测序, 因此也被称为高通量测序.现有平台主要包括Illumina Hiseq、MiSeq、NovaSeq 6000等.二代测序的进步极大推动了微生物组学的发展, 2006年,
The occurrence of potato common scab correlates with the community composition and function of the geocaulosphere soil microbiome
1
2019a
... (2)微生物与植物生长和健康.宏基因组学和宏代谢组学也被广泛用于探究微生物与宿主、环境之间的相互关系, 进而了解生物间相互作用的机制(
gcMeta: a global catalogue of metagenomics platform to support the archiving, standardization and analysis of microbiome data
1
2019b
...
Metatranscriptomics reveals unique microbial small RNAs in the ocean?s water column
1
2009
... 二代测序的原理是边合成边测序, 通过捕捉新合成的末端的标记来确定DNA的序列, 该技术在保持测序准确度的同时, 主要解决了Sanger法测序通量低的问题, 二代测序可同时对几万到几百万条DNA序列进行测序, 因此也被称为高通量测序.现有平台主要包括Illumina Hiseq、MiSeq、NovaSeq 6000等.二代测序的进步极大推动了微生物组学的发展, 2006年,
Microbial diversity in the deep sea and the underexplored “rare biosphere”
1
2006
... 微生物组学技术被广泛应用于揭示复杂环境中的微生物群落(包括多样性和群落组成)及其与周围环境或宿主之间的关系, 其研究对象涵盖土壤(
Microbial metabolomics
1
2011
... (4)微生物宏代谢组学(microbial metabolomics): 对微生物在特定生理时期内所有低分子量代谢物(包括代谢中间产物、激素、信号分子和次生代谢产物等)进行定性和定量分析, 并研究其与环境之间的相互作用(
Global diversity and geography of soil fungi
1
2014
... 微生物组学技术被广泛应用于揭示复杂环境中的微生物群落(包括多样性和群落组成)及其与周围环境或宿主之间的关系, 其研究对象涵盖土壤(
A communal catalogue reveals Earth’s multiscale microbial diversity
2
2017
... 微生物组学技术被广泛应用于揭示复杂环境中的微生物群落(包括多样性和群落组成)及其与周围环境或宿主之间的关系, 其研究对象涵盖土壤(
... 参考数据库的发展是限制微生物组数据分析的重要因素.据估计, 因缺乏参考基因组数据, 测序所得的宏基因组序列, 有7%-60%无法被准确分类(
Absolute quantitation of microbiota abundance in environmental samples
1
2018
... 基于标记基因的扩增子是大部分微生物组研究的基础, 但其样本检测步骤较多, 高通量测序读长较短、存在系统偏好性等问题, 且建库过程中使用了PCR扩增, 会导致某些序列可能被测了多次, 而有些量少的序列则无法被大量扩增, 不仅如此, PCR过程中可能会引入错配碱基, 从而造成信息的丢失.针对这些不足, 研究人员比较了不同文库制备方法, 利用标准样品详细分析扩增过程, 揭示了PCR扩增中造成错误和偏差的来源, 并提出了条件优化后的基于两步法的PCR扩增方法, 改进后的新方法能检测到更多现有方法中经常无法检测的类群, 同时提高微生物组研究的准确性(
Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil
1
2011
... 近年来, 从单细胞水平上分析微生物的生理代谢的单细胞技术迅速发展, 其中单细胞成像技术可将检测结果可视化, 能较好反映环境微生物类群、丰度及其功能活性(
Soil pH mediates the balance between stochastic and deterministic assembly of bacteria
1
2018
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
An invitation to the marriage of metagenomics and metabolomics
1
2008
... (1)微生物宏基因组学(microbial metagenomics): 通过提取环境微生物的全部DNA, 研究其群落组成、遗传信息及其与所处环境的协同进化关系(
Are multi-omics enough?
2
2016
... 参考数据库的发展是限制微生物组数据分析的重要因素.据估计, 因缺乏参考基因组数据, 测序所得的宏基因组序列, 有7%-60%无法被准确分类(
... 微生物组学的发展也离不开多组学结合的研究策略, 包括微生物分离培养、生态学鉴定和代谢产物分析等.尽管每种组学方法都对微生物组学研究提供了有价值的信息, 但仅仅基于DNA测序的方法分析微生物群落的多样性和群落组成, 并不能很好地研究微生物组的功能特征(
Host genotype and age shape the leaf and root microbiomes of a wild perennial plant
1
2016
... (2)微生物与植物生长和健康.宏基因组学和宏代谢组学也被广泛用于探究微生物与宿主、环境之间的相互关系, 进而了解生物间相互作用的机制(
Environmental microbial community proteomics: status, challenges and perspectives
3
2016
... (3)微生物宏蛋白质组学(microbial metaproteomics): 定性和定量地分析环境微生物在特定环境条件和特定时间下的全部蛋白质组分(
... 参考数据库的发展是限制微生物组数据分析的重要因素.据估计, 因缺乏参考基因组数据, 测序所得的宏基因组序列, 有7%-60%无法被准确分类(
... 微生物组学的发展也离不开多组学结合的研究策略, 包括微生物分离培养、生态学鉴定和代谢产物分析等.尽管每种组学方法都对微生物组学研究提供了有价值的信息, 但仅仅基于DNA测序的方法分析微生物群落的多样性和群落组成, 并不能很好地研究微生物组的功能特征(
质谱仪技术发展与应用
1
2009
... 质谱技术是宏蛋白质组学和宏代谢组学的核心技术, 通常结合色谱技术对复杂样品中的蛋白质或代谢物进行分析(
质谱仪技术发展与应用
1
2009
... 质谱技术是宏蛋白质组学和宏代谢组学的核心技术, 通常结合色谱技术对复杂样品中的蛋白质或代谢物进行分析(
宏组学方法在污水处理系统中的应用进展
1
2019
... 应用代谢组学对大量代谢物进行全面分析严重依赖于分离技术的发展.代谢组学常用的分析方法有气相色谱-质谱联用(GC-MS)、液相色谱-质谱联用(LC-MS)、核磁共振(NMR)和傅里叶转换红外光谱(FTIR).其中, GC-MS、LC-MS和NMR是最常用的手段, 能在一次运行中检测多种代谢物.GC-MS是一种高分辨率的色谱分离方法, 广泛用于挥发性和半挥发性有机物的分析, 适用于检测初级代谢产物脂肪酸、碳水化合物等, 大多数基于质谱的宏代谢组学研究均使用GC-MS进行, 但GC-MS对那些难挥发的物质分析效果不佳.LC-MS则是检测非挥发性化合物最常用的方法, 样品无需衍生化, 可直接检测代谢物, 比如黄酮类化合物、生物碱等次级代谢物, 以及氨基酸、碳水化合物等初级代谢物, 是分离分析复杂有机混合物的有效手段.除了质谱技术, NMR无需大量的样品制备和分馏工作, 可迅速检测样品中最丰富的代谢物, 也可以检测出质谱难以电离的化合物, 但与质谱技术相比, 其灵敏性较差、覆盖率低(
宏组学方法在污水处理系统中的应用进展
1
2019
... 应用代谢组学对大量代谢物进行全面分析严重依赖于分离技术的发展.代谢组学常用的分析方法有气相色谱-质谱联用(GC-MS)、液相色谱-质谱联用(LC-MS)、核磁共振(NMR)和傅里叶转换红外光谱(FTIR).其中, GC-MS、LC-MS和NMR是最常用的手段, 能在一次运行中检测多种代谢物.GC-MS是一种高分辨率的色谱分离方法, 广泛用于挥发性和半挥发性有机物的分析, 适用于检测初级代谢产物脂肪酸、碳水化合物等, 大多数基于质谱的宏代谢组学研究均使用GC-MS进行, 但GC-MS对那些难挥发的物质分析效果不佳.LC-MS则是检测非挥发性化合物最常用的方法, 样品无需衍生化, 可直接检测代谢物, 比如黄酮类化合物、生物碱等次级代谢物, 以及氨基酸、碳水化合物等初级代谢物, 是分离分析复杂有机混合物的有效手段.除了质谱技术, NMR无需大量的样品制备和分馏工作, 可迅速检测样品中最丰富的代谢物, 也可以检测出质谱难以电离的化合物, 但与质谱技术相比, 其灵敏性较差、覆盖率低(
The past, present and future of microbiome analyses
2
2016
... 质谱技术是宏蛋白质组学和宏代谢组学的核心技术, 通常结合色谱技术对复杂样品中的蛋白质或代谢物进行分析(
... ).传统的蛋白分析方法有凝集素亲和层析和Western蛋白印迹分析, 但一次实验仅能分析一到几个蛋白, 无法满足大规模分析的需求.20世纪70年代, 双向电泳的出现, 使得细胞系或细胞器的大量蛋白质能展示在双向凝胶上, 形成了狭义上的蛋白质组学概念, 该技术能对复杂蛋白质混合样品进行定性和定量分析, 但该技术对部分低丰度蛋白、极端等电点的蛋白以及一些疏水性蛋白分离效果不佳.近年来, 随着多重色谱分离与质谱联用技术的发展, 分离法在蛋白质组学分析中的应用日益广泛, 与传统双向凝胶电泳相比, 其分离效果要更优, 可以得到更为精确的蛋白质信息(
中国湖泊微生物组研究
1
2017
... 微生物与其所处环境构成了复杂的生态系统, 是地球上生物多样性的重要组成部分.微生物组(microbiome)是指一个特定环境或生态系统中全部微生物及其遗传信息的集合, 其内涵包括了微生物与其环境和宿主的相互作用(
中国湖泊微生物组研究
1
2017
... 微生物与其所处环境构成了复杂的生态系统, 是地球上生物多样性的重要组成部分.微生物组(microbiome)是指一个特定环境或生态系统中全部微生物及其遗传信息的集合, 其内涵包括了微生物与其环境和宿主的相互作用(
Ammonia- oxidizing bacteria rather than archaea respond to short-term urea amendment in an alpine grassland
1
2017
... (2)微生物对全球变化的响应.微生物对环境变化的响应较为敏感, 是全球气候变化的调节器, 因此, 研究微生物组对于环境变化的响应及反馈具有重要意义, 其结果可为预测全球变化背景下生态系统结构和功能的演变提供理论依据.利用微生物组学技术, 可探究土壤微生物群落对全球气候变化(
Emerging trends for microbiome analysis: from single-cell functional imaging to microbiome big data
1
2017
...
Soil fungal diversity in natural grasslands of the Tibetan Plateau: associations with plant diversity and productivity
2
2017
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
... (2)微生物与植物生长和健康.宏基因组学和宏代谢组学也被广泛用于探究微生物与宿主、环境之间的相互关系, 进而了解生物间相互作用的机制(
Phylogenetic imprint of woody plants on the soil mycobiome in natural mountain forests of eastern China
1
2019
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
环境微生物学的组学技术应用和突破
1
2013
... 生物信息学是伴随着人类基因组计划发展而来的学科, 包含了生物信息的获取、加工、存储、分配、分析、解释等在内的所有方面, 它综合运用数学、计算机科学和生物学的各种工具来阐明和理解大量数据所包含的生物学意义(
环境微生物学的组学技术应用和突破
1
2013
... 生物信息学是伴随着人类基因组计划发展而来的学科, 包含了生物信息的获取、加工、存储、分配、分析、解释等在内的所有方面, 它综合运用数学、计算机科学和生物学的各种工具来阐明和理解大量数据所包含的生物学意义(
宏蛋白质组学研究策略及应用
1
2009
... 从复杂环境中获得高保真性和高质量的样本是开展微生物组学研究的第一步.然而, 样品获取方法上的偏差仍是阻碍微生物组学研究的巨大挑战(
宏蛋白质组学研究策略及应用
1
2009
... 从复杂环境中获得高保真性和高质量的样本是开展微生物组学研究的第一步.然而, 样品获取方法上的偏差仍是阻碍微生物组学研究的巨大挑战(
Salinity is a key determinant for soil microbial communities in a desert ecosystem
2
2019
... 二代测序的原理是边合成边测序, 通过捕捉新合成的末端的标记来确定DNA的序列, 该技术在保持测序准确度的同时, 主要解决了Sanger法测序通量低的问题, 二代测序可同时对几万到几百万条DNA序列进行测序, 因此也被称为高通量测序.现有平台主要包括Illumina Hiseq、MiSeq、NovaSeq 6000等.二代测序的进步极大推动了微生物组学的发展, 2006年,
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
Effects of short-term warming and altered precipitation on soil microbial communities in alpine grassland of the Tibetan Plateau
1
2016
... (1)微生物生物地理.生物地理学是研究生物多样性时空分布的科学, 长期以来, 对生物地理分布特征的研究一直是生态学研究的重点, 包括多样性产生和维持机制(
生物信息学研究现状及发展趋势
1
2012
... 生物信息学是伴随着人类基因组计划发展而来的学科, 包含了生物信息的获取、加工、存储、分配、分析、解释等在内的所有方面, 它综合运用数学、计算机科学和生物学的各种工具来阐明和理解大量数据所包含的生物学意义(
生物信息学研究现状及发展趋势
1
2012
... 生物信息学是伴随着人类基因组计划发展而来的学科, 包含了生物信息的获取、加工、存储、分配、分析、解释等在内的所有方面, 它综合运用数学、计算机科学和生物学的各种工具来阐明和理解大量数据所包含的生物学意义(
Degradation pathways of 1-methylphenanthrene in bacterial Sphingobium sp. MP9-4 isolated from petroleum-contaminated soil
3
2017
... 微生物学研究大致可分为三个阶段.第一阶段: 在20世纪70年代以前, 主要采用传统的微生物分离培养技术获得菌株, 并进行一系列繁冗的生理生化分析, 因此人们对于微生物的认识基本停留在形态观察、描述、分类及生理学阶段.第二阶段: 从20世纪80年代开始, BIOLOG技术、磷脂脂肪酸法、DNA指纹图谱、基因芯片等分子生物学技术的兴起实现了不依赖于微生物培养, 而直接对环境微生物群落进行分析, 开创了微生物分子生态学研究的新时代.值得注意的是, 在DNA指纹图谱等技术的发展过程中, 还出现了第一代测序技术, 即Sanger法(
... 质谱技术是宏蛋白质组学和宏代谢组学的核心技术, 通常结合色谱技术对复杂样品中的蛋白质或代谢物进行分析(
... (3)环境污染微生物修复.微生物组学技术在环境污染治理方面也表现出了巨大潜力, 包括有机污染物降解、重金属转化和生物污染等领域.例如, 微生物组学技术可用于研究环境中多环芳烃(PAHs)的微生物降解机制.首先, 可以先通过对受到PAHs污染的土壤进行宏基因组学研究, 鉴定能降解PAHs的微生物及其关键基因(
中国土壤微生物组: 进展与展望
2
2017
... 微生物数量庞大、种类繁多, 是地球生物化学循环过程的驱动者, 是工农业生产、医药卫生和环境保护等关键领域的核心资源, 因此微生物组研究已成为新一轮科技革命的战略高地(
... 微生物与其所处环境构成了复杂的生态系统, 是地球上生物多样性的重要组成部分.微生物组(microbiome)是指一个特定环境或生态系统中全部微生物及其遗传信息的集合, 其内涵包括了微生物与其环境和宿主的相互作用(
中国土壤微生物组: 进展与展望
2
2017
... 微生物数量庞大、种类繁多, 是地球生物化学循环过程的驱动者, 是工农业生产、医药卫生和环境保护等关键领域的核心资源, 因此微生物组研究已成为新一轮科技革命的战略高地(
... 微生物与其所处环境构成了复杂的生态系统, 是地球上生物多样性的重要组成部分.微生物组(microbiome)是指一个特定环境或生态系统中全部微生物及其遗传信息的集合, 其内涵包括了微生物与其环境和宿主的相互作用(

{kind=link}
{kind=link}