0 引言
【研究意义】磷是植物生长发育必需营养元素之一,磷低效利用限制大豆产量和品质。酸性磷酸酶能够提高土壤有机磷利用效率、改良植物磷素营养性状,因此,研究该类基因的表达调控对揭示其作用机制具有重要意义。植物基因表达受一系列顺式作用元件与反式作用因子的调控,而植物基因启动子在基因转录调控中起关键作用,通过分析基因启动子表达特性能够为揭示酸性磷酸酶基因调控机制提供理论依据[1-4]。【前人研究进展】紫色酸性磷酸酶(purple acid phosphatase,PAP)广泛存在于植物、动物、微生物,具有5个保守基序、7个高度保守氨基酸残基和1个金属离子双核中心。LI等[5]分离得到拟南芥29个PAPs基因,半定量PCR发现,低磷条件下7个PAPs基因(AtPAP7—AtPAP13)诱导表达;HUR等[6]研究发现,OsPAP2仅受低磷环境影响而与其他元素无关;肖凯等[7-8]将MtPAP1转入拟南芥,植酸盐条件下,超表达植株生物产量、无机磷与全磷含量明显提高;WANG等[9]将AtPAP15转入大豆,在酸性土壤中3个转基因株系产量性状均明显优于野生型;BOZZO等[10]发现番茄酸性磷酸酶可水解胞外磷脂化合物释放出无机磷;LIANG等[11]发现菜豆酸性磷酸酶PvPAP3可利用胞外ATP作为磷源。关于大豆紫色酸性磷酸酶基因,LI等[12]利用已测序大豆基因组数据,通过同源比对方法进行预测,认为大豆基因组中可能存在35个紫色酸性磷酸酶基因。迄今为止,已克隆并开展功能研究的仅有4个(GmPAP1—GmPAP4),其中GmPAP1、GmPAP2通过半定量分析显示低磷条件下诱导表达,但其功能并未深入开展,对GmPAP3的研究发现,该基因仅受盐胁迫诱导,而不受低磷胁迫诱导,是一个与大豆耐盐相关的基因[13]。GmPAP4是河北农业大学农学院大豆育种创新团队前期克隆,定量PCR结果显示植酸磷条件下该基因诱导高效表达,并证实其超表达能显著提高转基因拟南芥有机磷的利用效率[14]。而关于紫色酸性磷酸酶启动子,目前主要在拟南芥进行了研究。ROBINSON等[15]分析AtPAP12启动子发现,正常磷条件下,转基因植株GUS不表达,而低磷诱导下转基因植株GUS染色明显,说明AtPAP12启动子受低磷诱导表达。对AtPAP10研究发现,正常磷条件下转AtPAP10:GUS植株根系表达主要集中在维管组织,而低磷诱导下的GUS表达遍布整个根系[16]。【本研究切入点】前人关于紫色酸性磷酸酶启动子研究主要集中于拟南芥、水稻等模式植物,大豆中未开展相关研究,本文作者前期对大豆紫色酸性磷酸酶基因GmPAP4提高植物有机磷利用功能进行了验证,然而关于其如何调控发挥作用还有待深入研究。【拟解决的关键问题】本研究通过克隆GmPAP4上游启动子,分析启动子序列,转基因拟南芥分析启动子的组织表达和不同磷环境下启动子活性的定量检测,了解GmPAP4的调控模式,为今后相关转录因子的研究奠定基础。1 材料与方法
1.1 试验材料
大豆品种中黄15、拟南芥(Arabidopsis thaliana)Columbia、农杆菌GV3101、植物表达载体pBI121由河北农业大学农学院大豆育种创新团队提供。大肠杆菌Top10、pMD19-T vector、限制性内切酶等HindⅢ和BamHⅠ等购自宝生物工程(大连)有限公司,X-gluc购自Sigma,其他化学试剂购自生工生物工程(上海)股份有限公司。1.2 大豆叶片基因组DNA的提取
采用CTAB法提取大豆叶片基因组DNA,1%琼脂糖凝胶电泳检测DNA质量,Nanodrop1000测定DNA浓度。1.3 GmPAP4启动子克隆
依据GmPAP4 cDNA序列(GenBank No. HQ162477),利用phytozome检索其上游序列[17],设计特异引物PAP4-pro-F/R(表1),以中黄15叶片DNA为模板,采用LaTaq(TaKaRa)进行PCR扩增(扩增体系与程序参照说明书);扩增产物经1%琼脂糖凝胶电泳检测,回收目的片段并与pMD19-T(TaKaRa)载体连接,转化大肠杆菌感受态细胞Top10、蓝白斑筛选,同时利用PAP4-pro-F/R进行菌液PCR筛选阳性克隆并通过测序分析获得序列正确的单克隆。1.4 GmPAP4启动子生物信息学分析
利用PlantCARE(http://bioinformatics.psb.ugent. be/webtools/plantcare/html/)、PLACE(https://sogo.dna. affrc.go.jp/)分析GmPAP4启动子调控元件。1.5 GmPAP4启动子驱动GUS表达载体构建
通过HindⅢ和BamHⅠ双酶切,将PAP4-pro替换pBI121载体上的35S启动子以构建GmPAP4启动子驱动GUS的表达载体。经T4 DNA连接酶连接,转化大肠杆菌感受态细胞Top10,菌液涂布于固体LB平板(含Kan 100 μg·mL-1),37℃倒置培养12 h后,挑取单菌落,接种于LB液体培养基(含Kan 100 μg·mL-1)。提取质粒,利用HindⅢ和BamHⅠ酶切鉴定重组植物表达载体PAP4-pro-GUS。1.6 农杆菌介导拟南芥遗传转化及分子检测
采用冻融法将PAP4-pro-GUS转入农杆菌菌株GV3101,浸花法转化拟南芥,收获成熟种子,并于MS固体培养基(含Kan 100 μg·mL-1)筛选阳性植株,提取阳性植株叶片DNA,利用跨载体引物PAP4-pro-GUS-F/R(表1)进行PCR检测,通过自交获得T3转PAP4-pro-GUS拟南芥,并用于后续的GUS染色和GUS表达分析。1.7 转基因拟南芥GUS染色与活性测定
GUS染色植株生长条件:转基因拟南芥T3种子播种于MS培养基。GUS染色方法:分别取相同时期的根、茎、叶、花组织,将各组织置于适量X-gluc(Sigma)染色液,37℃孵育12—16 h,并用75%、80%、100%乙醇进行梯度脱色,最后于奥林巴斯显微镜BX51(Japan)下观察GUS染色情况[17]。试验选取3个T3转基因株系,每个转基因株系取10株用于GUS染色,重复3次。GUS活性测定植株生长条件:转基因拟南芥T3种子播种于MS培养基,MS培养基设置2个磷素处理(适磷以KH2PO4为磷源;低磷以Phytate为磷源,两处理P浓度均为500 μmol·L-1,其他营养元素浓度不变)。植株生长20 d(温度24℃,光照16 h,黑暗8 h)后,取根系组织进行GUS活性测定[18]。GUS活性测定具体方法:提取转基因拟南芥根组织,液氮研磨,将约100 mg研磨样品置于1.5 mL离心管,吸取500 μL植物总蛋白提取液(康为世纪),震荡混匀,4℃孵育15 min,随后4℃ 12 000 r/min离心,取上清液即为提取的植物蛋白。通过BSA法测定提取蛋白的浓度,以4-甲基伞形酮酰-β-葡萄糖醛酸苷(MUG)为底物,在激发光365 nm,发射光455 nm条件下,测定GUS活性。试验选取3个T3转基因株系,每个转基因株系取10株用于GUS活性测定,重复3次。
1.8 转基因拟南芥GUS实时定量PCR检测
将转基因拟南芥种子播种于MS培养基,MS培养基设置2个磷素处理(适磷以KH2PO4为磷源;低磷以Phytate为磷源,两处理P浓度均为500 μmol·L-1,其他营养元素浓度不变)。植株生长20 d(温度24℃,光照16 h,黑暗8 h)后,取其根提RNA进行实时定量PCR。具体方法为设计实时定量PCR引物(基因引物为GUS-RT-F/R,内参引物为eEF1a-F/R,表1),采用SYBR®GreenⅠ荧光染料法进行实时定量PCR并通过2-△△CT法进行基因表达水平相对定量分析[19]。反应程序为95℃ 30 s;95℃ 10 s,58℃ 10 s,72℃ 15 s,40个循环;72℃ 10 min。试验选取3个T3转基因株系, 每个转基因株系取10株用于定量PCR,重复3次。Table 1
表1
表1本试验所用引物
Table 1PCR primer sequences in this study
| 引物名称 Primer name | 引物序列 Primer sequence (5′-3′) | 用途 Function |
|---|---|---|
| PAP4-pro-F/R | F:AAGCTTTAACGGTGAAGGAC R:GGATCCAATAACAGTAACAGTAAC | GmPAP4启动子克隆 Cloning of GmPAP4 promoter |
| PAP4-pro-GUS-F/R | F:CTACGAAGCGAAGCGGAA AAAG R:GCAATAACATA CGGCGTGACAT | 转基因植株检测 Detection of transgenic plants |
| GUS-RT-F/R | F:ATACCGAAAGGTTGGGCAGG R:CGGCAATAACATACGGCGTG | 荧光定量GUS GUS gene in qPCR |
| eEF1α-F/R | F:TACCTCCCAGGCTGATTGTG R:GGGTTGTAGCCGACCTTCTT | 荧光定量内参基因 Reference gene in qPCR |
新窗口打开
1.9 数据分析
采用SPSS 19.0分析软件,利用one-way ANOVA方法对试验中所有数据进行P<0.05水平的数据分析[20]。2 结果
2.1 大豆GmPAP4启动子克隆与生物信息学分析
根据设计的特异引物对中黄15的DNA进行PCR扩增,获得GmPAP4上游启动子,经测序显示该序列长度为2 049 bp,与williams 82大豆参考基因组相同。将起始密码子ATG的第一个碱基A定义为+1,通过PLACE和PlantCARE进行在线预测,结果显示,GmPAP4启动子除含有真核生物启动子必须的核心元件,还有其他调控元件:(1)组织特异调控元件:as1(根系特异表达调控元件)、Skn-1_motif(胚乳特异表达调控元件);(2)应答元件:TC-rich repeats(逆境胁迫反应调控元件)、Box-W3(真菌应答相关调控元件);(3)结合位点:MBS(MYB转录因子的结合位点)等(图1)。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1GmPAP4启动子调控元件预测
-->Fig.1Prediction of regulatory elements in GmPAP4 promoter sequence
-->
2.2 启动子表达载体构建及转基因拟南芥获得
利用HindⅢ/BamHⅠ将获得的GmPAP4启动子片段(PAP4-pro,2 049 bp)替换pBI121载体上的35S-pro构建GmPAP4启动子驱动GUS表达载体(PAP4-pro-GUS,图2-a),经转化大肠杆菌Top10,HindⅢ/BamHⅠ对重组质粒进行双酶切鉴定,表明PAP4-pro已替换pBI121中的35S-pro(图2-b)。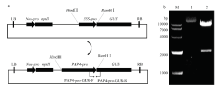 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2PAP4-pro-GUS载体构建a:PAP4-pro-GUS构建示意图。b:PAP4-pro-GUS HindⅢ/BamHⅠ双酶切检测,M:DNA分子量标准DL10000,1:未经HindⅢ/BamHⅠ双酶切PAP4-pro-GUS;2:经HindⅢ/BamHⅠ双酶切PAP4-pro-GUS
-->Fig. 2Construction of PAP4-pro-GUS a: Procedure of PAP4-pro-GUS construction. b: Detection of PAP4-pro-GUS digested with HindⅢ and BamHⅠ, M: DNA marker DL10000, 1: PAP4-pro-GUS undigested with HindⅢ and BamHⅠ; 2: PAP4-pro-GUS digested with HindⅢ and BamHⅠ
-->
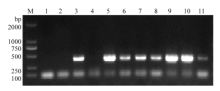
将重组表达载体PAP4-pro-GUS导入根癌农杆菌GV3101中,并通过农杆菌介导法将该载体转入拟南芥,利用跨载体引物PAP4-pro-GUS-F/R引物PCR(图2-a)扩增转PAP4-pro-GUS拟南芥,电泳结果显示目的片段大小为400 bp,与预期结果一致(图3)。通过自交加代最终获得转PAP4-pro-GUS拟南芥T3株系。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3转PAP4-pro-GUS拟南芥PCR检测M:DNA分子量标准DL2000;1:水对照;2:野生型对照;3:质粒对照;4—11:转PAP4-pro-GUS拟南芥
-->Fig. 3Detection of transgenic Arabidopsis plants with PAP4- pro-GUS by PCRM: DNA marker DL2000; 1: Water control; 2: Wild type control; 3: Plasmid control; 4-11: Transgenic Arabidopsis with PAP4-pro-GUS
-->
2.3 GmPAP4启动子组织器官表达特性分析
为了解GmPAP4启动子在不同时期和组织中的表达活性,分别对转基因拟南芥T3的根、茎、叶、花进行GUS染色。结果显示,GUS染色遍布转基因拟南芥整个根系且染色较深(图4-e),茎、叶中仅有微管组织有较明显GUS染色(图4-f,图4-g),花瓣微管组织中也能观察到微弱GUS染色(图4-h),而萌发种子未发现GUS染色(数据未列出)。这些结果预示着GmPAP4可能主要在根中表达,参与根系周围植酸磷的分解利用。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4转PAP4-pro-GUS拟南芥不同组织GUS染色分析
-->Fig. 4GUS staining in different tissues of transgenic Arabidopsis with PAP4-pro-GUS
-->
2.4 GmPAP4启动子植酸磷应答分析
为研究不同磷处理条件下GmPAP4启动子表达特性,分别采用适磷、植酸磷两种磷条件处理转PAP4-pro-GUS拟南芥T3植株,并测定其根系GUS表达及活性。定量PCR结果显示,与适磷处理相比,植酸磷处理条件下转基因拟南芥根系GUS表达提高了1.3倍(P<0.05,图5-a);同时GUS活性测定显示,与适磷处理相比植酸磷处理条件下转基因拟南芥根系GUS活性提高了1.9倍(P<0.05,图5-b)。结果表明,GmPAP4启动子能够对外界环境中的植酸磷处理作出响应,为揭示植酸磷处理条件下GmPAP4诱导表达提供了理论依据。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5不同磷处理下转PAP4-pro-GUS拟南芥根系GUS基因表达及活性测定a:不同磷处理条件下转PAP4-pro-GUS拟南芥根系GUS相对表达量;b:不同磷处理条件下转PAP4-pro-GUS拟南芥根系GUS活性测定;Pi:KH2PO4;Po:Phytate;*:P<0.05水平差异显著
-->Fig. 5GUS expression and activity measurement in roots of transgenic Arabidopsis with PAP4-pro-GUS under different phosphorus treatmentsa: Relative expression of GUS in roots of transgenic Arabidopsis plants with PAP4-pro-GUS under different phosphorus treatments; b: GUS activity measurement in roots of transgenic Arabidopsis plants with PAP4-pro-GUS under different phosphorus treatments; Pi: KH2PO4; Po: Phytate; *: The difference is significant at 0.05 level
-->
3 讨论
紫色酸性磷酸酶是一类水解酶,低磷条件下,植物根部的酸性磷酸酶活性提高,使植株根际周围更多的有机磷催化变为无机磷供植物利用。植物基因启动子作为关键调控元件,其序列分布着多种与基因功能密切相关的调控元件,使基因能够对外界环境作出及时响应。目前关于该类基因及其启动子研究主要限于模式植物,大豆中研究还有待开展[21-25]。前人通过对AtPAP15启动子研究发现,转基因植株根、茎、叶等组织都能观察到GUS染色,胁迫反应试验显示,未发现该启动子在低磷条件下表达活性显著提高[20]。DEL VECCHIO等[26]对AtPAP25启动子研究发现,该启动子仅在叶片表达,且低磷条件下GUS染色增强。OsPAP10c启动子研究发现,该启动子主要在植株根部表达,且低磷条件能诱导基因增强表达[27]。河北农业大学农学院大豆育种创新团队克隆了大豆紫色酸性磷酸酶基因GmPAP4,该基因具有分解有机磷的活性,实时定量PCR分析显示该基因在根部特异表达,且能够响应有机磷胁迫[14]。本研究克隆了GmPAP4启动子,通过转PAP4-pro-GUS拟南芥GUS染色结果显示,该启动子表达活性在不同组织存在较大差别,其中以转基因植株根系表达活性最高,同时发现,植酸磷胁迫条件下的转基因拟南芥根系GUS表达和活性都显著高于适磷处理,这一结果与OsPAP10c启动子表达特性类似。GNATATNC序列,作为磷饥饿诱导基因启动子上游的关键元件,能够与PHR1(作为磷调控网络中的核心调控因子)特异结合,使基因对低磷环境作出响应。拟南芥AtPHR1缺失抑制拟南芥中低磷应答相关基因表达,进一步研究发现,AtPHR1与下游磷饥饿诱导基因启动子序列上GNATATNC序列结合影响了磷相关基因的表达[28]。ZHANG等[24]对水稻27个PAP基因启动子生物信息学分析显示,有12个PAP启动序列中含有PIBS元件,且多位于启动子上游1 500 bp以内,如OsPAP10c启动子上游1 500 bp序列中含2个PIBS元件,同时多数基因启动子中PIBS元件个数与根系基因低磷诱导强度成正相关(各基因表达提高范围为2.7—61.5倍)。然而GmPAP4启动子上游2 049 bp序列中未发现PIBS元件,前期试验表明,低磷胁迫下GmPAP4表达最高提高了4倍,因此推测GmPAP4可能不受PHR1的调控,而启动子序列中的防御胁迫应答元件或许在诱导表达中发挥了关键作用。这与上述前人研究结果不一致,还需更多的试验数据作为支撑。
植物基因启动子按照表达模式不同,可分为组成型(如花椰菜花叶病毒启动子、玉米Ubiquitin启动子及根癌农杆菌胭脂碱合成酶基因启动子)[29-31]、特异型(如根特异、花特异、种子特异启动子)[32-34]和诱导型(拟南芥rd29A、小麦Em基因启动子)[35-36],其中以组成型启动子在植物基因工程中应用最多;但也伴随着一些问题,如持续过表达外源基因引起的异源蛋白大量增加带来的植物代谢平衡问题等。因此,采用组织特异型或诱导型基因启动子,实现外源基因的精准控制更有利于转基因植株的生长。本研究克隆的GmPAP4启动子中含有根特异表达调控元件和胁迫应答元件,若能够通过后续试验验证并分离这些特异表达调控序列,可为植物基因工程提供新的特异型基因启动子元件。
4 结论
获得大豆紫色酸性磷酸酶GmPAP4上游2 049 bp的启动子片段,并构建了融合GUS的植物表达载体PAP4-pro-GUS。该启动子主要在根部表达,且受低磷诱导。The authors have declared that no competing interests exist.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}