0 引言
【研究意义】叶绿体是植物进行光合作用的关键场所,是细胞内具有自主遗传信息的重要细胞器,普遍存在于陆地植物、藻类和部分原生生物中[1]。与核基因组相比,叶绿体基因组较小,结构稳定,序列高度保守[2],遗传重组率低,且属于母系遗传,后代遗传稳定[3-4],其结构和序列信息在揭示物种起源、进化演变及不同物种之间亲缘关系等方面具有重要价值[1]。因此,可从叶绿体的角度对葡萄属(Vitis)植物进行系统发育研究,而寻找一个快速、高效、便捷获得葡萄属植物叶绿体基因组的方法,是展开其研究的前提。【前人研究进展】自1986年烟草(Nicotiana tabacum)[5]和地钱(Marchantia polymorpha)[6]的叶绿体基因组序列公布以来,科研工作者对不同物种叶绿体基因组结构及其变异规律越来越关注。传统上多采用先分离叶绿体并提取cpDNA,再通过Sanger测序或高通量测序的方法,最终拼接、组装得到植物的叶绿体基因组[7-9],但是此种方法多受叶绿体分离及cpDNA提取困难的限制,应用范围很狭小;另一种传统方法是利用叶绿体基因组保守序列设计特异引物,并对其全基因组DNA进行PCR扩增,对扩增产物进行Sanger测序,最终拼接、组装得到完整的叶绿体基因组[10-12],此方法不仅耗时长、操作繁琐、费用高,而且组装得到的叶绿体基因组序列不一定完整。近年来,随着高通量测序和生物信息技术的飞速发展,对植物全基因组DNA进行重测序,并对其数据进行相关分析,可快速、高效、便捷地得到植物叶绿体基因组全序列。利用此方法已成功组装出丹参(Salvia miltiorrhiza)[13]、人参(Panax ginseng)[14]、朝鲜蓟(Globe artichoke)[15]等的叶绿体基因组。葡萄属于葡萄科(Vitaceae)葡萄属(Vitis),是起源最古老的植物之一,也是中国重要的栽培果树,其营养价值高,经济效益好,具有重要的研究与开发利用价值[16]。葡萄属植物集中分布在3个起源中心,即欧洲–西亚分布中心、北美分布中心和东亚分布中心[17]。按照地理起源又分为3个种群,即美洲种群、东亚种群和欧亚种群。近年来,人们采用RAPD分子标记[18]、SRAP分子标记[19]、ISSR分子标记和叶绿体部分片段[20]等方法对葡萄的起源演化进行了研究,但是迄今为止仍有诸多问题尚未阐明,如野生葡萄与栽培种之间的进化关系[21],东亚种群中各个种之间的亲缘关系[22]等。科研人员发现叶绿体基因组的结构和序列信息可应用于高等植物复杂进化关系的系统发育研究。如HUANG等[23]利用叶绿体全基因组研究了山茶属(Camellia)13个种之间的亲缘关系;NIE等[7]基于叶绿体全基因组研究了菊科(Asteraceae)中假藿香蓟属(Ageratina)中的紫荆泽兰(Ageratina adenophora)与其他5个属植物之间的亲缘关系;JANSEN等[24]通过‘黑比诺’的叶绿体基因组研究了葡萄科(Vitaceae)植物与蔷薇类植物的系统发育关系,组装得到了‘黑比诺’的叶绿体基因组,并对其基因结构、基因顺序、GC含量和重复序列(正向重复序列和反向重复序列)进行了分析,但是并未对叶绿体基因组的其他特征,如串联重复序列、散在重复序列、SSRs和密码子偏好性进行分析,因此欧亚种葡萄叶绿体基因组的特征还有待补充。此外,获得‘黑比诺’叶绿体基因组序列的过程繁琐,而随着高通量测序技术的迅猛发展,可建立一种快速、高效获得植物叶绿体基因组的方法,为后续植物叶绿体基因组的特征分析奠定基础。叶绿体基因组可为解决葡萄属植物的物种起源、进化演变及亲缘关系等问题提供新的切入点。目前,葡萄属植物叶绿体基因组的相关研究多集中在rbcL和trnH-psbA等部分序列上[25-27],而如何得到葡萄的叶绿体全基因组,从叶绿体全基因组角度研究葡萄属植物的进化和系统发育成为大家关注的焦点。【本研究切入点】欧亚种葡萄‘赤霞珠’是酿造红葡萄酒的优良传统品种,在中国的栽培面积大,具有重要的经济价值,目前,在其生理生化方面已有诸多的研究报道,但关于其分子遗传机制的研究相对较少。因此,对‘赤霞珠’叶绿体基因组的研究可为其分子遗传机制提供信息。然而葡萄叶片中含有各种色素及单宁类物质[28]其叶绿体的分离及cpDNA的提取就非常困难,且根据叶绿体基因组保守序列设计特异引物,进行PCR扩增,对扩增产物进行Sanger测序的方法,耗时长、操作繁琐、费用高。因此,传统获得叶绿体基因组的方法并不适用于葡萄属植物。JANSEN等[24]利用已发表‘黑比诺’的公共BAC文库信息,首次得到其叶绿体基因组序列,但是此研究并未直接利用高通量测序技术,也未建立适于葡萄属植物叶绿体基因组组装及特征分析的方法流程。【拟解决的关键问题】本研究旨在摸索得到一套适用于葡萄属植物完整叶绿体基因组组装及特征分析的方法,补充欧亚种葡萄叶绿体基因组特征分析中缺失的部分,为葡萄属植物的进化和系统发育研究提供方法指导。1 材料与方法
1.1 试验时间、地点
试验于2016年4月—8月在中国农业科学院郑州果树研究所农业部果树育种技术重点实验室完成。1.2 试验材料
‘赤霞珠’嫩梢上幼叶采自中国农业科学院郑州果树研究所国家果树种质郑州葡萄圃。用锡箔纸包裹,经液氮速冻后-80℃保存备用。1.3 全基因组DNA的提取及测序
用植物基因组DNA提取试剂盒(TIANGEN Beijing China)提取‘赤霞珠’全基因组DNA,并送样测序。样品经北京诺和致源生物信息科技有限公司检测合格后,采用Illumina HiSeq PE150双末端测序策略进行建库测序,建库类型为350 bp DNA小片段文库,测序深度为10倍,样品所出数据量是5.2 G。1.4 叶绿体基因组组装
通过HiSeq PE150测序平台对样品进行测序,产生的原始数据(Raw Data)存在一定比例低质量短序列(reads),为了提高后续分析的可靠性,对Raw Data进行如下处理:(1)过滤某个位点N含量≥80%的tile里的所有reads;(2)截取read1、read2中高质量区域序列,具体为:正常GC数据保留质量值>20且碱基含量>40%的cycle,异常GC数据保留质量值>2且碱基含量>40%的cycle;(3)过滤低质量的reads,具体为:正常GC数据保留质量值>20且碱基含量>40%的reads,异常GC数据保留质量值>2且碱基含量>40%的reads;(4)过滤N值含量大于10%的reads;(5)当adapter序列与reads比对上15 bp或以上,错配数≤3时,滤掉此对reads;(6)当一对reads完全比对上其他的reads,则过滤冗余的reads,从而得到全基因组的有效数据(Clean Data)。以发表的拟南芥(NC 000932)和‘黑比诺’(DQ 424856)叶绿体基因组序列为参考,从Clean Data中抽提葡萄叶绿体reads,并用SOAPdenovo[29]2.04软件(http://soap.genomics.o rg.cn/soap denovo.html)进行组装,经多次调整参数后获得最优组装结果。使用GapCloser[29]1.12软件(http://so ap.genomics.org.cn/ soapdenovo.html)对组装结果进行内洞修补,最后去除冗余的短序列得到最后的组装结果。1.5 叶绿体基因组特征分析
用DOGMA[30]软件(http://dogma.ccbb.utexas.edu/)预测编码基因和非编码RNA,其中编码蛋白预测Identity阈值设置为40,其他参数为默认值;用BLAST[31]局部比对软件结合NR(http://www. ncbi. nlm.nih.gov/)、KEGG(http://www.genome.jp/kegg/)、COG(http://www.ncbi.nlm.nih.gov/COG/)、GO(http:// geneontology.org/)和Swiss-Prot(http://www.ebi. ac.uk/uniprot/)数据库对基因进行功能注释;用RepeatMasker[32]3-3-0(http://www.repeatmasker.org/)软件预测散在重复序列,TRF[33]4.04(http://tandem.bu. edu/trf/trf.html)软件预测串联重复序列;用OGDRAW[34]软件呈现‘赤霞珠’叶绿体基因组序列图;用MISA[35](MIcroSAtellite identification tool)软件分析SSR;用EMBOSS 6.4.0(http://emboss.open-bio.org/)分析蛋白编码基因密码子偏好性;用MEGA[36]6.0软件构建进化树。2 结果
2.1 全基因组测序与叶绿体基因组组装
基于高通量测序技术得到全基因组Raw data 5.2 G,去掉低质量reads后,得到Clean data 5.1 G,全基因组的GC(%)含量为38.62%,有效数据的Q20(%)为95.88,有效数据的Q30(%)为90.68。以拟南芥和‘黑比诺’叶绿体基因组为参考,并用BLASTN软件同全基因组的Clean data进行比对,从中抽提葡萄叶绿体reads。基于Phred/Phrap软件更适用于小基因组片段(如,BAC等),而SOAPdenovo软件更适用于Illumina测序数据,也适用于组装各种大小的基因组,且对测序错误率较为敏感的特点,采用SOAPdenovo短序列软件对抽提出的叶绿体reads进行初步组装,共得到6条Scaffolds(表1)。6条Scaffold去掉重叠区域(overlap)后,初步得到1条Scaffold,将全基因组reads比对回完整的Scaffold上,再根据reads的paired-end和overlap关系,对组装结果进行局部组装和优化,最后使用GapCloser软件对组装结果进行补洞得到1条完整Scaffold,即叶绿体基因组序列(图1)。Table 1
表1
表1‘赤霞珠’叶绿体reads组装结果
Table 1Assembly results of chloroplast reads in Cabernet Sauvignon
| Scaffold (num) | 一致性 Identity (%) | E值 E-value | 组装长度 Align length (bp) |
|---|---|---|---|
| 1 | 100 | 0 | 49338 |
| 2 | 100 | 0 | 32809 |
| 3 | 100 | 0 | 26354 |
| 4 | 100 | 0 | 26235 |
| 5 | 100 | 0 | 19310 |
| 6 | 100 | 0 | 6742 |
新窗口打开
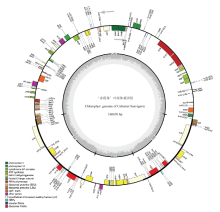 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1‘赤霞珠’叶绿体基因组序列图
外圈基因逆时针方向转录,内圈基因顺时针方向转录,不同的颜色表明基因不同的功能,小圈中深灰色表示GC含量,浅灰色表示AT含量
-->Fig. 1Sequence map of the Cabernet Sauvignon chloroplast genome
Genes drawn outside of the circle are transcribed counter-clockwise, while genes shown on the inside of the circle are transcribed clockwise. Genes belonging to different functional groups are color-coded. The darker gray in the inner circle indicates GC content, while the lighter gray corresponds to AT content
-->
将抽提的葡萄叶绿体reads比对到组装好的‘赤霞珠’叶绿体基因组序列上,统计组装序列的GC含量和reads覆盖深度,判断组装结果是否正常。理想情况下,GC-depth分布均呈泊松分布。分析结果表明覆盖度达99.99%,平均测序深度是1 700倍,可见组装效果非常好。
2.2 叶绿体基因组结构分析
‘赤霞珠’叶绿体基因组是共价闭合的双链环状分子,包含LSC、SSC、IRA和IRB 4个区段(图1)。序列全长160 676 bp,其中LSC区段长89 134 bp,SSC区段长19 072 bp,2个IR区段均为26 236 bp,且GC含量为37.43%。注释结果(表2)表明,‘赤霞珠’叶绿体基因组共有154个基因,包括99个蛋白编码基因,47个tRNA基因和8个rRNA基因。其中,20个基因含有2个拷贝,包括12个蛋白编码基因(psaB、ycf1、orf56、orf42、ycf68、rps12_3end、rps7、ndhB、ycf15、ycf2、rpl23和rpl2)、4个tRNA基因(tRNA-Lys、tRNA-Gly、tRNA-Pro和tRNA-Asn)和4个rRNA基因(rrn4.5、rrn5、rrn16和rrn23);3个tRNA基因(tRNA-Arg、tRNA-Ser和tRNA-Thr)含有3个拷贝;另外tRNA-Val、tRNA-Ile和tRNA-Ala含有4个拷贝;tRNA-Met和tRNA-Leu含有5个拷贝。蛋白编码基因序列总长89 574 bp,占叶绿体基因组序列的55.75%,GC含量为38.44%;tRNA基因序列总长2 960 bp,占叶绿体基因组序列的1.84%,GC含量为51.81%;rRNA基因序列总长9 036 bp,占叶绿体基因组序列的5.63%,GC含量为55.51%。在蛋白编码基因中,8个基因含有内含子(intron),其中6个基因(atpF、rpoC1、psaA、rpl2、ndhB和ndhA)仅含有1个内含子,另外2个基因(ycf3和clpP)则含有2个内含子。
Table 2
表2
表2‘赤霞珠’叶绿体基因组的基因列表
Table 2List of genes found in Cabernet Sauvignon chloroplast genome
| 基因分类 Category for genes | 基因分组 Group of genes | 基因名称 Name of genes |
|---|---|---|
| 表达相关基因 Self replication | 核糖体RNA基因 Ribosomal RNAs | 16S rRNA, 23S rRNA, 4.5S rRNA, 5S rRNA |
| 转运RAN基因 Transfer RNAs | tRNA-His, tRNA-Lys, tRNA-Gln, tRNA-Serd, tRNA-Gly, tRNA-Argd, tRNA-Cys, tRNA-Asp, tRNA-Tyr, tRNA-Glu, tRNA-Thrd, tRNA-Metf, tRNA-Leuf, tRNA-Phe, tRNA-Val, tRNA-Trp, tRNA-Pro, tRNA-Ilee, tRNA-Alae, tRNA-Asn | |
| 核糖体小亚基基因 Ribosomal protein (SSU) | rps16, rps2, rps14, rps4, rps18, rps11, rps8, rps3, rps19, rps7c, rps12_3endc, rps12, rps15 | |
| 核糖体大亚基基因 Ribosomal protein (SSU) | rpl33, rpl20, rpl36, rpl14, rpl16, rpl22, rpl2ac, rpl23c, rpl32 | |
| RNA聚合酶亚基基因 RNA polymerase | rpoC2, rpoC1a, rpoB, rpoA | |
| 光合作用相关基因 Genes for photosynthesis | 光合系统Ⅰ基因 Photosystem Ⅰ | psaBc, psaI, psaJ, psaC |
| 光合系统Ⅱ基因 Photosystem Ⅱ | psaAa, psbK, psbI, psbM, psbD, psbC, lhbA, psbJ, psbL, psi_psbT, psbT, psbH, psbN | |
| 细胞色素复合物基因 Cytochrome b/f complex | petN, petA, psbF, psbE, petG, petB, petD, petL | |
| ATP合酶基因 ATP synthase | atpA, atpFa, atpH, atpI, atpE, atpB | |
| 依赖ATP的蛋白酶单元p基因 ATP-dependent protease subunit p gene | clpPb | |
| 二磷酸核酮糖羧化酶大亚基基因 RubiscoCO large subunit | rbcL, rbcLr | |
| NADH脱氢酶基因 NADH dehydrogenase | ndhJ, ndhK, ndhC, ndhBac, ndhF, ndhD, ndhE, ndhG, ndhI, ndhAa, ndhH | |
| 其他基因 Other genes | 成熟酶基因 Maturase | matK |
| 包裹膜蛋白基因 Envelop membrane protein | ycf10, cemA | |
| 乙酰辅酶A羧化酶亚基基因 Subunit of acetyl-CoA-carboxylase | accD | |
| c型细胞色素合成基因 c-type cytochrome synthesis ccsA gene | ccsA | |
| 转录起始因子基因 Translation initiation factor IF-1 | infA | |
| 未知功能基因 Genes of unknown function | 假定叶绿体阅读框 Hypothetical chloroplast reading frames | ycf3b, ycf2c, ycf4, ycf15c, ycf68c, ycf1c |
| 开放性阅读框 ORFs | orf42c, orf56c, orf574 |
新窗口打开
按功能分类的基因在叶绿体基因组上的分布各异。
蛋白编码基因在叶绿体基因组的4个区段上均有分布;tRNA和rRNA基因的分布是不均匀的,其中,28个tRNA基因分布在LSC区段,18个tRNA基因分布在IR区段,仅有1个tRNA基因分布在SSC区段;所有的rRNA基因均分布在IR区段。
2.3 叶绿体基因组重复序列分析
重复序列包括串联重复序列(tandem repeat sequences)和散在重复序列(dispersed repeats)两大类。在‘赤霞珠’叶绿体基因组中共预测得到37个串联重复序列和53个散在重复序列。其中串联重复序列长度均在9—42 bp,绝大部分(35个)在11—42 bp,占叶绿体基因组序列的0.83%;散在重复序列包括19个长末端重复序列(LTR)、13个DNA转座子(DNA transposons)和4个长散在重复序列(LINE),它们的平均长度分别为142、115和64 bp,剩余的为未知重复序列,占叶绿体基因组序列的5.33%。这些重复序列可以开发成标记为种群的进化研究提供指导。2.4 叶绿体基因组SSRs开发
对‘赤霞珠’叶绿体基因组SSRs开发的参数设置如下:(1)1-10, 2-6, 3-5, 4-5, 5-5和6-5,即1个碱基重复≥10次;2个碱基重复≥6次;3个碱基重复≥5次;4个碱基重复≥5次;5个碱基重复≥5次;6个碱基重复≥5次。(2)2个SSR之间的最小距离设置为100 bp,若距离小于100 bp,则2个SSRs序列组成一个复合微卫星。结果表明,‘赤霞珠’叶绿体基因组含有50个SSR位点,其中,49个SSRs均由A或T组成,仅有一个SSR由C组成,这表明SSRs的碱基组成偏向使用A/T碱基。从SSRs的分布区段上看,39个SSRs位于LSC区段,7个SSRs位于SSC区段,而IR区域仅有4个SSRs,这表明SSRs在叶绿体基因组上的分布是不均匀的;从SSRs类型上看,除了大多数普通SSRs外,还得到6个复合微卫星,最大的是(TA)6有118 bp;从SSRs的碱基组成上看,有1个3碱基重复的SSR、1个2碱基重复的SSR和48个单碱基重复SSRs。
2.5 叶绿体基因组密码子偏好性
‘赤霞珠’叶绿体基因组中蛋白编码基因所对应的密码子偏好使用A/T碱基,其中第一个、第二个和第三个碱基为A/T碱基的密码子分别占总密码子数的53.90%、61.34%和69.43%。编码亮氨酸(L)的密码子使用频率最高,其次为异亮氨酸(I)和丝氨酸(S),而编码半胱氨酸(C)的密码子使用频率最低。2.6 系统发育分析
先前的科研工作者利用单基因和多基因对蔷薇类植物进行系统发育研究,得到7个主要分支,但是这些分支之间的关系仍未解决[37-40],其中就包括葡萄科。葡萄科的系统分类地位已争议多年,《克朗奎斯特分类法》[41]将其归在鼠李目(Rhamnales)下,而《APG分类法》[42]将其单列为不属于任何目的独立科。因此本研究从GenBank下载前人已发表的鼠李目(Vitales)、桃金娘目(Myrtales)、葫芦目(Cucurbitales)和豆目(Fabales)共24个物种的叶绿体基因组,同本研究的‘赤霞珠’叶绿体基因组序列进行比对。将蛋白编码序列比对结果导出至MEGA 6.0软件,采用邻接法(Neighbor-Joining Method)构建系统发育树(图2)。自举值(Bootstrap value)是基于500次抽样重复。结果表明,‘赤霞珠’与‘黑比诺’、夏葡萄和圆叶葡萄的亲缘关系最近,并与葡萄科的蛇葡萄(Ampelopsis glandulosa)和三叶青(Tetrastigma hemsleyanum)在同一分支。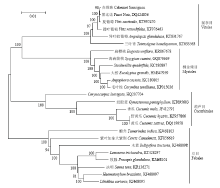 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2基于蛋白编码基因用邻接法构建25个物种的系统进化树
-->Fig. 2Phylogenetic tree of 25 species based on chloroplast protein-coding genes using neighbor-joining method (NJ)
-->
2.7 葡萄科内6种植物叶绿体基因组特征差异
表3表明,葡萄科内6种植物的叶绿体基因组序列长度为159 889—161 090 bp,它们的叶绿体基因组均表现为典型的环状四分体结构,且6种植物所对应的大单拷贝区和小单拷贝区的长度差异不大,而反向重复序列的长度差异较大,这可能是6种植物叶绿体基因组长度存在差异的原因。从叶绿体基因组的基因数目上看,‘赤霞珠’叶绿体基因组中的蛋白编码基因和tRNA基因总数最多,基因的拷贝数也最多。与‘黑比诺’、夏葡萄、圆叶葡萄、蛇葡萄和三叶青相比,在‘赤霞珠’叶绿体基因组中组装得到额外的10个叶绿体蛋白编码基因,它们是1hbA、rbcLr、ycf10、psi_psbT、rps12_3end、ycf15、ycf68、orf42、orf56和orf574。此外,葡萄科内6种植物叶绿体基因组中的基因结构、基因顺序和GC含量(%)是高度相似的,这与大多数已测被子植物叶绿体基因组类似,表明了叶绿体基因组序列的高度保守性[43-44]。Table 3
表3
表3葡萄科内6种植物叶绿体基因组特征
Table 3Characteristic analysis of six kinds of plants in Vitaceae
| 赤霞珠 Cabernet Sauvignon | 黑比诺 Vitis vinifera | 夏葡萄 Vitis aestivalis | 圆叶葡萄 Vitis rotundifolia | 蛇葡萄 Ampelopsis glandulosa | 三叶青 Tetrastigma hemsleyanum | |
|---|---|---|---|---|---|---|
| GenBank登录号 GenBank numbers | — | DQ424856 | KT997470 | KF976463 | KT831767 | KT033563 |
| 基因组大小 Size (bp) | 160 676 | 160 928 | 160 913 | 160 891 | 161 090 | 159 889 |
| 大单拷贝区长度 LSC (bp) | 89 134 | 89 147 | — | — | 89 266 | — |
| 小单拷贝区长度 SSC (bp) | 19 072 | 19 065 | — | — | 19 036 | — |
| 反向重复序列长度 IR (bp) | 26 235 | 26 358 | — | — | 26 394 | — |
| 基因总数 Number of total genes | 121 | 113 | 114 | 113 | 113 | 110 |
| 蛋白编码基因数 Number of protein-coding genes | 82 (12) | 79 (6) | 80 (6) | 80 (4) | 79 (6) | 78 (8) |
| tRNA基因数 Number of tRNA genes | 35 (12) | 30 (7) | 30 (7) | 29 (8) | 30 (7) | 28 (7) |
| rRNA基因数 Number of rRNA genes | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) | 4 (4) |
| GC含量 Overall GC content (%) | 37.4 | 37.4 | 37.4 | 37.4 | 37.4 | 37.6 |
新窗口打开
3 讨论
本研究采用高通量测序技术对‘赤霞珠’全基因组DNA进行重测序,并以拟南芥以及亲缘关系很近的欧亚种葡萄‘黑比诺’叶绿体基因组为参考,成功组装出其完整的叶绿体基因组。传统上植物叶绿体基因组的获取,如铁线蕨(Adiantum capillus-veneris L.)[45]、红藻(Cyanidioschyzon merolae)[46]、菝葜(Smilax china L.)[9]和紫荆泽兰(Ageratina adenophora)[7]等植物,是采用先分离叶绿体再提取cpDNA,并结合Sanger测序或高通量测序技术的方法,最终拼接、组装得到完整植物叶绿体基因组序列,但是此方法并不适于大范围使用,这是因为高等植物叶片往往含有较高含量的色素及单宁类物质,使得其叶绿体分离及cpDNA的提取较为困难[28]。另一种获得植物叶绿体基因组的传统方法是利用叶绿体基因组保守序列设计特异引物,并对其全基因组DNA进行PCR扩增,对扩增产物进行Sanger测序,最终拼接、组装得到完整的叶绿体基因组[10-12],但此方法不仅耗时长、操作繁琐、费用高,而且组装得到的叶绿体基因组序列不一定完整。而本研究采用高通量测序技术对植物全基因组DNA进行重测序的方法,克服了以上缺点,不需要分离植物叶绿体和提取cpDNA,只需提取其全基因组DNA,进行高通量测序,选取合适的叶绿体参考基因组,将所测得的全基因组序列与叶绿体参考基因组BLASTN比对,提取出相关的叶绿体reads,再用SOAPdenovo短序列软件对这些序列组装,根据序列的双末端和重叠序列的关系,进一步对组装结果进行局部组装和优化,最后使用GapCloser软件对组装结果进行补洞得到完整的叶绿体基因组。利用此方法已成功组装出长春花(Catharanthus roseus)[47]、凤梨(Ananas comosus)[48]和稗草(Echinochlon)[49]等的叶绿体基因组。与传统方法相比,此方法不需分离叶绿体和提取cpDNA,缩短了试验时间、降低了劳动强度和缩减了费用,并且极大的提高了试验的可行性。在成功组装出‘赤霞珠’叶绿体基因组后,本研究又从GenBank下载前人已发表的鼠李目(Vitales)、桃金娘目(Myrtales)、葫芦目(Cucurbitales)和豆目(Fabales)共24个物种的叶绿体基因组,同‘赤霞珠’叶绿体基因组做系统发育研究。结果表明,‘赤霞珠’与‘黑比诺’的亲缘关系最近,但是它们叶绿体基因组序列长度仍存在差异,造成这些长度差异的原因可能是:(1)全基因组数据来源不同,‘赤霞珠’全基因组数据是通过高通量测序得到的,‘黑比诺’全基因组数据来源于在线的BAC文库,这是用鸟枪法得到的;(2)组装二者叶绿体基因组的软件不同,‘赤霞珠’叶绿体基因组组装用的是主流的SOAPdenovo软件,而‘黑比诺’用的是现在不常用的Phred/Phrap 软件。此外,二者叶绿体基因组的基因种类和数量也存在差异,可能是因为在进行叶绿体基因组基因注释时,二者所用的数据库不同,‘赤霞珠’叶绿体基因组注释时用的是Swiss-Prot、NR、KEGG、COG、GO共5个数据库,‘黑比诺’叶绿体基因组注释时只用了自定义数据库(custom database)。本研究组装得到‘赤霞珠’叶绿体基因组的同时也补充了欧亚种葡萄‘黑比诺’叶绿体基因组研究中所缺少的密码子偏好性、重复序列和SSRs特征分析部分,可为欧亚种葡萄叶绿体基因组的研究提供更为详细、完善的数据。
本研究通过采用高通量测序的方法得到植物全基因组DNA数据,并以拟南芥和‘黑比诺’叶绿体基因组为参考,利用BLASTN序列比对抽提得到的葡萄叶绿体基因组序列占全基因组序列的8%。虽然叶绿体基因组序列只占全基因组序列的8%,但是已足够用以组装叶绿体基因组。此方法简单、高效,但是应用此方法要注意以下几点:(1)在该物种的科内外或属内外选择合适的参考基因组,以便从全基因组序列中得到相关的叶绿体基因组序列;(2)所选的参考基因组必须与所研究物种的亲缘关系很近;(3)在高等植物中,一些叶绿体基因会转移到细胞核中,因此在从全基因组序列中提取分离得到的叶绿体序列可能会来自细胞核[50]。
4 结论
采用高通量测序技术对植物全基因组DNA进行重测序的方法,选择合适叶绿体参考基因组,并结合相关的生物信息技术,成功组装出‘赤霞珠’的完整叶绿体基因组。与传统获得叶绿体基因组的方法相比,此法不需要分离叶绿体和提取cpDNA,大大缩短了试验时间、降低了劳动强度,并且极大的提高了试验的可行性。同葡萄科内另5种植物相比,在‘赤霞珠’叶绿体基因组中组装得到额外的10个叶绿体蛋白编码基因,它们是1hbA、rbcLr、ycf10、psi_psbT、rps12_3end、ycf15、ycf68、orf42、orf56和orf574;‘赤霞珠’叶绿体基因组的基因结构、基因顺序、GC含量和密码子偏好性均与典型的被子植物叶绿体基因组类似。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}