0 引言
【研究意义】牦牛作为青藏高原地区特有的反刍动物,对高海拔、低氧、低温等恶劣生活环境具有良好适应能力,能够为当地牧民提供乳、肉、皮毛及燃料等不可或缺的生产生活资料[1],是高原畜牧业最重要的畜种。作为反刍动物,牦牛能够进行反刍再次咀嚼饲草并通过瘤胃内微生物菌群发酵,将难以消化的纤维素转化为可以被小肠吸收的蛋白质、氨基酸、糖类、酸类和脂类等[2-3]。研究发现反刍动物瘤胃内栖息着大量微生物,这些微生物在动物胃肠道中扮演着重要角色[4]。青藏高原作为全球为数不多的未受大规模环境污染的地方之一,生态环境仍处于比较原始的状态。高原牧区由于畜牧医疗技术的相对落后及抗生素等药物的慎用,保证了牦牛瘤胃中的微生物群落是较接近于原始状态,未受到人为干扰的。因此,牦牛是研究瘤胃微生物群落结构的理想模式动物。【前人研究进展】近年来,随着高通量测序技术的不断发展及成熟,该技术在探究及鉴定环境微生物群落中也得到了广泛的应用,这种方法突破了微生物分离培养的限制,能够全面分析自然界的微生物类群及充分挖掘基因资源[5-8]。然而,对于这种基于核酸的分子生物学技术,DNA提取质量的好坏会直接影响到微生物群落结构的分析结果[9-16]。在现有的研究中,李旦等[17]对瘤胃微生物总DNA提取方法进行了比较,但其所提取DNA能否用于高通量测序并未得到验证。刘薇等[18-19]对奶牛瘤胃微生物总DNA的提取方法进行了研究与优化,徐文华等[20]对秦川牛瘤胃微生物DNA提取方法进行了比较。然而关于牦牛瘤胃微生物的提取方法尚未见相关报道。【本研究切入点】由于采食环境的特殊性,与规模舍饲养殖的牛群相比,牦牛瘤胃内往往含有大量的腐殖质和泥沙,这些物质会对DNA提取产生影响并增加基因组提取的难度。【拟解决的关键问题】因此探索一套适用于牦牛瘤胃内容物宏基因组的提取方法对于牦牛瘤胃微生物群落的鉴定具有重要意义。1 材料与方法
1.1 主要试剂
NaCl、KH2PO4、Na2HPO4、KCl、CTAB、SDS及溶菌酶均购自德国Sigma公司;氯仿、苯酚、异戊醇、异丙醇等均为国产分析纯;QIA amp DNA Stool Mini Kit 购于德国QIAGEN公司;λ-HindⅢ digested Marker及PCR Taq酶购自天根生化科技(北京)有限公司。1.2 瘤胃内容物的采集与处理
牦牛选自海拔3 700 m左右的阿坝州红原县(北纬N32°47′35.50″, 东经E102°32′34.32″)。所选牦牛均为检验检疫合格及未饲喂任何精饲料和抗生素的4岁左右公牦牛。于2014年9月在红原县高原屠宰场,在目标牦牛刚屠宰后取出的瘤胃背部切开一小口,手戴一次性无菌手套,持灭菌的50 mL离心管,探入瘤胃内容物中部,打开离心管盖,随机取瘤胃内容物装入离心管,取样过程中可搅动瘤胃内容物使样品更均匀,离心管充满后立刻盖严,并迅速取出,做好标记和记录,立即投入干冰中冻存,及时带回实验室于-80℃冰箱中保存。1.3 瘤胃细菌菌体获得
向3 g牦牛瘤胃样品(3头牦牛瘤胃内容物混合物)中加入10 mL经4℃预冷PBS缓冲液,待样品融化后涡旋振荡1 min,4℃、1 000 r/min离心5 min,吸取上清液;样品重复洗涤3次;将4次所得上清液于4℃、12 000 r/min离心10 min,弃掉离心管上层清液,向每管底部沉淀各加入1.7 mL TE缓冲液,混匀后吸取液体至同一10 mL灭菌离心管中,充分震荡混匀,-80℃保存备用。1.4 瘤胃细菌基因组DNA提取
瘤胃微生物DNA的提取最关键的是如何有效地裂解细胞,本试验结合了瘤胃的特点及目前常用的裂解方法,具体为两种物理方法:珠磨法(bead-beating)、反复冻融法(freeze-thaw);两种化学方法:表面活性剂十二烷基磺酸钠(sodium dodecyl sulfate,SDS)提取液、溴化十六烷甲基铵(cetyltrimethyl ammonium bromide,CTAB)提取液,两种酶解法:溶菌酶(lysozyme)、蛋白酶K(proteinase K);组合生成了9种不同的瘤胃样品DNA提取方法(表1),即方法1(CTAB+SDS+Lysozyme+无特殊物理处理法)、方法2(CTAB+SDS+Lysozyme+反复冻融法)、方法3(CTAB+SDS+Lysozyme+珠磨法)、方法4(CTAB+ Lysozyme+无特殊物理处理法)、方法5(CTAB+ Lysozyme+反复冻融法)、方法6(CTAB+Lysozyme+珠磨法)、方法7(SDS+Lysozyme+无特殊物理处理法)、方法8(SDS+Lysozyme+反复冻融法)、方法9(SDS+Lysozyme+珠磨法)。同时以QIA amp DNA Stool Mini Kit(方法10)为对照,来比较它们的DNA提取效率,每种方法均取样品600 µL(方法10按说明书进行提取),所有方法均经过蛋白酶K处理。每种方法进行3次重复。Table 1
表1
表1基因组DNA提取方法汇总表
Table 1Summary table of genomic DNA extraction methods
| 裂解方式 Pyrolysis method | 基因组提取方法 DNA extraction methods | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| 十二烷基硫酸钠 CTABa | + | + | + | + | + | + | - | - | - | - |
| 溴化十六烷甲基铵 SDSb | + | + | + | - | - | - | + | + | + | + |
| 蛋白酶K Proteinase Kc | + | + | + | + | + | + | + | + | + | + |
| 溶菌酶Lysozymed | + | + | + | + | + | + | + | + | + | - |
| 反复冻融Freeze-thaw | - | + | - | - | + | - | - | + | - | - |
| 珠磨Bead-beating | - | - | + | - | - | + | - | - | + | - |
| DNA纯化柱 | - | - | - | - | - | - | - | - | - | + |
新窗口打开
样品经上述各方法处理后,分别加入溶菌酶3 mg,混匀后于37℃孵育1 h;加入等体积氯仿:异戊醇(24﹕1),抽提2次,4℃、12 000 r/min离心10 min;收集上清液,加入等体积苯酚﹕氯仿﹕异戊醇(25﹕24﹕1)混匀,4℃、12 000 r/min离心3 min ;吸取上清液,加入等体积异丙醇,室温放置10 min,12 000 r/min离心10 min。弃上清,沉淀加入70%冰乙醇洗涤2次,自然干燥。加入50 µL DEPC水,-20℃保存。
1.5 DNA浓度、纯度及片段完整性测定
采用分度光度计对提取的DNA溶液纯度、浓度进行检查,以0.8%琼脂糖凝胶电泳,λDNA作为Marker,电压为150V,电泳时间为45 min,对提取的DNA片段完整性进行检查。1.6 16S rRNA PCR扩增
将每种方法的3个样品混合形成1个DNA pool后进行PCR扩增。合成细菌V3到V4区带有barcode的16S rRNA 基因特异性引物:338F(5′-ACTCCT ACGGGAGGCAGCA-3′)和806R(5′-GGACTACHV GGGTWTCTAAT-3′)。PCR反应体系(50 µL):95℃预变性5 min;94℃变性45 s,56℃退火45 s,72℃延伸30 s,30个循环,72℃延伸10 min。PCR产物用2%琼脂糖凝胶电泳检测,使用AxyPrep DNA凝胶回收试剂盒(AXYGEN公司)切胶回收PCR产物,纯化的PCR产物经送至深圳华大基因研究院(BGI)进行16S rRNA高通量测序。1.7 16S rRNA高通量测序及数据处理
基于Illumina MiSeq PE300(Illumina, USA)测序平台,在保证上样浓度一致,测序量相同的基础上,对牦牛瘤胃细菌基因组进行16S rRNA高通量测序。Miseq测序得到的raw reads首先根据overlap关系进行拼接,同时对序列质量进行质控和过滤。序列过滤后,将那些具有高度相似性(97%)的序列归为一个OTU(Operational Taxonomic Unit),运用blast比对程序在RDP(Ribosomal Database Project)数据库进行物种分类学分析。根据以上分类学信息,估算出样品稀释性曲线(Rarefaction curves)[21-22]、Ace、Chao[22]菌群丰度指数及Shannon、Simpson[23]菌群多样性指数。2 结果
2.1 不同DNA提取方法效率比对
表2是采用不同提取方法得到的牦牛瘤胃细菌基因组DNA的产量和纯度的结果。结果表明,方法3及方法6提取的DNA具有较高的浓度及纯度,方法7—10缺乏CTAB阳离子去污剂,提取的DNA量显著低于其他方法(P<0.05)。另外,同样的化学及生物酶裂解条件下,结合珠磨法及反复冻融法能够显著地增强细胞裂解效率,提高DNA产量。表3可见优质序列的长度分布集中于301—400bp,达到99.48%的比例。从图1可以看出,DNA产物在23 130 bp左右出现一条较亮条带,证明提取DNA产物较完整。另外,方法3及方法6的DNA产物的条带较亮,证明回收的DNA产物浓度较大。16S rRNA PCR扩增结果显示,方法2和8所提取的样品经多次试验,PCR 产物目的条带太弱或未检测到。其余8种方法提取的样品PCR 产物目的条带大小正确,浓度合适,符合高通量测序的要求。Table 2
表2
表2不同提取方法DNA产量及纯度
Table 2Yield and purity of DNA extracted by each method
| 方法Method | 产量Yield (ng·μL-1) | OD260/280 |
|---|---|---|
| 1 | 336.61±7.22 | 1.60±0.08 |
| 2 | 455.30±6.55 | 1.53±0.12 |
| 3 | 527.48±5.62 | 1.85±0.13 |
| 4 | 324.12±6.88 | 1.72±0.12 |
| 5 | 384.81±7.82 | 1.79±0.21 |
| 6 | 525.55±11.22 | 1.88±0.11 |
| 7 | 59.71±12.11 | 1.82±0.21 |
| 8 | 39.03±0.23 | 1.65±0.12 |
| 9 | 49.64±0.75 | 1.81±0.15 |
| 10 | 62.05±0.24 | 1.81±0.15 |
新窗口打开
Table 3
表3
表3优质序列的长度分布
Table 3Length distribution of valid sequences
| 长度Length (bp) | 序列数Sequences | 百分比Percent (%) |
|---|---|---|
| 1-300 | 2 | 0.00 |
| 301-400 | 170338 | 99.48 |
| 401-500 | 890 | 0.52 |
| 501-600 | 1 | 0.00 |
新窗口打开
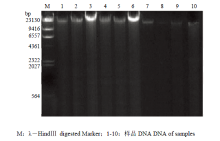 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1不同提取方法总DNA产物电泳
-->Fig. 1Electrophorotogram of total DNA of each methods
-->
2.2 16S rRNA高通量测序
由于高通量测序技术对DNA产量、纯度及片段完整性的要求限制,方法2及方法8不能够成功地进行高通量测序分析。其余8个方法中,高通量扩增分析大约生成了191 349条原始数据。质控过滤及去杂后,并根据序列末端的box序列校正序列方向,然后按照barcode标签序列识别并区分样品得到有效序列171 231条(表4)。可以看出各方法原始测序序列结果与各方法DNA产量具有一致性。Table 4
表4
表4不同提取方法DNA产物相关序列信息
Table 4Specific sequence information of each method
| 方法 Method | 序列数 Sequences | 碱基数 Bases (bp) | 平均片段长度 Average length (bp) |
|---|---|---|---|
| 1 | 25465 | 10022673 | 393.59 |
| 3 | 26080 | 10270236 | 393.8 |
| 4 | 23216 | 9134524 | 393.46 |
| 5 | 20325 | 7997392 | 393.48 |
| 6 | 29763 | 11722005 | 393.84 |
| 7 | 10446 | 4114615 | 393.89 |
| 9 | 15781 | 6211301 | 393.59 |
| 10 | 20155 | 7944212 | 394.16 |
新窗口打开
使用97%相似度的OTU,利用mothur做稀释性曲线分析并作图(图2),可知,随着测序深度的增加,曲线逐步趋于平坦,说明测序数据量合理并达到饱和,该数据量能够完整反映样品的菌群种类。
2.3 OTU聚类、分类学分析、多样性指数分析
经过OTU聚类数据统计、分类学和多样性指数分析,评价DNA提取方法效率。生成的Ace、Chao指数通常用于估计OTU数目指数;Shannon、Simpson指数则用于估计微生物多样性,Shannon值越大反映了样品中微生物多样性越丰富。从表5中可以看出,方法6和10包含的细菌较丰富。根据分类学分析结果,可以得知一个或多个样品在各分类水平上的比对情况。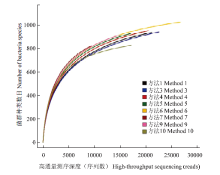 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图216S rRNA高通量测序稀释性曲线(97%相似水平)
-->Fig. 2Rarefaction curves of the rumen microbial community based on the 16S rRNA gene sequencing (at 0.97 identity level)
-->
Table 5
表5
表5OTU值及多样性指数
Table 5Specific OTUs and OTU-based diversity index
| 方法 Method | OTU | 多样性指数Diversity index (0.97) | |||
|---|---|---|---|---|---|
| Ace | Chao | Shannon | Simpson | ||
| 1 | 937 | 1069 | 1055 | 5.24 | 0.0143 |
| 3 | 942 | 1093 | 1119 | 5.24 | 0.0131 |
| 4 | 953 | 1084 | 1081 | 5.22 | 0.0177 |
| 5 | 929 | 1055 | 1052 | 5.4 | 0.0115 |
| 6 | 1026 | 1126 | 1131 | 5.46 | 0.0101 |
| 7 | 824 | 983 | 978 | 5.47 | 0.0097 |
| 9 | 914 | 1055 | 1062 | 5.44 | 0.011 |
| 10 | 829 | 898 | 920 | 5.48 | 0.0102 |
新窗口打开
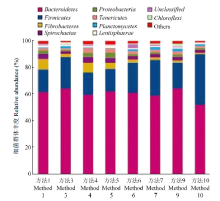
本试验依据样品群落结构分析,生成了不同样品间细菌群体结构柱状图(图3),该柱状图反映了两个信息:样品中含有何种细菌;样品中各细菌的序列数,即各细菌的相对丰度。分析结果显示牦牛瘤胃中丰度较高的细菌群是拟杆菌Bacteroidtes (64%)、厚壁菌Firmicutes(20%)、螺旋体Spirochaetae(2.3%)和变形菌Proteobacteri(1.8%),纤维杆菌Fibrobacter(1.7%)。同时,笔者还发现一些细菌群体在不同样品间所占比例存在一定差异,这可能是由于提取方法的不同导致细菌裂解效率存在一定差异。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3牦牛瘤胃细菌群体结构图(在门分类水平上)
-->Fig. 3Bacterial community structures of samples from yak rumen (at the phylum level)
-->
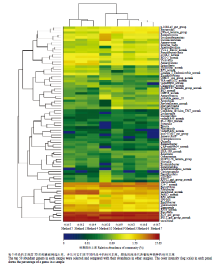
样品生成的菌体群体结构热点图(图4)中,颜色深浅代表数值大小,根据颜色变化直接判定DNA提取方法的优劣:蓝色条框(较低丰度)越少则反映这种方法提取效率越高。因此,方法6是较理想的DNA提取方法。另外,统观方法3、6和9,证明了珠磨法在微生物裂解过程中起着重要作用,并且重复性较好。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4基于样品丰度前50的细菌的热点图(在属分类水平上)
-->Fig. 4Heat map analysis of bacterial community based on top 50 genera
-->
2.4 不同提取方法对革兰氏阳性菌提取效果比对
革兰氏阳性菌一般具有较厚的细胞壁难以破裂,造成了DNA提取的难度。因此,根据不同方法对革兰氏阳性菌裂解效率来判定DNA提取方法的优劣[24]。图5列出了不同提取方法提取革兰氏阳性菌效率,从图5中可以看出,方法6裂解革兰氏阳性菌细胞壁的能力比其他方法相对较高,尤其是对丁酸弧菌的裂解能力比其他方法好。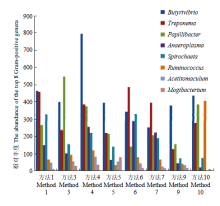 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5瘤胃丰度较高的8种革兰氏阳性菌在样品间的分布
-->Fig. 5The abundance of the top 8 Gram-positive genera in the different samples top 50 genera
-->
3 讨论
试验结果发现,珠磨法及反复冻融能够显著提高DNA产量,其中珠磨法裂解效果要优于冻融法。这是因为珠磨法在击打细菌细胞壁过程中,能够将DNA及蛋白质结合体打破,从而释放出更多的DNA。对于方法8和方法9而言,即便同时采用了珠磨及反复冻融法,但提取的DNA量显著低于其他方法,说明CTAB阳离子去污剂在DNA提取过程中具有重要作用。利用PCR扩增分析微生物群体构成的方法,DNA数量的多少并不是评价提取方法优劣的关键因素。例如,方法7和方法9 所提取DNA含量较低,但是菌落结构分析显示他们具有相似的群体结构。值得一提的是方法2提取的DNA含量较高,但其DNA却不能顺利地进行高通量测序分析,推测可能是由于该方法产生的切割力较强(CTAB+SDS+反复冻融法),易使样品微生物DNA断裂为较小碎片,而这些片段不能作为16S rRNA PCR目标区域的有效扩增模板所致。方法8提取的DNA不能进行高通量测序分析,是由于其DNA浓度没有达到Illumina MiSeq PE300(Illumina,USA)测序平台的最低浓度标准。16S rRNA测序后将那些具有高度相似性(97%)的序列归为一个OTU(Operational Taxonomic Unit),样品中OTU数目及基于OTU值生成的多样性指数可以用于评价DNA提取方法的优劣,通常,OTU值及Shannon多样性指数越大,则说明样品中具有较丰富的微生物群体。
因此,那些采用了珠磨法裂解的方法(方法3和6)具有较理想的裂解能力,它们能够有效地提取到不同微生物种类的DNA。QIAamp DNA Stool Mini Kit能够裂解不同微生物的细胞壁从而提取DNA,但由于后续纯化过程中部分DNA流失,导致了样品具有较少的OTU,却具有较大的Shannon多样性指数。所以,使用QIAamp DNA Stool Mini Kit提取的DNA生成的细菌群落结构不能真实反应牦牛瘤胃细菌分布情况。
本试验结果生成了不同样品间细菌群体结构柱状图及Heat-map,根据分析图可以看出不同提取方法生成的群体结构图存在着一定的差异,这可能与不同提取方法对不同细菌细胞壁裂解能力的差异性相关。革兰氏阳性菌具有较厚的细胞壁,并往往形成孢子,造成了细胞壁裂解难度大,如果提取裂解方法不理想,则得到革兰氏阳性菌的数量及种类会很少。因此可根据鉴定到的革兰氏阳性菌多少来判断DNA提取方法的有效性[24]。比较发现方法6中革兰氏阳性菌丰度较高,推测出方法6能够有效地裂解革兰氏阳性菌细胞壁。另外,由于其他方法的裂解能力较弱,生成的群体结构图相对于方法6实际上低估了革兰氏阳性菌含量而高估了革兰氏阴性菌含量。
目前,牦牛瘤胃内微生物还没有得到足够的关注,关于牦牛瘤胃微生物的文献较少,没有完整的阐述牦牛瘤胃菌群宏基因的提取方法。本试验除了筛选理想的牦牛瘤胃菌群DNA提取方法,另一个重要目的就是初步鉴定牦牛瘤胃内的细菌群体结构。将测序结果运用blast程序进行比对分析,归类结果显示:牦牛瘤胃体内至少包括21门、35纲、75科、112属类细菌。主要细菌门类包括拟杆菌(64%)和厚壁菌(20%),螺旋体(2.3%),变形菌(1.8%)及纤维杆菌 (1.7%)。GUO等之前的研究中得出牦牛瘤胃菌群主要分为两个大类,即厚壁菌(45.9%)和拟杆菌(39.68%),还存在少量的变形菌(4.85%)和螺旋体(2.47%),与本试验的结果相比,主要菌群的种类一致,在菌群结构上有一定的差异,这说明牦牛之间瘤胃基本微生物菌群结构大体一致,但由于地域不同、牧草种类不同以及采样季节不同可能会造成具体的菌群比例差异[25]。牦牛瘤胃细菌的种类与黄牛相比存在着较大差异:在门和属的水平上分别高于黄牛10.5%、105.5%。由于饮食及栖息环境的显著差异,牦牛瘤胃内可能含有一些特有细菌物种。然而,牦牛体内的主要细菌门类(拟杆菌、厚壁菌、螺旋体、变形菌、纤维杆菌)和黄牛相似,暗示了这些细菌在反刍动物体内代谢中的普遍具有重要功能[26-29]。
纤维杆菌与瘤胃球菌在瘤胃中是主要负责分解纤维的菌种。因为牦牛采食较为粗糙的草料,所以猜测在牦牛的瘤胃中,分解纤维的菌种数量更多。本试验的分析结果显示,牦牛瘤胃中的纤维杆菌大约占1.7%,与黄牛相似,但瘤胃球菌(0.4%)明显少于黄牛(5.0%),这一明显的差异可作为今后研究的重点。
普氏菌属是一类具有多种功能的菌种,它促进最初的蛋白质分解并协同其他分解纤维素的菌种发挥作用,提高机体对纤维素的分解能力。在成年奶用牛和黄牛的瘤胃微生物分析中可见普氏菌属是一类很重要的菌种,大约占有40%—50%。但是,在本试验中,牦牛瘤胃微生物菌群分析可见,普氏菌属仅占有15%,与黄牛差异明显,这可能与日常饮食有关。奶用牛和黄牛主要饲喂高质量的草料和谷物,而牦牛主要饲喂较为粗糙的草料,这提示饮食结构对瘤胃微生物菌群结构有一定的影响。PETRI[30]等的研究中也提到,不同的饮食结构,普氏菌属所占比例有明显的变化,饲喂草料、混合饲料、高谷物饲料的黄牛,普氏菌属所占比例分别为8.9%、12.8%和31.6%。
另外,高通量测序结果显示牦牛瘤胃体内有大量的未被描述和鉴定的细菌。例如, RC9_gut_group及BS11_gut_group_norank 菌种分别占整个微生物菌体的13.12%,10.10%。这些微生物可能在牦牛瘤胃体内扮演着重要的生理及生态角色。
4 结论
本试验首次对牦牛瘤胃细菌宏基因组提取方法进行了比较研究。方法6(CTAB+Lysozyme+ 珠磨法)提取的DNA可用于16S rRNA高通量测序及其他分子生物学操作,被证明是较理想的牦牛瘤胃细菌基因组提取方法,QIAamp DNA Stool Extraction Kit试剂盒提取的瘤胃DNA不适宜用于分析细菌菌群结构。另外,牦牛瘤胃内存在大量未鉴定的微生物,这些微生物将是下一步研究的重点方向。因为不同的饮食结构,牦牛与黄牛的瘤胃微生物菌群结构有着明显的差异,牦牛瘤胃中有大量的未被描述的细菌,它们的特殊功能和所扮演的生态角色仍然是未知的,有待在今后的研究中进一步发掘。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}