 ,1, 范小雪,1, 蒋海宾1, 王杰1, 冯睿蓉1, 张文德1, 余岢骏1, 隆琦1, 蔡宗兵1, 熊翠玲1, 郑燕珍1,2, 陈大福1,2, 付中民1,2, 徐国钧1,2, 郭睿,1,2
,1, 范小雪,1, 蒋海宾1, 王杰1, 冯睿蓉1, 张文德1, 余岢骏1, 隆琦1, 蔡宗兵1, 熊翠玲1, 郑燕珍1,2, 陈大福1,2, 付中民1,2, 徐国钧1,2, 郭睿,1,2MicroRNA-Mediated Cross-Kingdom Regulation of Apis mellifera ligustica Worker to Nosema ceranae
DU Yu,1, FAN XiaoXue,1, JIANG HaiBin1, WANG Jie1, FENG RuiRong1, ZHANG WenDe1, YU KeJun1, LONG Qi1, CAI ZongBing1, XIONG CuiLing1, ZHENG YanZhen1,2, CHEN DaFu1,2, FU ZhongMin1,2, XU GuoJun1,2, GUO Rui,1,2通讯作者:
责任编辑: 岳梅
收稿日期:2020-06-2接受日期:2020-06-22网络出版日期:2021-04-16
| 基金资助: |
Received:2020-06-2Accepted:2020-06-22Online:2021-04-16
作者简介 About authors
杜宇,E-mail:
范小雪,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (12391KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
杜宇, 范小雪, 蒋海宾, 王杰, 冯睿蓉, 张文德, 余岢骏, 隆琦, 蔡宗兵, 熊翠玲, 郑燕珍, 陈大福, 付中民, 徐国钧, 郭睿. 微小RNA介导意大利蜜蜂工蜂对东方蜜蜂微孢子虫的跨界调控[J]. 中国农业科学, 2021, 54(8): 1805-1820 doi:10.3864/j.issn.0578-1752.2021.08.019
DU Yu, FAN XiaoXue, JIANG HaiBin, WANG Jie, FENG RuiRong, ZHANG WenDe, YU KeJun, LONG Qi, CAI ZongBing, XIONG CuiLing, ZHENG YanZhen, CHEN DaFu, FU ZhongMin, XU GuoJun, GUO Rui.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】意大利蜜蜂(Apis mellifera ligustica,简称意蜂)作为重要的经济昆虫和授粉昆虫在养蜂生产、科学研究和生态多样性维持等方面具有不可替代的价值[1]。作为群居性昆虫,蜜蜂易遭受多种病原微生物的侵袭,其中东方蜜蜂微孢子虫(Nosema ceranae)是一种专性侵染成年蜜蜂中肠上皮细胞的单细胞真菌病原,可导致蜜蜂微孢子虫病,该病原还能与其他生物因子或非生物因子共同胁迫蜜蜂,严重危害蜜蜂健康和养蜂生产[2]。人们对于蜜蜂与东方蜜蜂微孢子虫的相互作用进行了较多研究[3,4,5],但对背后的分子机制还知之甚少。因此,探究微小RNA(microRNA,miRNA)介导意蜂工蜂对东方蜜蜂微孢子虫的跨界调控,不仅可为明确相关分子机制提供理论依据,也能加深对二者间互作的理解。【前人研究进展】在蜂群中,东方蜜蜂微孢子虫的孢子通过粪-口或口-口途径被蜜蜂宿主摄入体内,病原增殖高度依赖宿主细胞的物质和能量供应[3,4,5]。长期的协同进化使二者间形成了独特的互作关系,东方蜜蜂微孢子虫能抑制蜜蜂的免疫反应,引起消化系统紊乱,缩短蜜蜂寿命,并影响其定位、学习记忆和归巢能力等[3]。但对于蜜蜂能否跨界调控东方蜜蜂微孢子虫,相关研究还很滞后。MiRNA是一类长度约为18—25 nt的高度保守的单链非编码RNA(non-coding RNA,ncRNA),可通过靶向mRNA的3′ UTR抑制mRNA的翻译或使其降解,从而发挥转录后水平的调控作用[6]。近期研究发现,miRNA不仅在原生细胞中发挥功能,还能在物种之间相互传播,促进不同物种之间的串扰、交流或信号干扰[7,8,9,10]。2012年,ZHANG等首次证实植物来源的miR-168a可通过胃肠道吸收进入哺乳动物的肝细胞,通过抑制小鼠低密度脂蛋白受体的表达适配器蛋白1(LDLRAP1),减弱血浆中低密度脂蛋白的清除[7]。该团队还发现植物蜂粮来源的miR-162a通过抑制蜜蜂幼蜂的卵巢和整体的生长发育,阻止幼虫分化为蜂王并诱导趋向工蜂的发育过程[8]。目前被广泛认可的外源RNA介导的调控机制主要分为两种,一种是在秀丽隐杆线虫(Caenorhabditis elegans)[11]、赤拟谷盗(Tribolium castaneum)[12]和褐飞虱(Nilaparvata lugens)[13]等物种体内的系统性RNA干扰(RNAi)缺陷(SID)跨膜通道介导的远源dsRNA摄取,进而导致体内基因表达沉默,例如XU等[13]发现褐飞虱中外源性dsRNA通过siRNA途径触发基因沉默,SID-1是褐飞虱系统性RNAi必需的蛋白;另一种是miRNA可通过脱落囊泡(SV)、外泌体及凋亡小体等微囊泡(MV)腔室的包裹作用,以保护其在另一物种体内不被外源RNase酶降解,并进入到细胞体内发挥跨界调控基因表达的作用[14,15]。MiRNA作为关键的效应因子在宿主-病原互作中扮演关键角色[16,17]。CUI等[9]发现球孢白僵菌(Beauveria bassiana)可将bba-milR-1装载进囊泡并转运到斯氏按蚊(Anopheles stephensi)的细胞内,跨界调控宿主基因CLIPB9和Spz4的表达,球孢白僵菌在侵染前期通过下调Spz4的表达抑制Toll信号通路,而在侵染后期通过下调CLIPB9的表达以逃避黑化反应。MAYORAL等[10]通过印迹杂交证实沃尔巴克氏体(Wolbachia)的miRNA存在于埃及伊蚊(Aedes aegypti)纯化的细胞层中,并作为效应因子调节埃及伊蚊Dhc表达,促进自身增殖。蜜蜂与微孢子虫的跨界调控研究迄今仅有一例报道,HUANG等[18]合成东方蜜蜂微孢子虫Dicer的siRNA并饲喂给被该病原侵染的西方蜜蜂(Apis mellifera),通过深度测序和比较分析发现在侵染后1—6 d分别有7条宿主miRNA和5条病原miRNA发生差异表达,进一步推测东方蜜蜂微孢子虫miRNA可能被转运到宿主细胞质调控宿主的新陈代谢和免疫应答。笔者团队前期已对东方蜜蜂微孢子虫侵染意蜂工蜂过程中宿主的侵染应答机制和病原的侵染机制进行了进行一系列探索,系统解析了意蜂工蜂中肠的mRNA差异表达谱和免疫应答[19],miRNA差异表达谱及调控网络[20],差异表达lncRNA的多种调控方式及潜在功能[21],以及东方蜜蜂微孢子虫的高表达基因[22]、可变剪接基因[23]、差异基因[24]、差异miRNA的表达谱[25]。【本研究切入点】目前,有关意蜂与东方蜜蜂微孢子虫之间的跨界调控研究极为有限。笔者团队前期已对东方蜜蜂微孢子虫侵染意蜂工蜂过程中宿主的miRNA差异表达谱和病原的mRNA差异表达谱分别进行解析,可为进一步探究宿主差异表达miRNA(differentially expressed miRNA,DEmiRNA)跨界调控病原差异表达mRNA(DEmRNA)提供必要的数据基础。【拟解决的关键问题】通过生物信息学方法预测意蜂工蜂中肠DEmiRNA靶向结合的东方蜜蜂微孢子虫DEmRNA,对靶DEmRNA进行数据库注释和相关分析,进一步构建宿主DEmiRNA与病原DEmRNA的调控网络,并对调控网络中的病原DEmRNA进行分析和探讨,以期在组学水平解析DEmiRNA介导意蜂工蜂对东方蜜蜂微孢子虫的跨界调控,为阐明背后的分子机制打下基础。1 材料与方法
试验于2017年9月至2019年10月在福建农林大学动物科学学院(蜂学学院)蜜蜂保护实验室完成。1.1 供试生物材料
意蜂工蜂取自福建农林大学动物科学学院(蜂学学院)教学蜂场。东方蜜蜂微孢子虫感染的意蜂外勤蜂取自福州市闽侯县荆溪源安养蜂场。1.2 意蜂工蜂中肠的miRNA组学数据来源
笔者团队前期通过Percoll不连续密度梯度离心法对东方蜜蜂微孢子虫孢子进行纯化,并对意蜂工蜂进行饲喂接种及中肠样品的剖取[21]。前期已分别抽提东方蜜蜂微孢子虫侵染7 d和10 d的工蜂中肠样品(AmT1、AmT2)和未受侵染的工蜂中肠样品(AmCK1、AmCK2)的总RNA,并委托广州基迪奥生物科技有限公司通过Illumina MiSeq平台对建好的cDNA文库进行单端测序。笔者团队已对测序数据进行过滤和质控[26]:(1)剔除原始读段(raw reads)中含5′接头序列、含polyA、低质量的reads和剪切掉3′接头序列后的<18或>30个核苷酸的序列,得到高质量的有效序列标签(clean tags);(2)利用Bowite软件[27]将获得的clean tags比对GeneBank及Rfam(11.0)数据库,过滤比对上rRNA、scRNA、snoRNA和tRNA的clean tags,得到未注释的tags(unannotated tags);(3)比对东方蜜蜂微孢子虫参考基因组(assembly ASM98816v1)(
笔者团队前期已利用miRDeep2软件[28]将上述剩余的mapped tags与miRBase数据库中收录的miRNA前体序列进行比对,获得已知miRNA序列。同时,将未比对上的tags比对基因组,得到可能的前体序列,根据tags在前体序列上的分布信息和前体结构能量信息,采用贝叶斯模型经打分实现novel miRNA的鉴定。利用每百万标签序列(tags per million,TPM)公式(TPM=T×106/N,T表示miRNA的tags,N表示总miRNA的tags)对miRNA进行表达量的归一化处理。按照|log2fold change (FC)|≥1且P≤0.05的标准筛选AmCK1 vs AmT1和AmCK2 vs AmT2比较组的显著性DEmiRNA,用于本研究中靶向东方蜜蜂微孢子虫的mRNA和DEmRNA的预测和分析。
1.3 东方蜜蜂微孢子虫的mRNA组学数据来源
笔者团队前期按照1.2中的方法对意蜂工蜂进行饲喂接种及中肠样品制备,并利用基于链特异性cDNA建库的RNA-seq技术对接种的中肠样品进行测序,得到同时包含宿主数据和病原数据的混合mRNA组学数据[29]。将上述混合数据连续比对核糖体数据库、西方蜜蜂基因组(assembly Amel_4.5)和东方蜜蜂微孢子虫基因组(assembly ASM98816v1),筛滤得到处于侵染过程的病原mRNA组学数据[20]。其中,将侵染8日龄(即侵染后7 d)工蜂中肠内的东方蜜蜂微孢子虫设为NcT1(NcT1-1、NcT1-2和NcT1-3为3个生物学重复),侵染11日龄(即侵染后10 d)工蜂中肠内的东方蜜蜂微孢子虫设为NcT2(NcT2-1、NcT2-2和NcT2-3为3个生物学重复)。笔者团队前期也已利用基于链特异性cDNA建库的RNA-seq技术对东方蜜蜂微孢子虫的纯净孢子(NcCK:NcCK-1、NcCK-2和NcCK-3)进行深度测序,获得了高质量的mRNA组学数据[30]。测序原始数据已上传NCBI SRA数据库,Bioproject号分别为PRJNA395264(NcCK)和PRJNA406998(NcT1和NcT2)。笔者团队前期已采用FPKM(Fragments Per Kilobase of transcript per Million fragments mapped)算法计算和归一化基因表达量;利用edgeR软件[31]筛选NcCK vs NcT1和NcCK vs NcT2比较组的显著性DEmRNA,筛选标准为|log2FC|≥1且P≤0.05。上述病原DEmRNA可用于本研究中宿主显著性DEmiRNA的靶向预测及分析。
1.4 意蜂工蜂中肠显著性DEmiRNA靶向东方蜜蜂微孢子虫mRNA和DEmRNA的预测及分析
利用TargetFinder软件[32]预测AmCK1 vs AmT1和AmCK2 vs AmT2比较组中显著性DEmiRNA靶向结合的东方蜜蜂微孢子虫mRNA,以及NcCK vs NcT1和NcCK vs NcT2比较组的显著性DEmRNA,采用默认参数。利用OmicShare在线工具集合(www. omicshare.com)的相关工具对上述靶标mRNA和DEmRNA进行GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库注释,采用默认参数。1.5 意蜂工蜂中肠显著性DEmiRNA与东方蜜蜂微孢子虫mRNA和DEmRNA的调控网络构建及分析
根据1.4中预测出的意蜂工蜂中肠显著性DEmiRNA与东方蜜蜂微孢子虫mRNA和显著性DEmRNA的靶向结合关系,构建二者之间的调控网络,并利用Cytoscape软件[33]可视化调控网络。根据前人在微孢子虫和笔者所在课题组在东方蜜蜂微孢子虫方面的研究结果[34,35,36,37,38,39],孢壁蛋白、极管蛋白、蓖麻毒素B凝集素、糖酵解/糖异生途径以及ABC转运蛋白和ATP/ADP转位酶与微孢子虫的侵染和增殖活动关系密切,筛选与上述蛋白和途径相关的病原DEmRNA及存在靶向结合关系的宿主显著性DEmiRNA,并构建、分析及可视化调控网络。2 结果
2.1 意蜂工蜂中肠DEmiRNA靶向东方蜜蜂微孢子虫mRNA和DEmRNA的预测及分析
AmCK1 vs AmT1比较组中84条显著性DEmiRNA靶向结合东方蜜蜂微孢子虫的1 620条mRNA,其中宿主的48条显著上调miRNA和36条显著下调miRNA分别靶向病原的1 345和1 046条mRNA。进一步分析发现,宿主的47条显著上调miRNA可靶向NcCK vs NcT1比较组中病原的584条显著下调mRNA,34条显著下调miRNA可靶向病原的265条显著上调mRNA(图1-A、1-B)。AmCK2 vs AmT2比较组中107条显著性DEmiRNA共靶向结合东方蜜蜂微孢子虫的1 717条mRNA,其中宿主的56条显著上调miRNA和51条显著下调miRNA分别靶向病原的1 260和1 317条mRNA。进一步分析发现,宿主的52条显著上调miRNA可靶向NcCK vs NcT2比较组中病原的587条显著下调mRNA,49条显著下调miRNA可靶向病原的336条显著上调mRNA(图1-C、1-D)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1意蜂工蜂中肠显著性DEmiRNA靶向东方蜜蜂微孢子虫显著性DEmRNA的调控网络
A:AmCK1 vs AmT1中显著下调miRNA及其靶向NcCK vs NcT1中显著上调mRNA的调控网络Regulatory network of significantly down-regulated miRNAs in AmCK1 vs AmT1 and their target significantly up-regulated mRNAs in NcCK vs NcT1;B:AmCK1 vs AmT1中显著上调miRNA靶向NcCK vs NcT1中显著下调mRNA的调控网络Regulatory network of significantly up-regulated miRNAs in AmCK1 vs AmT1 and their target significantly down-regulated mRNAs in NcCK vs NcT1;C:AmCK2 vs AmT2中显著下调miRNA靶向NcCK vs NcT2中显著上调mRNA的调控网络Regulatory network of significantly down-regulated miRNAs in AmCK2 vs AmT2 and their target significantly up-regulated mRNAs in NcCK vs NcT2;D:AmCK2 vs AmT2中显著上调miRNA靶向NcCK vs NcT2中显著下调mRNA的调控网络Regulatory network of significantly up-regulated miRNAs in AmCK2 vs AmT2 and their target significantly down-regulated mRNAs in NcCK vs NcT2
Fig. 1Regulatory networks of significant DEmiRNAs in A. m. ligustica workers’ midguts and their target significant DEmRNAs in N. ceranae
进一步分析发现,AmCK1 vs AmT1和AmCK2 vs AmT2比较组包含8条共同显著上调miRNA,可靶向NcCK vs NcT1和NcCK vs NcT2比较组中92条共同显著上调mRNA和144条共同显著下调mRNA;此外,1条共同显著下调miRNA可靶向病原的10条共同显著上调mRNA和16条共同显著下调mRNA(图2)。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2AmCK1 vs AmT1和AmCK2 vs AmT2比较组的共同显著上调(下调)miRNA靶向NcCK vs NcT1和NcCK vs NcT2比较组的共同显著下调(上调)mRNA的调控网络
Fig. 2Regulatory networks of common significantly up-regulated (down-regulated) miRNAs in AmCK1 vs AmT1 and AmCK2 vs AmT2 and their target significantly down-regulated (up-regulated) mRNAs in NcCK vs NcT1 and NcCK vs NcT2
2.2 意蜂工蜂中肠DEmiRNA靶向东方蜜蜂微孢子虫mRNA和DEmRNA的功能注释
GO数据库注释结果显示,AmCK1 vs AmT1比较组中显著性DEmiRNA靶向东方蜜蜂微孢子虫的mRNA可注释到25个功能条目,包括代谢进程(312)、催化活性(279)和结合(274)等;AmCK2 vs AmT2比较组中显著性DEmiRNA靶向东方蜜蜂微孢子虫的mRNA可注释到25个功能条目,包括代谢进程(322)、催化活性(284)和细胞进程(277)等。AmCK1 vs AmT1中显著上调miRNA靶向NcCK vs NcT1中的584条显著下调mRNA,涉及代谢进程(84)和催化活性(82)等19个功能条目;宿主的显著下调miRNA靶向病原的265条显著上调mRNA,涉及代谢进程(70)和结合(63)等22个功能条目。AmCK2 vs AmT2中显著上调miRNA靶向NcCK vs NcT2中病原的587条显著下调mRNA,涉及代谢进程(76)和催化活性(75)等20个功能条目;宿主的显著下调miRNA靶向病原的336条显著上调mRNA,涉及代谢进程(76)和结合(69)等23个功能条目。进一步分析结果显示,AmCK1 vs AmT1和AmCK2 vs AmT2比较组8条共同显著上调miRNA可靶向NcCK vs NcT1和NcCK vs NcT2比较组中144条共同显著下调mRNA,涉及代谢进程(20)等18个功能条目(图3);而1条宿主的共同显著下调miRNA可靶向10条病原的共同显著上调mRNA,涉及代谢进程(4)等13个功能条目(图3)。括号内的数字表示注释在该条目的mRNA数量。
图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3AmCK1 vs AmT1和AmCK2 vs AmT2比较组的共同显著上调(下调)miRNA靶向NcCK vs NcT1和NcCK vs NcT2共同显著下调(上调)mRNA的GO数据库注释
1:代谢进程Metabolic process;2:细胞进程Cellular process;3:单一组织进程Single-organism process;4:应激反应Response to stimulus;5:信号Signaling;6:生物调节进程Biological regulation process;7:定位Localization;8:生物调节Biological regulation;9:细胞成分组织或生物合成Cellular component organization or biogenesis;10:细胞Cell;11:细胞组件Cell part;12:细胞器Organelle;13:细胞膜组件Cell membrane part;14:细胞膜Cell membrane;15:高分子复合物Macromolecular complex;16:细胞器组件Organelle part;17:催化活性Catalytic activity;18:结合Binding
Fig. 3GO database annotation of significantly down-regulated (up-regulated) mRNAs in NcCK vs NcT1 and NcCK vs NcT2 targeted by significantly up-regulated (down-regulated) miRNAs in AmCK1 vs AmT1 and AmCK2 vs AmT2
2.3 意蜂工蜂中肠显著性DEmiRNA靶向东方蜜蜂微孢子虫mRNA和显著性DEmRNA的通路注释
KEGG数据库注释结果显示,AmCK1 vs AmT1比较组中显著性DEmiRNA靶向东方蜜蜂微孢子虫的mRNA可注释到84条通路,包括代谢途径(107)、次生代谢物的生物合成(40)和核糖体(39)等;AmCK2 vs AmT2比较组中显著性DEmiRNA靶向东方蜜蜂微孢子虫的mRNA可注释到84条通路,包括代谢途径(118)、核糖体(50)和次生代谢物的生物合成(48)等。AmCK1 vs AmT1中显著上调miRNA靶向NcCK vs NcT1中的显著下调mRNA可注释到代谢途径(35)和核糖体在真核生物中的生物合成(17)等66条通路;宿主的显著下调miRNA靶向病原的显著上调mRNA可注释到代谢途径(29)和次生代谢物的生物合成(16)等64条通路。AmCK2 vs AmT2中显著上调miRNA靶向NcCK vs NcT2中的显著下调mRNA可注释到代谢途径(34)和细胞周期(15)等64条通路;宿主的显著下调miRNA靶向病原的显著上调mRNA可注释到代谢途径(35)和次生代谢物的生物合成(19)等65条通路。此外,AmCK1 vs AmT1和AmCK2 vs AmT2比较组的8条共同显著上调miRNA可靶向NcCK vs NcT1和NcCK vs NcT2比较组中144条共同显著下调mRNA,可注释到代谢途径(10)和RNA转运(5)等38条通路;1条宿主的共同显著下调miRNA可靶向10条病原的共同显著上调的mRNA,可注释到代谢途径(2)和抗生素的生物合成(1)等7条通路。括号内的数字表示注释在该通路的mRNA数量。
2.4 意蜂工蜂中肠显著性DEmiRNA及其靶向的东方蜜蜂微孢子虫毒力因子/侵染因子相关DEmRNA的调控网络
在AmCK1 vs AmT1比较组中,分别有13条显著上调miRNA(miR-8212-y、miR-374-y和miR-590-y等)和9条显著下调miRNA(miR-291-y、miR-409-y和miR-326-y等)靶向4条ABC转运蛋白编码基因相关的显著DEmRNA,分别有7条显著上调miRNA(ame-miR-6052、miR-501-y和miR-767-x等)和1条显著下调miRNA(miR-381-y)靶向3条ATP/ADP转位酶编码基因相关的显著DEmRNA(图4-A、表1、表2)。分别有9条显著上调miRNA(miR-193-y、ame-miR-193和miR-590-y等)和1条显著下调miRNA(miR-451-x)靶向NcCK vs NcT1比较组中的3条极管蛋白编码基因相关的显著DEmRNA,分别有5条显著上调miRNA(miR-941-y、miR-16-y和novel-m0007-5p等)和1条显著下调miRNA(miR-291-y)靶向1条孢壁和锚定吸盘复合蛋白编码基因和1条孢壁蛋白编码基因相关的显著DEmRNA,分别有8条显著上调miRNA(ame-miR-6052、miR-8212-y和miR-144-x等)和6条显著下调miRNA(miR-409-y、miR-294-y和miR-291-x等)靶向7条蓖麻毒素B凝集素编码基因相关的显著DEmRNA(图4-B、表1、表2)。分别有21条显著上调miRNA(miR-8232-x、miR-144-x和miR-767-x等)和10条显著下调miRNA(miR-291-y、miR-381-y和miR-462-y等)靶向10条糖酵解/糖异生途径相关的显著DEmRNA(图4-C、表1、表2)。此外,分别有7条显著上调miRNA(miR-224-x、novel-m0007-5p和miR-16-y等)和2条显著下调miRNA(miR-155-x和miR-291-x)靶向MAPK信号通路相关的5条显著DEmRNA(表1、表2)。图4
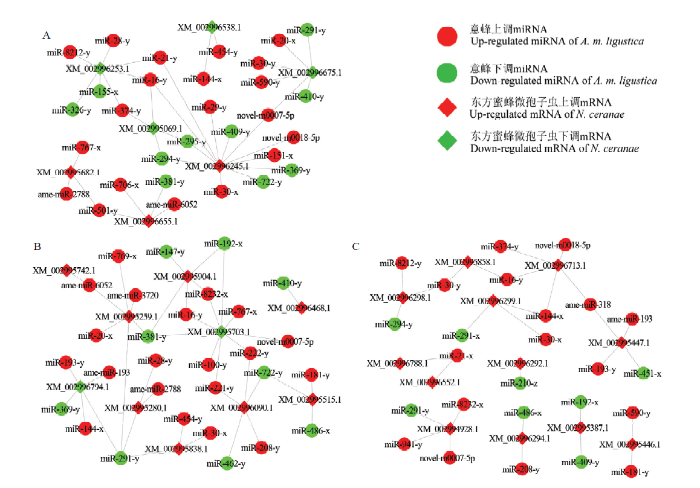 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4AmCK1 vs AmT1比较组中显著性DEmiRNA及其靶向的NcCK vs NcT1比较组中毒力因子/侵染因子相关DEmRNA的调控网络
A:宿主的显著性DEmiRNA与病原的ABC转运蛋白、ATP/ADP转位酶相关DEmRNA的调控网络Regulatory network of host significant DEmiRNAs and pathogen DEmRNAs associated with ABC transporter and ATP/ADP translocase;B:宿主的显著性DEmiRNA与病原的蓖麻毒素B凝集素、孢壁蛋白和极管蛋白相关DEmRNA的调控网络Regulatory network of host significant DEmiRNAs and pathogen DEmRNAs associated with ricin B lectin, spore wall protein and polar tube protein;C:宿主的显著性DEmiRNA与病原的糖酵解/糖异生途径相关DEmRNA的调控网络Regulatory network of host significant DEmiRNAs and pathogen DEmRNAs associated with glycolysis/gluconeogenesis pathway
Fig. 4Regulatory networks of significant DEmiRNAs in AmCK1 vs AmT1 and their target DEmRNAs associated with virulence factor/infection factor in NcCK vs NcT1
Table 1
表1
表1AmCK1 vs AmT1中显著上调miRNA靶向NcCK vs NcT1中东方蜜蜂微孢子虫的毒力因子/侵染因子相关下调mRNA的信息概要
Table 1
| 差异表达miRNA DEmiRNA | 差异表达mRNA DEmRNA | 差异表达mRNA的log2FC log2FC of DEmRNA | 差异表达mRNA的P值 P value of DEmRNA | Nr数据库描述 Description in Nr database |
|---|---|---|---|---|
| ame-miR-6052 | XM_002996297.1 | -10.4546 | 0.0802 | 蓖麻毒素B凝集素 Ricin B lectin |
| miR-196-x | XM_002996297.1 | -10.4546 | 0.0802 | 蓖麻毒素B凝集素 Ricin B lectin |
| miR-16-y | XM_002995069.1 | -1.7206 | 2.6275E-25 | ABC转运蛋白 ABC transporter |
| miR-16-y | XM_002996253.1 | -3.6539 | 1.2369E-118 | ABC转运蛋白 ABC transporter |
| miR-20-x | XM_002996675.1 | -1.9081 | 5.2746E-22 | ABC转运蛋白 ABC transporter |
| miR-21-y | XM_002996253.1 | -3.6539 | 1.2369E-118 | ABC转运蛋白 ABC transporter |
| miR-28-y | XM_002996253.1 | -3.6539 | 1.2369E-118 | ABC转运蛋白 ABC transporter |
| miR-30-y | XM_002996675.1 | -1.9081 | 5.2746E-22 | ABC转运蛋白 ABC transporter |
| miR-374-y | XM_002995069.1 | -1.7206 | 2.6275E-25 | ABC转运蛋白 ABC transporter |
| miR-374-y | XM_002996253.1 | -3.6539 | 1.2369E-118 | ABC转运蛋白 ABC transporter |
| miR-590-y | XM_002996675.1 | -1.9081 | 5.2746E-22 | ABC转运蛋白 ABC transporter |
| miR-8212-y | XM_002996253.1 | -3.6539 | 1.2369E-118 | ABC转运蛋白 ABC transporter |
| novel-m0007-5p | XM_002996675.1 | -1.9081 | 5.2746E-22 | ABC转运蛋白 ABC transporter |
| miR-144-x | XM_002996538.1 | -3.6489 | 8.2034E-116 | ATP/ADP转位酶 ATP/ADP translocase |
| miR-454-y | XM_002996538.1 | -3.6489 | 8.2034E-116 | ATP/ADP转位酶 ATP/ADP translocase |
| ame-miR-193 | XM_002996794.1 | -1.1738 | 1.1394E-08 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| miR-100-y | XM_002995703.1 | -2.3880 | 1.2790E-52 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| miR-144-x | XM_002996794.1 | -1.1738 | 1.1394E-08 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| miR-16-y | XM_002995703.1 | -2.3880 | 1.2790E-52 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| miR-193-y | XM_002996794.1 | -1.1738 | 1.1394E-08 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| miR-221-y | XM_002995703.1 | -2.3880 | 1.2790E-52 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| miR-222-y | XM_002995703.1 | -2.3880 | 1.2790E-52 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| miR-767-x | XM_002995703.1 | -2.3880 | 1.2790E-52 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| miR-8232-x | XM_002995703.1 | -2.3880 | 1.2790E-52 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| novel-m0007-5p | XM_002995703.1 | -2.3880 | 1.2790E-52 | 糖酵解/糖异生 Glycolysis/gluconeogenesis |
| miR-16-y | XM_002996061.1 | -5.8896 | 2.4289E-21 | MAPK信号通路 MAPK signaling pathway |
| miR-29-y | XM_002995842.1 | -2.6200 | 1.1211E-28 | MAPK信号通路 MAPK signaling pathway |
| novel-m0007-5p | XM_002995842.1 | -2.6200 | 1.1211E-28 | MAPK信号通路 MAPK signaling pathway |
新窗口打开|下载CSV
Table 2
表2
表2靶向NcCK vs NcT1中病原毒力因子/侵染因子相关下调DEmRNA的AmCK1 vs AmT1中宿主显著上调miRNA的信息概要
Table 2
| 差异表达miRNA ID DEmiRNA ID | 差异表达miRNA的log2FC log2FC of DEmiRNA | 差异表达miRNA的P值 P value of DEmiRNA | AmCK1组中的TPM值 TPM in AmCK1 group | AmT1组中的TPM值 TPM in AmT1 group |
|---|---|---|---|---|
| ame-miR-6052 | 11.7271 | 0.0021 | 0.0010 | 3.3900 |
| miR-196-x | 2.3757 | 0.0012 | 3.2800 | 17.0233 |
| miR-16-y | 2.7925 | 0.0421 | 0.5100 | 3.5333 |
| miR-20-x | 1.9111 | 0.0500 | 2.2733 | 8.5500 |
| miR-21-y | 2.2041 | 0.0000 | 12.6300 | 58.1967 |
| miR-28-y | 1.6298 | 0.0072 | 7.7467 | 23.9733 |
| miR-30-y | 2.1421 | 0.0034 | 6.3900 | 28.2067 |
| miR-374-y | 4.7301 | 0.0009 | 0.2033 | 5.3967 |
| miR-590-y | 3.6286 | 0.0325 | 0.1900 | 2.3500 |
| miR-8212-y | 11.4683 | 0.0036 | 0.0010 | 2.8333 |
| novel-m0007-5p | 2.9010 | 0.0223 | 0.4900 | 3.6600 |
| miR-144-x | 4.2291 | 0.0307 | 0.3933 | 7.3767 |
| miR-454-y | 3.5531 | 0.0306 | 0.2033 | 2.3867 |
| ame-miR-193 | 3.1393 | 0.0039 | 0.9167 | 8.0767 |
| miR-100-y | 3.3495 | 0.0003 | 1.0367 | 10.5667 |
| miR-193-y | 2.8593 | 0.0049 | 0.8833 | 6.4100 |
| miR-221-y | 1.3376 | 0.0444 | 9.6667 | 24.4300 |
| miR-222-y | 1.8682 | 0.0000 | 100.1467 | 365.6167 |
| miR-767-x | 4.7167 | 0.0011 | 0.2033 | 5.3467 |
| miR-8232-x | 3.7159 | 0.0226 | 0.1900 | 2.4967 |
| miR-29-y | 1.5049 | 0.0014 | 29.6800 | 84.2333 |
新窗口打开|下载CSV
在AmCK2 vs AmT2比较组中,分别有8条显著上调miRNA(miR-28-y、miR-8924-y和miR-8212-y等)和10条显著下调miRNA(miR-142-y、miR-8159-x和miR-2184-x等)可靶向3条ABC转运蛋白编码基因相关的显著DEmRNA;分别有6条显著上调miRNA(novel-m0009-3p、miR-706-x和miR-1332-y等)和7条显著下调miRNA(miR-462-x、miR-144-x和miR-8159-x等)可靶向3条ATP/ADP转位酶编码基因相关的显著DEmRNA(图5-A)。分别有8条显著上调miRNA(miR-424-x、miR-318-y和miR-4217-y等)和7条显著下调miRNA(miR-144-x、miR-8159-x和miR-142-x等)可靶向NcCK vs NcT2比较组中的3条极管蛋白编码基因相关的显著DEmRNA;分别有7条显著上调miRNA(miR-182-x、miR-5119-y和miR-138-x等)和9条显著下调miRNA(miR-462-x、miR-142-y和miR-5112-x等)可靶向1条孢壁和锚定吸盘复合蛋白编码基因、2条孢壁蛋白前体编码基因及1条孢壁蛋白编码基因相关的显著DEmRNA;分别有5条显著上调miRNA(miR-547-x、miR-4577-y和miR-8212-y等)和9条显著下调miRNA(miR-144-x、miR-142-y和miR-223-y等)可靶向6条蓖麻毒素B凝集素编码基因相关的显著DEmRNA(图5-B)。分别有14条显著上调miRNA(miR-222-y、miR-221-z和miR-1332-y等)和19条显著下调miRNA(miR- 8159-x、miR-2184-x和miR-8271-y等)可靶向病原的12条糖酵解/糖异生途径相关的显著DEmRNA(图5-C)。此外,分别有5条显著上调miRNA(miR-4796-y、miR-1332-y和miR-4217-y等)和5条显著下调miRNA(miR-142-y、miR-8159-x和miR-731-x等)可靶向MAPK信号通路相关的5条显著DEmRNA。
图5
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5AmCK2 vs AmT2比较组中显著性DEmiRNA及其靶向的NcCK vs NcT2比较组中毒力因子/侵染因子相关DEmRNA的调控网络
A:宿主的显著性DEmiRNA与病原的ABC转运蛋白、ATP/ADP转位酶相关DEmRNA的调控网络Regulatory network of host significant DEmiRNAs and pathogen DEmRNAs associated with ABC transporter and ATP/ADP translocase;B:宿主的显著性DEmiRNA与病原的蓖麻毒素B凝集素、孢壁蛋白和极管蛋白相关DEmRNA的调控网络Regulatory network of host significant DEmiRNAs and pathogen DEmRNAs associated with ricin B lectin, spore wall protein and polar tube protein;C:宿主的显著性DEmiRNA与病原的糖酵解/糖异生途径相关DEmRNA的调控网络Regulatory network of host significant DEmiRNAs and pathogen DEmRNAs associated with glycolysis/gluconeogenesis pathway
Fig. 5Regulatory networks of significant DEmiRNAs in AmCK2 vs AmT2 and their target DEmRNAs associated with virulence factor/infection factor in NcCK vs NcT2
3 讨论
随着相关研究的增多和深入,有关miRNA在宿主和病原互作中的媒介作用被广泛报道[16-17,40-42]。前期研究中,笔者团队一方面解析了意蜂工蜂中肠响应东方蜜蜂微孢子虫侵染的miRNA差异表达谱及DEmiRNA介导的宿主免疫应答[20];另一方面解析了东方蜜蜂微孢子虫侵染意蜂工蜂过程中的mRNA差异表达谱以及毒力因子、侵染因子及相关DEmRNA在病原侵染中的作用[24]。利用已获得的高质量miRNA和mRNA组学数据,本研究进一步探究miRNA介导意蜂工蜂与东方蜜蜂微孢子虫间的相互作用。东方蜜蜂微孢子虫能够控制蜜蜂的物质代谢、能量代谢和免疫防御等[43]。但关于蜜蜂是否能够通过差异表达miRNA调控东方蜜蜂微孢子虫基因表达的研究未见报道。本研究中,AmCK1 vs AmT1比较组中的显著上调miRNA靶向NcCK vs NcT1比较组中的显著下调mRNA可注释到11条碳水化合物代谢通路、8条脂质代谢通路、3条氨基酸代谢通路及2条能量代谢通路。AmCK2 vs AmT2比较组中显著上调miRNA靶向NcCK vs NcT2比较组中的显著下调mRNA可注释到10条碳水化合物代谢通路、8条脂质代谢通路、2条氨基酸代谢通路及2条能量代谢通路。此外,AmCK1 vs AmT1和AmCK2 vs AmT2比较组的共同显著上调miRNA靶向NcCK vs NcT1和NcCK vs NcT2比较组的共同显著下调mRNA可注释到8条碳水化合物代谢通路、2条脂质代谢通路以及2条能量代谢通路。以上结果表明被东方蜜蜂微孢子虫感染的意蜂工蜂中肠可能通过合成与分泌miRNA跨界抑制或降解病原相关mRNA,进而影响中肠细胞内寄生的病原的糖类、脂质、蛋白和遗传物质等物质代谢途径,以及氧化磷酸化和甲烷代谢等能量代谢途径,体现出二者之间存在密切的相互作用。
RNAi是一种RNA介导的特异性基因沉默机制,已被大量研究证实在动物、植物、昆虫和微孢子虫等生物中发挥基因表达调节作用,具有防控病虫害的应用潜力[12-13,44-45]。多数微孢子虫在进化过程已失去RNAi途径,但东方蜜蜂微孢子虫保留了该途径的3个关键基因即Dicer、Argonaute和RNA-dependent RNA polymerase(RdRp)的同源序列[46]。HUANG等[18]针对Dicer设计特异性siRNA,并混入饲料对东方蜜蜂微孢子虫感染的西方蜜蜂工蜂进行饲喂,发现病原的孢子载量显著降低,并且超过10%的病原蛋白编码基因发生显著性差异表达。本研究发现,在AmCK1 vs AmT1比较组中,宿主的miR-30-x、miR-30-y和miR-196-x显著上调表达且靶向NcCK vs NcT1比较组中病原2个显著下调表达的RdRp-1,包括XM_002995496.1(P=3.68E-34, log2FC=-2.6)和XM_002996709.1(P=3.39E-34, log2FC=-3.0),说明意蜂工蜂中肠可能通过差异表达相应的miRNA对病原的RNAi途径相关的部分mRNA进行表达调控,进而影响东方蜜蜂微孢子虫的RNAi途径。
孢壁蛋白与微孢子虫的孢子发芽与侵染能力密切相关,并通过促使孢子黏附宿主细胞以调节家蚕微孢子虫(Nosema bombycis)的感染过程[47,48]。本研究发现,AmCK2 vs AmT2比较组中宿主的2条显著上调miRNA靶向NcCK vs NcT2比较组中病原显著下调的孢壁蛋白9编码基因(图5),暗示被东方蜜蜂微孢子虫侵染的意蜂工蜂中肠可能通过上调部分miRNA的表达量加强对东方蜜蜂微孢子虫孢壁蛋白编码基因的抑制,从而一定程度上限制病原侵染和增殖。
蓖麻毒素B凝集素是一种通过识别并结合宿主细胞表面多糖蛋白配体和糖脂的半乳糖残基的凝集素蛋白,并促进病原体附着或感染宿主细胞[49,50]。LIU等[51]研究证实家蚕微孢子虫的蓖麻毒素在孢子黏附宿主细胞和提高孢子的感染性等方面扮演重要角色。本研究发现,AmCK1 vs AmT1和AmCK2 vs AmT2比较组中宿主的miR-196-x显著上调且均靶向NcCK vs NcT1和NcCK vs NcT2比较组中下调表达的蓖麻毒素B凝集素编码基因XM_002996297.1(表1、表2),暗示意蜂工蜂中肠在东方蜜蜂微孢子虫侵染的过程中通过上调表达miR-196-x对靶向的XM_002996297.1进行持续抑制,在病原侵染的不同时间点通过选择性差异表达不同的miRNA影响病原的蓖麻毒素B凝集素编码基因的表达,从而阻遏东方蜜蜂微孢子虫对宿主细胞的黏附和侵染。
在长期的协同进化过程中,微孢子虫的线粒体已逐渐退化消失,取而代之的是一种称为纺锤剩体(mitosome)的细胞器[35]。微孢子虫在寄主细胞中生存和增殖需要源源不断的能量和物质供给,既能通过糖酵解/糖异生途径将葡萄糖转化为丙酮酸[52],还可以通过ATP/ADP转位酶和ABC转运蛋白窃取宿主合成的能量和物质供自身能量需求[36]。本研究发现,AmCK1 vs AmT1比较组中宿主的miR-222-y等10条显著上调miRNA靶向NcCK vs NcT1比较组中病原的糖酵解/糖异生途径相关的2条显著下调mRNA(乙酰辅酶A合成酶编码基因XM_002995703.1和磷酸丙糖异构酶编码基因XM_002996794.1)(表1、表2、图4);AmCK2 vs AmT2比较组中宿主的miR-222-y和miR-221-z显著上调表达且均靶向NcCK vs NcT2比较组中病原的糖酵解/糖异生途径相关的显著下调表达的XM_002995703.1(图5)。上述结果表明在东方蜜蜂微孢子虫的侵染过程中,意蜂工蜂中肠可能通过上调miR-222-y和miR-221-z等miRNA的表达量,加强对病原糖酵解/糖异生途径中乙酰辅酶A合成酶编码基因(XM_002995703.1)的抑制,从而限制病原的能量代谢,影响病原的侵染与增殖。
ATP/ADP转位酶和ABC转运蛋白参与微孢子虫对宿主物质和能量的窃取[36]。ABC转运蛋白广泛存在于真核细胞,能够利用ATP的能量对胞内的糖、核苷酸、氨基酸、多肽、蛋白质等进行跨膜转运[53]。PALDI等[36]研究发现通过RNAi敲减东方蜜蜂微孢子虫的ATP/ADP转位酶基因可导致病原的增殖水平下降。本研究发现,AmCK1 vs AmT1比较组中宿主的miR-454-y和miR-144-x显著上调且靶向NcCK vs NcT1比较组中显著下调表达的ATP/ADP转位酶编码基因(XM_002996538.1)(表1、表2、图4);AmCK2 vs AmT2比较组中显著上调表达的miR-1332-y等4条miRNA靶向结合NcCK vs NcT2比较组中显著下调表达的ATP/ADP转位酶编码基因(XM_002996538.1)(图5)。此外,AmCK1 vs AmT1比较组中宿主的miR-16-y等9条显著上调miRNA靶向NcCK vs NcT1比较组中病原的3条显著下调表达的ABC转运蛋白编码基因(XM_002995069.1、XM_002996253.1和XM_002996675.1)(表1、表2、图4);AmCK2 vs AmT2比较组中宿主的6条显著上调miRNA靶向NcCK vs NcT2比较组中病原的2条显著下调表达的ABC转运蛋白编码基因(图5)。有趣的是,宿主的miR-28-y和miR-8212-y在2个比较组中均显著上调表达且均靶向2个比较组中病原的下调表达的ABC转运蛋白编码基因。以上结果说明意蜂工蜂中肠在东方蜜蜂微孢子虫侵染过程的不同时间点差异表达不同的miRNA对ATP/ADP转位酶编码基因进行跨界调控,从而抑制东方蜜蜂微孢子虫对宿主细胞的能量窃取;宿主也可能通过差异表达不同的miRNA、持续差异表达相同的miRNA对病原的ABC转运蛋白编码基因相关mRNA进行抑制,进而限制东方蜜蜂微孢子虫通过转运宿主的营养物质满足增殖所需。
MAPK信号通路与真菌的交配、菌丝侵染、附着胞形成、细胞壁完整性、胁迫反应和毒力等过程密切相关[54]。笔者团队前期研究发现蜜蜂球囊菌(Ascopshera apis)在侵染不同抗性蜜蜂幼虫的过程中,MAPK信号通路及富集基因的活跃程度存在差异,中华蜜蜂(Apis cerana cerana)幼虫可能通过抑制该信号通路影响蜜蜂球囊菌的增殖,而蜜蜂球囊菌可能通过激活该通路促进对意蜂幼虫的侵染,体现了二者互作的复杂性[55,56]。本研究发现,AmCK1 vs AmT1比较组中宿主的3条显著上调miRNA(novel-m0007- 5p、miR-29-y和miR-16-y)靶向NcCK vs NcT1比较组中病原MAPK信号通路相关的2条显著下调mRNA(HECTD蛋白编码基因XM_002995842.1和磷脂酰肌醇-4-磷酸-5-激酶编码基因XM_002996061.1)(表1、表2);AmCK2 vs AmT2比较组中宿主显著下调表达的miR-8159-x和miR-316-x共同靶向NcCK vs NcT2比较组中病原显著下调表达的HECTD蛋白编码基因XM_002995842.1。上述结果说明意蜂工蜂中肠在东方蜜蜂微孢子虫侵染过程的不同时间点通过差异表达不同的miRNA抑制病原的MAPK信号通路,从而影响病原在宿主细胞内的环境适应,以及病原的细胞壁完整性和毒力等方面。
4 结论
在东方蜜蜂微孢子虫侵染意蜂工蜂中肠的过程中,宿主DEmiRNA与病原DEmRNA之间存在复杂的靶向结合关系和潜在的调控关系,宿主的DEmiRNA可能通过调控病原的RNAi途径、毒力因子、糖酵解/糖异生途径、ATP/ADP移位酶、ABC转运蛋白及MAPK信号通路相关DEmRNA的表达影响病原的侵染和增殖。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 2]
[本文引用: 2]
URLPMID:14744438 [本文引用: 1]
URLPMID:21931358 [本文引用: 2]
URLPMID:28829772 [本文引用: 2]
URLPMID:31541102 [本文引用: 2]
[本文引用: 2]
URLPMID:22981770 [本文引用: 1]
[本文引用: 2]
DOI:10.1111/imb.12051URLPMID:23937246 [本文引用: 3]

The brown planthopper (BPH), Nilaparvata lugens, is a major rice pest in Asia, and accumulated evidence indicates that this species is susceptible to RNA interference (RNAi); however, the mechanism underlying RNAi and parental RNAi has not yet been determined. We comprehensively investigated the repertoire of core genes involved in small interfering RNA (siRNA) and micro-RNA (miRNA) pathways in the BPH by comparing its newly assembled transcriptome and genome with those of Drosophila melanogaster, Tribolium castaneum and Caenorhabditis elegans. Our analysis showed that the BPH possesses one drosha and two Dicer (dcr) genes, three dsRNA-binding motif protein genes, two Argonaute (ago) genes, two Eri-1-like genes (eri-1), and a Sid-1-like gene (sid-1). Additionally, we report for first time that parental RNAi might occur in this species, and siRNA pathway and Sid-1 were required for high efficiency of systemic RNAi triggered by exogenous dsRNA. Furthermore, our results also demonstrated that the miRNA pathway was involved in BPH metamorphosis as depletion of the ago1 or dcr1 gene severely impaired ecdysis. The BPH might be a good model system to study the molecular mechanism of systemic RNAi in hemimetabolous insects, and RNAi has potential to be developed to control this pest in agricultural settings.
[本文引用: 1]
DOI:10.1124/pr.112.005983URLPMID:22722893 [本文引用: 1]
Both eukaryotic and prokaryotic cells release small, phospholipid-enclosed vesicles into their environment. Why do cells release vesicles? Initial studies showed that eukaryotic vesicles are used to remove obsolete cellular molecules. Although this release of vesicles is beneficial to the cell, the vesicles can also be a danger to their environment, for instance in blood, where vesicles can provide a surface supporting coagulation. Evidence is accumulating that vesicles are cargo containers used by eukaryotic cells to exchange biomolecules as transmembrane receptors and genetic information. Because also bacteria communicate to each other via extracellular vesicles, the intercellular communication via extracellular cargo carriers seems to be conserved throughout evolution, and therefore vesicles are likely to be a highly efficient, robust, and economic manner of exchanging information between cells. Furthermore, vesicles protect cells from accumulation of waste or drugs, they contribute to physiology and pathology, and they have a myriad of potential clinical applications, ranging from biomarkers to anticancer therapy. Because vesicles may pass the blood-brain barrier, they can perhaps even be considered naturally occurring liposomes. Unfortunately, pathways of vesicle release and vesicles themselves are also being used by tumors and infectious diseases to facilitate spreading, and to escape from immune surveillance. In this review, the different types, nomenclature, functions, and clinical relevance of vesicles will be discussed.
[本文引用: 2]
DOI:10.1158/1078-0432.CCR-16-2936URLPMID:28143871 [本文引用: 2]
Purpose: Human papillomavirus 16 (HPV-16) is an important risk factor in head and neck cancer (HNC). Studies suggest that miRNAs play an important role in cancer; however, their role in HPV-mediated oncogenesis remains largely unknown. We investigated the role of miRNAs with HPV-16 as putative target in HPV-16-mediated cancers.Experimental Design: Using in silico tools, we identified miRNAs with putative binding sequences on HPV-16 miRNAs. Hsa-miR-139-3p was identified as best candidate miRNA by luciferase reporter assay and was found to be significantly downregulated in HPV-16-positive tissues and cell lines. Overexpression/inhibition studies were performed to determine the role of miRNA in regulating oncogenic pathways.Results: Hsa-miR-139-3p was found to target high-risk HPV-16 oncogenic proteins and revive major tumor suppressor proteins (p53, p21, and p16). This resulted in inhibition of cell proliferation and cell migration, cell-cycle arrest at G2-M phase and increased cell death of HPV-16-positive cells. Analysis of The Cancer Genome Atlas (TCGA) data showed decreased expression of Hsa-miR-139-3p in HPV-16-positive HNC and cervical cancer cases, and its higher expression correlated with better survival outcome in both cases. Increased DNA methylation of Hsa-miR-139-3p harboring gene PDE2A at its promoter/CpG islands was observed in HPV-16-positive tissues and cell lines, which further correlated with Hsa-miR-139-3p expression, suggesting its role in regulating Hsa-miR-139-3p expression. Furthermore, we observed an increased sensitization of Hsa-miR-139-3p overexpressed HPV-16-positive cells to chemotherapeutic drugs (cisplatin and 5-fluorouracil).Conclusions: HPV-16-mediated downregulation of Hsa-miR-139-3p may promote oncogenesis in HNC and cervical cancer. Clin Cancer Res; 23(14); 3884-95. (c)2017 AACR.
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 3]
[本文引用: 3]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1093/nar/gkr688URLPMID:21911355 [本文引用: 1]
microRNAs (miRNAs) are a large class of small non-coding RNAs which post-transcriptionally regulate the expression of a large fraction of all animal genes and are important in a wide range of biological processes. Recent advances in high-throughput sequencing allow miRNA detection at unprecedented sensitivity, but the computational task of accurately identifying the miRNAs in the background of sequenced RNAs remains challenging. For this purpose, we have designed miRDeep2, a substantially improved algorithm which identifies canonical and non-canonical miRNAs such as those derived from transposable elements and informs on high-confidence candidates that are detected in multiple independent samples. Analyzing data from seven animal species representing the major animal clades, miRDeep2 identified miRNAs with an accuracy of 98.6-99.9% and reported hundreds of novel miRNAs. To test the accuracy of miRDeep2, we knocked down the miRNA biogenesis pathway in a human cell line and sequenced small RNAs before and after. The vast majority of the >100 novel miRNAs expressed in this cell line were indeed specifically downregulated, validating most miRDeep2 predictions. Last, a new miRNA expression profiling routine, low time and memory usage and user-friendly interactive graphic output can make miRDeep2 useful to a wide range of researchers.
URLPMID:31516938 [本文引用: 1]
[本文引用: 1]
DOI:10.1093/bioinformatics/btp616URLPMID:19910308 [本文引用: 1]
SUMMARY: It is expected that emerging digital gene expression (DGE) technologies will overtake microarray technologies in the near future for many functional genomics applications. One of the fundamental data analysis tasks, especially for gene expression studies, involves determining whether there is evidence that counts for a transcript or exon are significantly different across experimental conditions. edgeR is a Bioconductor software package for examining differential expression of replicated count data. An overdispersed Poisson model is used to account for both biological and technical variability. Empirical Bayes methods are used to moderate the degree of overdispersion across transcripts, improving the reliability of inference. The methodology can be used even with the most minimal levels of replication, provided at least one phenotype or experimental condition is replicated. The software may have other applications beyond sequencing data, such as proteome peptide count data. AVAILABILITY: The package is freely available under the LGPL licence from the Bioconductor web site (http://bioconductor.org).
URLPMID:33857425 [本文引用: 1]
URLPMID:33876181 [本文引用: 1]
[本文引用: 1]
URLPMID:19503607 [本文引用: 2]
URLPMID:20622131 [本文引用: 4]
DOI:10.1111/1462-2920.12883URLPMID:25914091 [本文引用: 1]
Nosema ceranae is a microsporidian pathogen whose infections have been associated with recent global declines in the populations of western honeybees (Apis mellifera). Despite the outstanding economic and ecological threat that N. ceranae may represent for honeybees worldwide, many aspects of its biology, including its mode of reproduction, propagation and ploidy, are either very unclear or unknown. In the present study, we set to gain knowledge in these biological aspects by re-sequencing the genome of eight isolates (i.e. a population of spores isolated from one single beehive) of this species harvested from eight geographically distant beehives, and by investigating their level of polymorphism. Consistent with previous analyses performed using single gene sequences, our analyses uncovered the presence of very high genetic diversity within each isolate, but also very little hive-specific polymorphism. Surprisingly, the nature, location and distribution of this genetic variation suggest that beehives around the globe are infected by a population of N. ceranae cells that may be polyploid (4n or more), and possibly clonal. Lastly, phylogenetic analyses based on genome-wide single-nucleotide polymorphism data extracted from these parasites and mitochondrial sequences from their hosts all failed to support the current geographical structure of our isolates.
URLPMID:16363121 [本文引用: 1]
DOI:10.1093/abbs/gmv140URLPMID:26837419 [本文引用: 1]
Nosema bombycis is an obligate intracellular parasitic fungus that utilizes a distinctive mechanism to infect Bombyx mori. Germination, an indispensible process through which microsporidia infect the host cells, is regarded as a key developmental turning point for microsporidia from dormant state to reproduction state. Thus, elucidating the transcriptome changes before and after germination is crucial for parasite control. However, the molecular basis of germination of microsporidia remains unknown. To investigate this germination process, the transcriptome of N. bombycis ungerminated spores and germinated spores were sequenced and analyzed. More than 60 million high-quality transcript reads were generated from these two groups using RNA-Seq technology. After assembly, 2756 and 2690 unigenes were identified, respectively, and subsequently annotated based on known proteins. After analysis of differentially expressed genes, 66 genes were identified to be differentially expressed (P
[本文引用: 1]
URLPMID:23308063 [本文引用: 1]
DOI:10.1016/j.jip.2014.07.002URLPMID:25038465 [本文引用: 1]
Many invasive pathogens effectively bypass the insect defenses to ensure the completion of their life cycle. Among those, an invasive microsporidian species, Nosema ceranae, can cause nosemosis in honeybees. N. ceranae was first described in the Asian honeybee Apis cerana and is suspected to be involved in Western honeybee (Apis mellifera) declines worldwide. The midgut of honeybees is the first barrier against N. ceranae attacks. To bring proteomics data on honeybee/N. ceranae crosstalk and more precisely to decipher the worker honeybee midgut response after an oral inoculation of N. ceranae (10days post-infection), we used 2D-DIGE (2-Dimensional Differential In-Gel Electrophoresis) combined with mass spectrometry. Forty-five protein spots produced by the infected worker honeybee group were shown to be differentially expressed when compared to the uninfected group; 14 were subsequently identified by mass spectrometry. N. ceranae mainly caused a modulation of proteins involved in three key host biological functions: (i) energy production, (ii) innate immunity (reactive oxygen stress) and (iii) protein regulation. The modulation of these host biological functions suggests that N. ceranae creates a zone of
[本文引用: 1]
URLPMID:12110901 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
URLPMID:28031263 [本文引用: 1]
DOI:10.1038/333426a0URLPMID:3374584 [本文引用: 1]
The three-dimensional structures of influenza virus haemagglutinins complexed with cell receptor analogues show sialic acids bound to a pocket of conserved amino acids surrounded by antibody-binding sites. Sialic acid fills the conserved pocket, demonstrating that it is the influenza virus receptor. The proximity of the antibody-binding sites suggests that antibodies neutralize virus infectivity by preventing virus-to-cell binding. The structures suggest approaches to the design of anti-viral drugs that could block attachment of viruses to cells.
[本文引用: 1]
DOI:10.1093/abbs/gmw093URLPMID:27649890 [本文引用: 1]
Nosema bombycis is an obligate intracellular parasitic fungus that utilizes a distinctive mechanism to infect Bombyx mori Spore germination can be used for host cell invasion; however, the detailed mechanism remains to be elucidated. The ricin-B-lectin (RBL) gene is significantly differentially regulated after N. bombycis spore germination, and NbRBL might play roles in spore germination and infection. In this study, the biological function of NbRBL was examined. Protein sequence analysis showed that NbRBL is a secreted protein that attaches to carbohydrates. The relative expression level of the NbRBL gene was low during the first 30 h post-infection (hpi) in BmN cells, and high expression was detected from 42 hpi. Gene cloning, prokaryotic expression, and antibody preparation for NbRBL were performed. NbRBL was detected in total and secreted proteins using western blot analysis. Subcellular localization analysis showed that NbRBL is an intracellular protein. Spore adherence and infection assays showed that NbRBL could enhance spore adhesion to BmN cells; the proliferative activities of BmN cells incubated with anti-NbRBL were higher than those in negative control groups after N. bombycis infection; and the treatment groups showed less damage from spore invasion. We therefore, propose that NbRBL is released during spore germination, enhances spore adhesion to BmN cells, and contributes to spore invasion.
URLPMID:18956766 [本文引用: 1]
Glucose is degraded to pyruvate via the so called
URLPMID:18956766 [本文引用: 1]
Glucose is degraded to pyruvate via the so called
URLPMID:1282354 [本文引用: 1]
URLPMID:22517321 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}