Transcriptome Analysis of Citrus reticulata Blanco, cv. Hongjv Infected with Alternaria alternata Tangerine Pathotype
TANG KeZhi, ZHOU ChangYongCitrus Research Institute, Southwest University, Chongqing 400712通讯作者:
收稿日期:2020-01-19接受日期:2020-03-27网络出版日期:2020-11-16
| 基金资助: |
Received:2020-01-19Accepted:2020-03-27Online:2020-11-16
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (2650KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
唐科志, 周常勇. 红橘响应褐斑病菌侵染的转录组学分析[J]. 中国农业科学, 2020, 53(22): 4584-4600 doi:10.3864/j.issn.0578-1752.2020.22.006
TANG KeZhi, ZHOU ChangYong.
开放科学(资源服务)标识码(OSID):

0 引言
【研究意义】由链格孢菌橘致病型(Alternaria alternata tangerine pathotype)引起的柑橘褐斑病(Alternaria brown spot,ABS)可危害柑橘的嫩梢、嫩叶及果实,常对部分宽皮柑橘生产带来严重问题[1,2,3]。澳大利亚于1903年首次报道该病害[4],随后在美国、哥伦比亚、土耳其、以色列等柑橘产地相继发现。2010年,在我国云南的栽培品种上首次鉴定到柑橘褐斑病菌[5],此后,在国内其他柑橘产区也陆续暴发该病[6,7,8]。近年来,柑橘褐斑病在敏感品种红橘(Citrus reticulata Blanco, cv. Hongjv)上的暴发已造成局部地区橘类产业巨大的经济损失。国内外对柑橘褐斑病菌部分功能基因研究已取得一定进展[9,10,11,12],但其流行规律、致病机理以及寄主应答机制仍不清楚,有待进一步深入研究。【前人研究进展】WANG等[13,14]对链格孢菌橘致病型菌株Z7进行了全基因组测序分析,证明柑橘褐斑病菌功能基因主要为ACT毒素合成基因、信号转导基因、次级代谢相关基因、ROS解毒相关基因等,并通过转录组测序研究了主要转录因子对菌株产孢及致病性的调控;WU等[15]通过对霜霉病菌(Plasmopara viticola)侵染葡萄前后的叶片样品进行RNA-seq测序,共获得15 249个候选的差异表达基因;XU等[16]通过RNA-seq研究了棉花受黄萎病菌(Verticillium dahlia)侵染后的转录组变化,共鉴定到3 442个与防御相关的基因,其中木质素合成相关基因的表达在抗病品系中表达显著上调,推测木质素可能在棉花抗病中发挥重要作用;WARD等[17]借助RNA-seq技术研究了覆盆子根腐病抗病及感病品系受到病原菌Phytophthora rubi侵染后的基因表达差异,结果显示WRKY转录因子、三羧酸循环、木质素合成及编码病程相关蛋白的基因等均表现为转录表达上调;WANG等[18]利用RNA-seq技术研究了葡萄柚的抗病及感病品系在黄龙病菌(Candidatus Liberibacter asiaticus)侵染后的转录表达差异,发现两个品系在转录因子、次级代谢、受体激酶及激素信号转导等多个途径相关基因均存在表达差异,通过后续验证,推测出与黄龙病菌抗性相关的两个基因DMR6-like和 NPR1-like;李湘龙等[19]通过RNA-seq技术分析水稻与稻瘟病菌(Magnaporthe oryzae)互作早期的转录表达谱,为研究稻瘟病菌效应蛋白基因及其功能打下了基础。【本研究切入点】RNA-seq可为植物响应胁迫过程中基因表达调控、蛋白质功能和代谢通路等研究提供大量信息,在植物病原菌的致病机理、植物对病原菌的胁迫应答,以及病原菌与植物寄主相互作用的机制机理等研究中应用十分广泛。但利用RNA-seq技术进行链格孢菌橘致病型与柑橘互作的研究尚未见报道。【拟解决的关键问题】利用RNA-seq技术对链格孢菌橘致病型侵染红橘前后的基因表达变化进行分析,找到红橘响应链格孢菌橘致病型侵染的关键基因,为柑橘的抗病育种提供理论依据。1 材料与方法
试验于2018—2019年在西南大学柑桔研究所国家柑桔苗木脱毒中心实验室完成。1.1 供试材料及样品采集
链格孢菌橘致病型菌株Z7由浙江大学李红叶教授提供,保存于国家柑桔苗木脱毒中心实验室。寄主材料为红橘,种植于国家柑桔苗木脱毒中心网室。取活化后的链格孢菌橘致病型菌株Z7于PDB培养基上,30℃、150 r/min摇菌48 h。收集单个菌丝球接种大小、叶龄和成熟度均一致的红橘离体叶片,将处理组和未接种菌丝的对照组叶片于28℃培养箱保湿培养。接种28 h后,叶片出现轻微反应,未见明显症状,此时采集样品,利用RNA-seq技术分析红橘对链格孢菌橘致病型的早期防御机制。每个样本同时取两份,一份送交公司测序,另一份液氮中速冻后保存于-80℃冰箱用于后期试验验证。对照和处理组分别设置3个生物学重复(对照样本名称分别为C0_1/2/3,接种菌株样本名称分别为ZC_1/2/3)。1.2 转录组测序
1.2.1 总RNA提取及质量检测 采用Trizol(Invitrogen)提取寄主叶片总RNA[11],并用RQ1 RNase-Free DNase(Promega)处理去除DNA。利用1.2%琼脂糖凝胶电泳进行RNA条带初步分析。使用分光光度计NanoDropTM(ThermoFisher)进行样本纯度的检测,通过OD260/280及OD260/230比值进行判断。采用Agilent 2100 Bioanalyzer(Agilent RNA 6000 Nano Kit)检测样品总RNA的浓度、完整性(RIN值、28S/18S)及片段大小,主要根据真核生物rRNA的完整性来判断,包括28S rRNA、18S rRNA及5.8S rRNA的峰值及电泳条带完整性。1.2.2 文库构建与上机测序 样品提取总RNA后,用Dynalbeads oligo(dT)磁珠富集有polyA尾巴的mRNA,加入fragment buffer将RNA片段化,再以片段化的mRNA为模板,采用随机的N6 primer进行反转录生成cDNA一链,再合成cDNA二链,纯化并经DNA末端修复,加碱基A,加测序接头,再经琼脂糖凝胶电泳回收目的片段,完成文库构建。利用BGISEQ-500平台采用双末端法(Pair-End)对文库进行测序。
1.3 转录组数据分析
1.3.1 原始测序数据处理 测序得到的原始序列中含有带接头的、低质量的reads,为了保证后续信息分析质量,需要对原始测序数据进行过滤,得到clean reads,以保证后续分析数据的可靠性。1.3.2 参考基因组比对 针对上述得到的clean reads,使用HISAT[20]将clean reads比对到甜橙参考基因组序列(http://www.ncbi.nlm.nih.gov/genome/10702),然后统计比对上的基因组的比对率以及reads在染色体上的分布,从整体上了解样品差异。
1.3.3 表达量分析及差异表达基因检测 使用Bowtie 2将clean reads比对到上述较为完整的参考序列,再使用RSEM计算基因和转录本的表达水平。使用PossionDis算法进行差异基因检测,参数为|log2 fold change (FC)|≥1,q-value≤0.01。
1.3.4 差异表达基因功能分析 利用GO数据库对差异表达基因进行功能分类,利用KEGG分析代谢途径,使用P-value<0.05作为显著富集基因的阈值,使用MapMan软件对差异基因进行图形化分析。
1.4 qRT-PCR验证
采用与转录组测序相同的样品材料,随机选取差异表达基因进行qRT-PCR分析,验证转录组测序分析数据的可靠性。通过DNAman软件设计引物,然后由英潍捷基(上海)贸易有限公司进行引物的合成,并通过电泳及qRT-PCR熔解曲线检测引物特异性。相关验证基因扩增引物序列见表1。使用2-ΔΔCT法计算基因的相对表达并换算成log2FC。
Table 1
表1
表1qRT-PCR验证基因选择及引物设计
Table 1
| 基因ID Gene ID | 引物序列 Primer sequence (5′-3′) | 产物长度 Length of product (bp) | |
|---|---|---|---|
| Cs4g12760 | F: TTCGTCTTGCTCTTCGGATAA | R: GCACTCCAACGGAATCTCTAA | 202 |
| Cs2g09310 | F: GGTGTCATTCCTCCTCCTACC | R: GCAGTTCCCTCGCCTATTCT | 198 |
| Cs5g15470 | F: CAGCCTTGTCGGTATGAGAA | R: CAACACCCAATCTTCCTTGAG | 151 |
| Cs5g21860 | F: GCTCTTGTGGGCATTCTTGC | R: CTCTCGTGTAGAAGCTCTTGCC | 142 |
| Cs7g20700 | F: ACGGTTCAGGCTCATTTCAG | R: TGGGATTTGGCATCATCAAT | 185 |
| Cs3g10430 | F: TGGAGACGTAGAGGCTGTCAA | R: CATACCAATATTTTGAGCATTTT | 121 |
| Cs6g07410 | F: GGAGAGTGGTGGAATGCTGAT | R: CACTTCGAGCGTGTAGGTTTG | 150 |
| Cs6g20850 | F: CCAGCAGGCATGAGAAATTA | R: TGACCATCGTGGGAACAGTA | 229 |
| Cs1g01180 | F: TGCAGTAGAAGTTGATGGTGATG | R: AATGAGCCGTTAGCGACAAG | 165 |
| Cs1g04680 | F: AGCTGCAAGGGTTTGGTTAG | R: GAATTTGCGTTTGGTGATGA | 150 |
| Cs8g13680 | F: GCCTATGCTTGCTGTTGTTTC | R: GAAGGCAGTCCATCCATACTTC | 194 |
| orange1.1t03118 | F: CAAGCTCTCCAGGCAAGAGT | R: GGTCCACGGCCATAGTAGAA | 247 |
| orange1.1t03618 | F: CGGGATGAACATTTGGTTTA | R: CTAGCCTTCTGATCTTGACACA | 184 |
| orange1.1t05311 | F: GCTGGGATATAACTCCTTCTCA | R: TTCCGCTAAACCAATCACTT | 172 |
| Cs2g06120 | F: GCACAAGGAAATGGGTTTGT | R: GAAACACGCTGGGATCACTT | 229 |
| Cs6g07400 | F: ACATGGCTGCAAGAGCATAC | R: CCATTGAGGTCCACCACTTA | 197 |
| orange1.1t00214 | F: ACGCTCTGTCCCTCAACAAG | R: CCGCTACTGCCTCCTGTATC | 171 |
| orange1.1t02319 | F: GGGATCTACTGCCGACACTC | R: CGACGACGACCTTTGATCTT | 245 |
| orange1.1t03603 | F: GGACAATGCTGATCCGAAAG | R: TCAACCAAGCCTCCTGAAAC | 203 |
| CitActin | F: CATCCCTCAGCACCTTCC | R: CCAACCTTAGCACTTCTCC | 191 |
新窗口打开|下载CSV
2 结果
2.1 与参考基因组比对结果
测序reads经过数据质控后,将各样品过滤后的clean reads与甜橙基因组进行比对分析,各样品的比对率及唯一比对率都相对比较均匀,详细的比对结果见表2。经测序得到的6个转录组文库中的数据与甜橙参考基因组进行比对后获得的mapped reads占总reads的74%以上,根据样品间分析可知,对照样品中的mapping率接近90%,说明在接菌后随着菌体在叶片内增殖,病原菌的转录本影响总的mapping。2.2 差异表达基因筛选
根据差异基因筛选条件q-value≤0.01,差异倍数 |log2FC|≥1,链格孢菌橘致病型接种红橘28 h后获得上调差异基因5 173个,下调差异基因为6 555个(图1)。Table 2
表2
表2比对参考基因组结果统计
Table 2
| 样本 Sample | 总序列数 Total reads | 比对上序列比例 Total mapped reads (%) | 比对上唯一位置序列比例 Unique mapped reads (%) | 双端比对上序列比例 Reads mapped in paired (%) |
|---|---|---|---|---|
| C0_1 | 47713764 | 90.30 | 86.99 | 84.18 |
| C0_2 | 54696950 | 88.96 | 85.64 | 82.20 |
| C0_3 | 52284118 | 89.93 | 86.59 | 83.36 |
| ZC_1 | 55857454 | 75.22 | 71.84 | 69.94 |
| ZC_2 | 53372222 | 74.18 | 70.98 | 68.37 |
| ZC_3 | 57202046 | 74.31 | 70.99 | 68.87 |
新窗口打开|下载CSV
图1
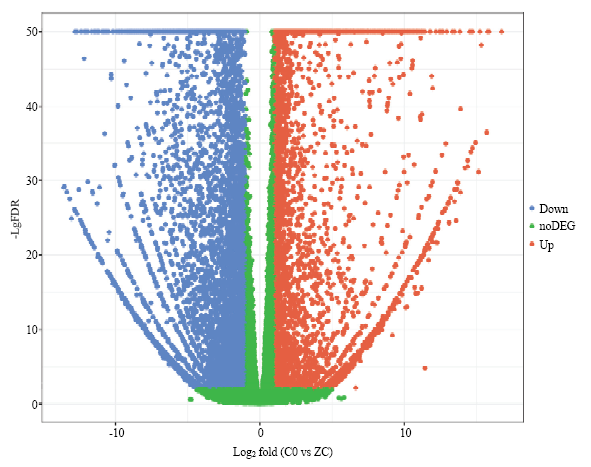 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1链格孢菌橘致病型侵染引起红橘差异表达基因火山图 图中每个点代表一个基因,蓝色和红色的点代表显著差异表达基因,红色的点表示其基因表达量上调,蓝色的点表示其基因表达量下调,绿色的点表示这些基因无显著差异
Fig. 1Volcano plot of DEGs of tangerine inoculated with A. alternata tangerine pathotype Each point in the figure represents a gene, and the blue and red dots represent significant differentially expressed genes (DEGs), the red dot indicates that the gene expression level is up-regulated, the blue dot indicates that the gene expression level is down-regulated, the green dot indicates that these genes are not significant difference
2.3 差异表达基因GO功能富集分析
红橘受链格孢菌橘致病型侵染的差异基因根据GO分类的规则分为3大类:分子功能、细胞组分和生物学过程(图2)。在生物学过程中差异基因主要属于蛋白磷酸化、代谢过程、氧化还原过程和跨膜运输。细胞组分中只包括两类与膜相关的条目。分子功能相关的基因富集最多,其中更多基因富集在蛋白结合、ATP结合、催化活性、蛋白激酶活性和氧化还原活性。图2
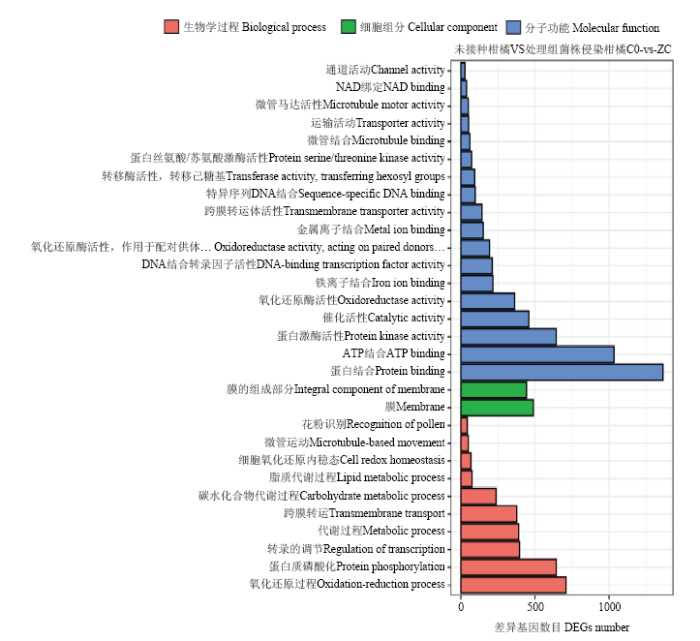 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2差异表达基因GO富集
Fig. 2DEGs GO enrichment
对所有富集的GO term进行筛选,筛选条件为多重检验校正q-value<0.05,利用WEGO通过比较所有富集的基因和差异基因占基因总数的百分比,可以得出哪些差异的比例高出一般基因富集的水平,如图3所示,在代谢过程、膜系统、抗氧化活性、转运及核酸绑定相关基因GO富集程度高于平均水平。
图3
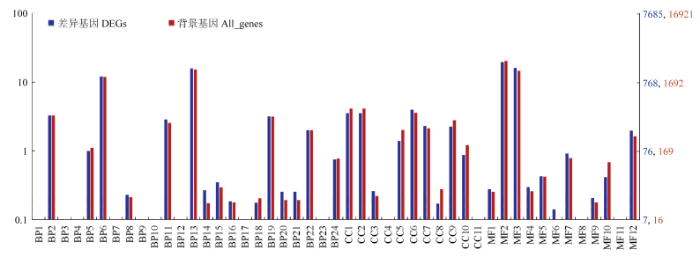 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3差异表达基因WEGO分类
横坐标表示GO的3个方面;左侧纵坐标表示基因数目的百分比;右侧纵坐标表示对应GO term的基因数X axis: Three aspects of GO; The left ordinate: The percentage of the number of genes; The right ordinate: The corresponding GO term gene numberBP1:生物黏附Biological adhesion;BP2:生物调节Biological regulation;BP3:碳利用Carbon utilization;BP4:细胞增殖Cell proliferation;BP5:细胞成分组成或发生Cellular component organization or biogenesis;BP6:细胞过程Cellular process;BP7:解毒Detoxification;BP8:发展过程Developmental process;BP9:增长Growth;BP10:免疫系统过程Immune system process;BP11:定位Localization;BP12:运动Locomotion;BP13:代谢过程Metabolic process;BP14:多生物过程Multi-organism process;BP15:多细胞生物过程Multicellular organismal process;BP16:生物过程负调控Negative regulation of biological process;BP17:氮利用Nitrogen utilization;BP18:生物过程正调控Positive regulation of biological process;BP19:生物过程调节Regulation of biological process;BP20:繁殖Reproduction;BP21:生殖过程Reproductive process;BP22:刺激反应Response to stimulus;BP23:节律过程Rhythmic process;BP24:信号Signaling;CC1:细胞Cell;CC2:细胞部分Cell part;CC3:细胞外区域Extracellular region;CC4:细胞外区域部分Extracellular region part;CC5:大分子复合物Macromolecular complex;CC6:膜Membrane;CC7:膜部分Membrane part;CC8:膜包围腔Membrane-enclosed lumen;CC9:细胞器Organelle;CC10:细胞器部分Organelle part;CC11:超分子复合物Supramolecular complex;MF1:抗氧化活性Antioxidant activity;MF2:绑定Binding;MF3:催化活性Catalytic activity;MF4:电子载体活性Electron carrier activity;MF5:功能分子调控Molecular function regulator;MF6:分子传感器活性Molecular transducer activity;MF7:核酸结合转录因子活性Nucleic acid binding transcription factor activity;MF8:营养库活性Nutrient reservoir activity;MF9:信号传感器活性Signal transducer activity;MF10:结构分子活性Structural molecule activity;MF11:转录因子活性、蛋白质结合Transcription factor activity, protein binding;MF12:转运活性Transporter activity
Fig. 3WEGO classification of DEGs
将差异基因分成表达上调和表达下调基因,图4显示在同病原菌互作过程中,红橘不同代谢途径的基因均呈现上调或下调,值得注意的是碳水化合物代谢过程中上调基因91个而下调基因146个,说明病原菌侵染直接破坏红橘的碳水化合物代谢。叶绿体相关的基因和调控光系统的基因也主要呈下调状态,说明光合作用也受到负面影响。微管相关基因也是以下调状态为主,说明细胞骨架也被病原菌攻击受损。DNA绑定的转录因子活性增强,说明大量转录因子被激活调控下游抗病基因表达,蛋白激酶活性也以上调为主,说明免疫相关的PTI和ETI中大量受体激酶也被病原菌激活。
图4
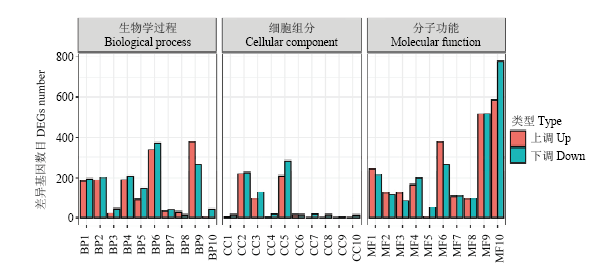 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4差异表达基因GO分类
BP1:跨膜运输Transmembrane transport;BP2:细胞氧化还原内稳态Cell redox homeostasis;BP3:转录调节、DNA模板化Regulation of transcription, DNA-templated;BP4:碳水化合物代谢过程Carbohydrate metabolic process;BP5:氧化还原过程Oxidation-reduction process;BP6:脂质代谢过程Lipid metabolic process;BP7:花粉识别Recognition of pollen;BP8:蛋白质磷酸化Protein phosphorylation;BP9:基于微管运动Microtubule-based movement;BP10:代谢过程Metabolic process;CC1:叶绿体Chloroplast;CC2:核Nucleus;CC3:微管Microtubule;CC4:膜Membrane;CC5:细胞外区域Extracellular region;CC6:光系统II Photosystem II;CC7:光系统II氧进化复合体Photosystem II oxygen evolving complex;CC8:光系统I反应中心Photosystem I reaction center;CC9:膜的外部成分Extrinsic component of membrane;CC10:膜的组成部分Integral component of membrane;MF1:催化活性Catalytic activity;MF2:DNA结合转录因子活性DNA-binding transcription factor activity;MF3:氧化还原酶活性Oxidoreductase activity;MF4:微管结合Microtubule binding;MF5:蛋白激酶活性Protein kinase activity;MF6:铁离子结合Iron ion binding;MF7:氧化还原酶活性,作用于配对供体,结合或还原分子Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular;MF8:ATP结合ATP binding;MF9:蛋白结合Protein binding;MF10:血红素结合Heme binding
Fig. 4GO classification of DEGs
2.4 差异表达基因KEGG功能富集分析
以KEGG Pathway为单位,应用超几何检验,分析鉴别差异表达基因中显著富集的代谢通路或信号转导途径。Pathway富集到的通路涉及到6 975个差异基因,对所有富集的KEGG Pathway进行筛选,共获得20条显著富集的Pathway,分别为代谢途径(ko01100)、次生代谢物合成(ko01110)、碳循环(ko01200)、糖酵解/糖异生(ko00010)、丙酮酸代谢(ko00620)、光合作用(ko00195)、柠檬酸循环(ko00020)、光合作用-天线蛋白质(ko00196)、脂肪酸降解(ko00071)、卟啉和叶绿素代谢(ko00860)、甘氨酸、丝氨酸和苏氨酸代谢(ko00260)、氨基糖和核苷酸糖代谢(ko00520)、谷胱甘肽代谢(ko00480)、α-亚麻酸代谢(ko00592)、苯丙烷类生物合成(ko00940)、植物激素信号转导(ko04075)、抗坏血酸代谢(ko00053)、双酚降解(ko00363)、柠檬烯和蒎烯的降解(ko00903)、二苯乙烯、二芳庚烯和姜辣素的生物合成(ko00945)(图5)。图5
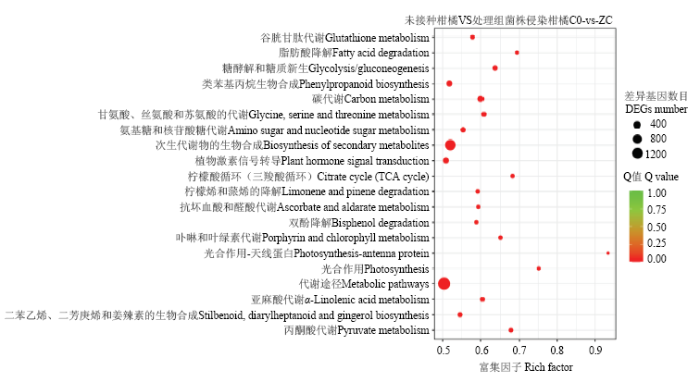 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5差异表达基因的KEGG通路分析
Fig. 5KEGG pathway analysis of DEGs
2.5 红橘响应链格孢菌橘致病型侵染差异表达基因
2.5.1 植物激素信号转导途径及相关基因 接种链格孢菌橘致病型28 h RNA-seq结果显示,红橘防御相关的植物激素信号转导途径中多个基因表达具有差异,乙烯(ET)、水杨酸(SA)和生长素(AUX)等重要防御相关的植物激素信号通路受到影响(图6)。图6
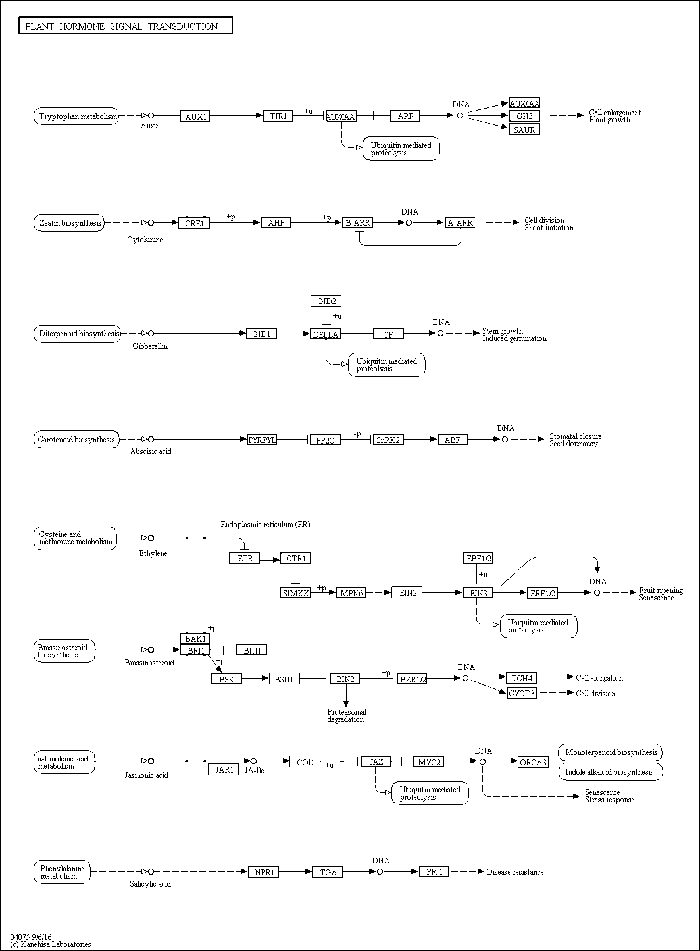 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6KEGG map分析植物激素信号转导相关的差异表达基因
Fig. 6DEGs involved in plant hormone signal transduction analyzed by KEGG map
在红橘响应链格孢菌橘致病型的激素信号转导中乙烯起了主导作用。首先,乙烯受体ETR的3个成员Cs9g08850、Cs5g33560和Cs5g14390被该菌不同程度激活,ETR下游的激酶CTR1中Cs6g15490、Cs3g17090表达量也随之升高,随后受MPK-MEKK将信号进一步放大,到信号下游Cs4g17960和Cs7g05390(编码EBF 1/2),Cs3g06940、Cs3g06930、orange1.1t04381、Cs2g29100(编码EIN3)和最终的转录调控因子乙烯响应因子ERF 1/2均表现出表达量升高趋势。
而在生长素信号中大部分的关键基因表达下调,AUX作为生长素信号的最上游,大量的红橘同源基因Cs8g16440、Cs3g05470、Cs2g05440、Cs3g05500、orange1.1t00508等均呈现下调表达趋势,而生长素响应蛋白编码基因Cs8g16440、Cs3g05470、Cs2g05440、Cs3g05500、orange1.1t00508等也随AUX信号诱导剧烈上升,在生长素信号途径重要转录因子家族中的生长素响应因子ARF是具有多个成员的基因家族,而红橘中绝大部分成员如Cs8g16440、Cs3g05470、Cs2g05440、Cs3g05500、orange1.1t00508、Cs5g32400都表现出受链格孢菌橘致病型诱导的负调控表达。
在SA合成-降解途径中,Cs2g18260编码的UGT74F1在体外将UDP-葡萄糖转移至水杨酸(形成葡糖苷)、苯甲酸、槲皮素和草酸盐。Cs2g18240对应S-腺苷-L-甲硫氨酸依赖性甲基转移酶超家族蛋白,具有甲基转移酶活性参与水杨酸合成,Cs5g21260(编码UGT74F1)由水杨酸、病毒、真菌和细菌诱导,参与色氨酸合成途径。这些参与水杨酸合成途径的基因受链格孢菌橘致病型诱导后均表达下调,推测在该菌侵染过程中有大量SA合成。在SA信号转导途径中大量的TGA转录因子同样受该菌诱导,而下游的PR基因不同成员既有表达上调也有表达下调,这些基因除了受SA调控外也受其他信号影响。
2.5.2 红橘响应链格孢菌橘致病型的转录调控 转录因子(TF)是开启基因转录过程的“主要开关”。图7显示本研究在差异表达基因中发现来自不同TF家族的多个成员。最具代表性的TF类是WRKY系列,在红橘中大概有50个成员,大部分成员均受链格孢菌橘致病型诱导正调控,其中WRKY22、WRKY33、WRKY72、WRKY75、WRKY28都相较于对照上调10倍以上,同时也包括WRKY13在内的一些成员响应该菌并表达下调。bZIP类转录因子能与WRKY蛋白发生互作,通常与WRKY参与相同的调控模式。转录组数据显示在差异表达基因中bZIP家族的大部分成员(Cs1g26010、Cs3g10860、Ccs7g29820)也受该菌侵染上调表达,只不过表达显著性稍低于WRKY转录因子。由于乙烯信号途径基因的大量表达,在转录因子中有一类称为乙烯响应因子的蛋白与该信号途径联系紧密,而差异表达分析也验证了相关的数据,在该菌侵染28 h后的样品中超过50%的ERF家族成员(Cs3g23270、Cs4g06410、Cs5g33540)表达上调,其他TF家族也不同形式的被诱导差异表达,包括NAC、MYB、ZF-HD、B3、HB、C2H2、TiFy和bHLH家族。
图7
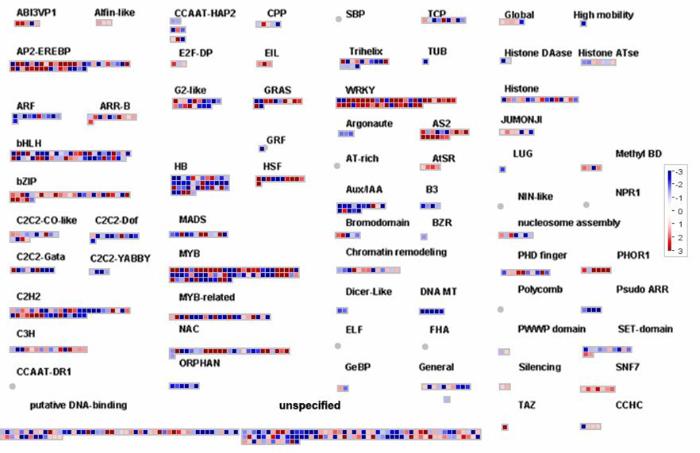 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7MapMan分析转录因子相关的差异表达基因
Fig. 7DEGs involved in transcription factor analyzed by MapMan
2.5.3 次级代谢产物生物合成相关基因 在响应链格孢菌橘致病型过程中,与黄酮醇、花青素、萜类化合物和生物碱生物合成相关的基因受该菌诱导产生剧烈变化(图8)。萜类合成中大部分基因表现下调,而黄酮类合成相关的差异表达基因既有表达上调又有表达下调,总体来说表达上调基因的数量和表达趋势均强于表达下调基因,其中黄烷酮-3-羟化酶(Cs1g03950)、2-oxoglutarate(Cs5g29930、Cs9g02930、Cs9g14520、Cs9g14500)和Fe(II)/ascorbate oxidase(orange1.1t03581、orange1.1t03576、orange1.1t03582)均在侵染过程中积累,UDP-糖基转移酶家族则在黄酮和花青素合成过程中表达下调。硫代葡萄糖苷有抗虫和抑菌的作用,在该菌的影响下总体呈现上调趋势,而生物碱相关基因和类胡萝卜素代谢相关基因表达趋势与此相反。此外,该菌还诱导了涉及木质素生物合成的基因PAL(Cs8g16290、Cs7g24940)、4CL(Cs5g06990)、HTC(Cs1g14450),表明在受侵染的组织中木质素沉积,木质化可作为限制病原菌定殖的有效防御机制,转录组数据呈现出感染组织中细胞壁活动剧烈变化,支持了植物在感染期间感知病原并激活防御机制的观点。
图8
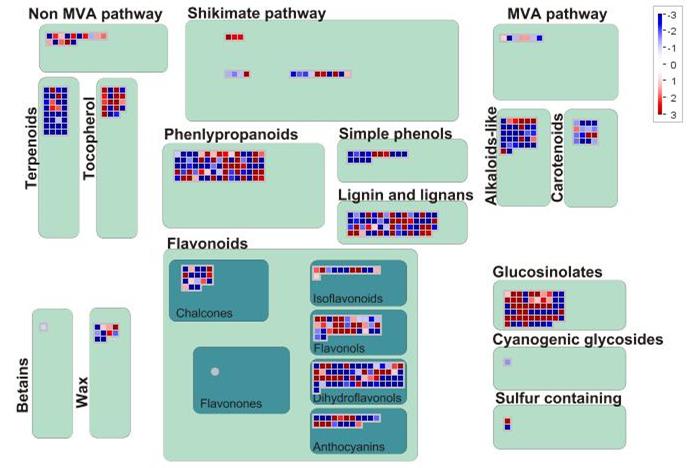 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图8MapMan分析次级代谢产物合成相关的差异表达基因
Fig. 8DEGs involved in secondary metabolite biosynthesis analyzed by MapMan
2.5.4 病原与植物互作途径相关基因 MAPK信号途径中CsMPK7、CsMKK3、CsMAP3Ka信号级联均受链格孢菌橘致病型激活表达,同时表达量上调的还有下游主要转录因子成员ERF、WRKY、bZIP、MYB。ETI中的R蛋白基因(Cs5g21850、Cs5g21860、orange1.1t03118),TIR-NBS-LRR类抗病蛋白编码基因(Cs1g26330)和NB-ARC类抗病蛋白编码基因(orange1.1t03734)均响应该菌正向调控表达,NB-LRR家族也是由该菌侵染诱导引起寄主细胞程序性死亡并产生过敏反应(HR)。
植物跨膜受体能感知细胞外微生物相关分子,跨膜受体对应不同的受体激酶,激酶有不同的胞外结构域对应识别不同的胞外分子。本研究结果显示,受感染的红橘显著富集受体激酶和受体蛋白质,在被链格孢菌橘致病型侵染28 h后,红橘多个LRR类受体基因(Cs2g04320、Cs2g04330、Cs6g10370)被激活,此外还有DUF受体激酶、LRK受体激酶、LysM类受体激酶、WAK受体激酶和凝集素受体激酶这些不同类型的激酶基因都有超过5个以上的家族成员被剧烈诱导。此外多种基因编码抗菌蛋白也在该菌激活下引起上调,这些基因属于病程相关(PR)超家族,包括多个家族组成的抗病群体构成植物诱导防御机制,编码PR基因的转录本PR-1、PR-2(β-1-3-葡聚糖酶)、PR-3、PR-4、PR-8、PR-11(几丁质酶)、PR-5(thaumatin)、PR-6(蛋白酶抑制剂)、PR-9(过氧化物酶)和PR-10(核糖核酸酶)在链格孢菌橘致病型侵染期间累积。高等植物中的保护酶系统在遭受胁迫过程中保护植物不受细胞内ROS造成的氧化损伤,调控抗氧化活性清除ROS对植物的伤害依赖于过氧化物酶(POD)家族蛋白。显然,在红橘受到链格孢菌橘致病型侵害的过程中需要积累大量的POD基因,因此Cs2g09310、Cs2g28680、Cs7g20700等22个POD成员基因均受到活性氧信号激发大量表达。其他上调防御基因包括转录因子、蛋白酶抑制剂和NADPH氧化酶负责产生超氧离子,这些结果证实柑橘内部启动了完全的防御反应(图9)。
图9
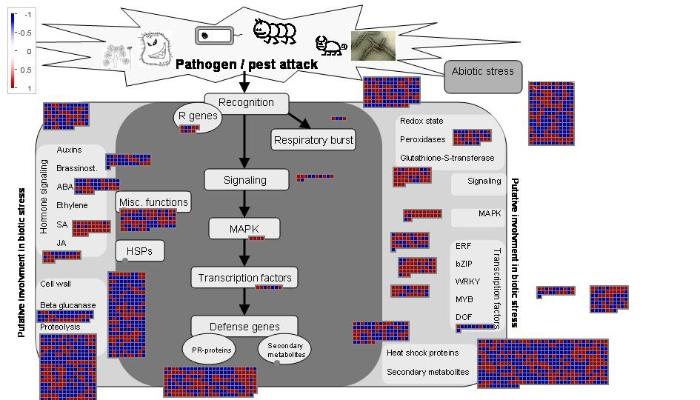 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图9MapMan分析寄主-病原互作相关的差异表达基因
Fig. 9DEGs involved in host-pathogen interaction analyzed by MapMan
2.6 qRT-PCR验证
为检测转录数据的真实性和可靠性,根据转录组的差异基因,选取了一些已报道明确与植物抗病相关的基因且在差异表达分析中log2FC接近10的成员,通过qRT-PCR技术检测其表达水平。由图10可以看出调控抗氧化活性的POD基因(Cs2g09310、Cs7g20700)和谷胱甘肽转移酶基因(orange1. 1t03618)在接种链格孢菌橘致病型的样品中均显著表达上调,细胞壁代谢相关的漆酶(Cs6g07410)和果胶转移酶(Cs3g10430)受该菌攻击也呈现上调趋势,PTI响应的受体激酶LRR(orange1.1t05311)也受该菌诱导,同样ETI调控的R基因NB-ARC 蛋白(Cs1g01180)也上调,几丁质酶(Cs5g21860、orange1.1t03118)作为主要的抗病蛋白上调倍数均超过10倍,转录因子NAC(Cs5g15470)、MYB(Cs4g12760)和WRKY(Cs6g20850)均为植物抗逆过程中重要的调控因子,qRT-PCR和转录数据显示所检测的3个转录因子均受该菌诱导显著上调,qRT-PCR结果与转录组显示基因表达变化趋势一致。图10
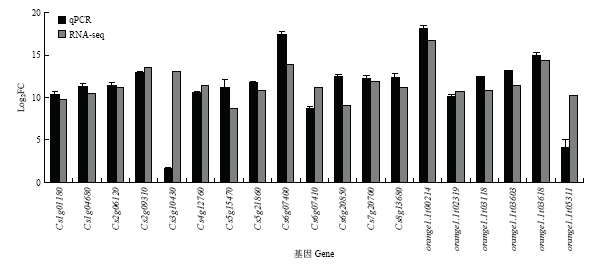 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图10差异表达基因qRT-PCR鉴定
Fig. 10Identification of DEGs by qRT-PCR
3 讨论
3.1 植物防御反应中的激素平衡
本研究中的MPK信号通路和乙烯信号途径均富集大量上调的差异基因,MPK家族成员接受链格孢菌橘致病型信号从而控制红橘ET信号合成,通过ET信号调控下游ERF类转录因子起到激活抗病基因的作用。本氏烟的Ca2+结合蛋白Calreticulin 3a在识别源自致病疫霉(Phytophthora infestans)的MAMP后参与ET的诱导产生,进一步证明ET信号传导是产生抗微生物毒素所必需的[21]。Ca2+信号(CDPK和CML)在本研究中被激活,因此Ca2+也参与响应链格孢菌橘致病型入侵信号并激活ET产生来调控抗病反应。通过MapMan和KEGG所注释的差异基因显示,红橘茉莉酸(JA)途径中的激素合成及信号转导相关基因均发生剧烈变化,其中编码JA生物合成所需12-氧代二亚油酸还原酶的7个基因全部上调,JA受链格孢菌橘致病型侵染后大量合成作为抗病信号激活下游基因表达。关于JA参与防御反应的报道很多,如通过激发子和病原体攻击增加内源性JA和茉莉酸甲酯(MeJA)水平,外源性应用JA或MeJA诱导防御反应基因表达[22,23]。本研究中的JA下游PR基因和相关次生代谢产物合成基因均受到不同程度诱导。芸薹生链格孢(A. brassicicola)感染上调JA生物合成和JA诱导的防御基因[24]。通过直接对拟南芥喷雾接种芸薹生链格孢分生孢子能激活JA相关基因表达[25]。JA在小麦冠腐病菌(Fusarium pseudograminearum)攻击期间延迟了小麦症状的产生[26],并且增强了对壳多胞(Stagonospora nodorum)感染的抗性[27],因此JA信号通路是植物对抗死体营养型真菌特别是链格孢真菌的主要信号转导途径。
3.2 转录因子调控植物防御反应
链格孢菌橘致病型侵染红橘的过程调控抗病基因表达的主要为WRKY和ERF转录因子,检测到的43个差异表达的WRKY转录因子中只有9个表达下调,其余成员均显著上调,其中WRKY22/28/72/75的log2FC接近10。由于受乙烯信号的刺激,乙烯响应因子ERF(Cs7g19640、Cs7g04300、Cs1g04680和Cs1g04650)显著上调并且超过10倍以上。WRKY转录因子可作为防御基因表达中的正或负调节因子,并且是不同防御信号途径的靶标[28]。拟南芥WRKY70参与防御途径之间的交互作用,对坏死细菌软腐菌(Pectobacterium carotovorum)的抗性起重要作用,作为SA响应转录的激活因子又是JA响应转录的抑制剂[29]。AtWRKY70在灰葡萄孢(Botrytis cinerea)感染中被诱导,并且wrky70突变体表现出对此菌的敏感性增强,却对死体营养型真菌芸薹生链格孢具有抗性[30]。本研究中红橘的WRKY70作为拟南芥AtWRKY的同源基因,也受链格孢菌橘致病型激活,说明WRKY70对植物抵御死体营养型真菌的机制有一定相似性。拟南芥wrky33突变体对上述两种菌均非常敏感,表明WRKY33是对这些死体营养型真菌防御的关键正调控因子[31]。WRKY33整合寄主信号传导以赋予对病原体攻击抗性的作用模式,并且WRKY33与MAP4及MPK4的底物MKS1在细胞核相互作用来实现对抗病信号的调控[32,33]。在菊花中也筛选到一个响应链格孢的转录因子基因CmWRKY33.1,通过在菊花中过表达证实该基因亦能增强菊花对该菌的敏感性[34]。过表达菊花的另一个WRKY转录因子基因CmWRKY15可激活ABA相关下游基因,增加过表达植株对细极链格孢(A. tenuissima)的敏感性[35]。在甜橙基因组中预计有超过50个WRKY转录因子,本试验接种链格孢菌橘致病型28 h后通过MapMan注释到WRKY成员中80%以上显著上调,红橘受该菌胁迫后激活转录因子的主要成员为WRKY,其转录的部分成员可能是红橘对链格孢菌橘致病型的感病基因。
APETALA2/乙烯响应元件结合因子(AP2/ERF)家族构成一个大型植物特异性转录因子家族,拟南芥中有140多个成员,水稻中有160个成员[36]。转录分析揭示AP2/ERF型转录因子家族成员在病原菌攻击后被强烈诱导。ERF亚家族成员显示出对GCC序列(AGCCGCC)的最大亲和力,并且参与对生物应激响应基因的调节,特别是与JA和ET信号传导途径相关的基因[37,38,39]。过表达ERF1的转基因拟南芥系足以赋予植株对灰葡萄孢、棉花枯萎病菌(F. oxysporum)和小不整球壳菌(Plectosphaerella cucumerina)的抗性[40,41]。ERF59/ORA59的过表达增强对灰葡萄孢的抗性,而RNAi-ORA59沉默的株系更易受影响[36]。ERF1和ERF59/ORA59似乎都是JA和ET信号通路的关键整合子[25]。ORA59被证明是抵消SA介导的JA/ET应答基因抑制的关键介质[42]。在链格孢菌橘致病型侵染过程中红橘的ERF家族成员受病原菌剧烈诱导,也说明红橘在响应该菌侵染的过程中ET水平改变作为信号刺激下游抗病基因表达来实现抗菌目的。
3.3 植物次级代谢产物介导的防御反应
苯丙烷代谢途径是参与植物防御反应的主要次级代谢产物通路之一,该途径主要通过诱导植物产生具有抗病原微生物活性的次级代谢产物[43]。ZHU等[44]在对苹果响应链格孢菌苹果致病型(A. alternata apple pathotype)的转录组研究中发现β-葡萄糖苷酶基因被诱导表达,该基因参加修饰多种植物抗毒素。氧脂素作为一种具有保护作用的分子,其相关的合成基因LOX也受该菌诱导。本研究中β-葡萄糖苷酶基因和LOX同源基因也受链格孢菌橘致病型诱导大量表达。另外,木质素合成相关的漆酶(Cs6g07400、Cs6g07410)、HCT(Cs7g29080)、COMT(Cs5g18010、orange1.1t05423、Cs5g24990、orange1.1t00578、Cs5g24910)表达量均超出对照6倍以上。DORIA等利用双向电泳研究柑橘响应该菌的蛋白质组学也发现木质素合成相关基因Caffeic acid 3-O-methyltransferase-like(COMT)和Caffeoyl-coa O-methyltransferase-like(CCOMT)显著上调表达[45]。烟草在抵御链格孢菌烟草致病型(A. alternata tobacco pathotype)时能响应激发子诱导的JA信号苯丙烷途径中feruloyl-CoA 6′-hydroxylase 1表达生成东莨菪素[46]。此外,在红橘响应链格孢菌橘致病型的过程中,咖啡因代谢途径中的差异基因(Cs2g30140和Cs7g23340)均显著上调,因此次生代谢产物能直接或间接地参与红橘对该菌的抗病过程。类异戊二烯或萜类化合物是一组用于生长和发育的化学物质,但也具有抵抗不同压力的特殊功能[47],烟草中的ERF转录因子受ET和JA信号调控,能激活下游的EAS12表达合成倍半萜类抗菌物质甜椒醇抑制链格孢菌烟草致病型[48]。水稻OsTPS19超表达帮助植株产生大量柠檬烯从而抵御稻瘟病菌[49]。正常状态下植物的萜类物质具有参与抑制病原菌生长并且在植物体内传递抗病信号的作用[50],然而红橘被链格孢菌橘致病型侵染过程中MapMan注释的萜类合成30个基因中22个呈现下调表达,而在激素信号中ET和JA信号又呈现出上调表达,表明该菌对红橘体内萜类骨架的破坏导致抗病的萜烯类物质合成受影响,造成萜类物质含量不足以抑制该菌导致感病。
3.4 红橘防御链格孢菌橘致病型侵染的ROS信号及ROS清除
ROS产生与清理之间的平衡会受到许多不利因素的干扰,植物受到严重胁迫时ROS平衡会被打破[51]。病原菌攻击会造成大量的ROS产生来抑制病菌在植物体内繁殖,病菌和植物体双方都通过清除ROS维持自身细胞活性,链格孢菌通过NADPH氧化酶和过氧化物酶耐受植物产生的ROS保证其毒力[10]。NADPH氧化酶是植物-病原体互作过程中产生ROS的主要因子之一,通过将电子从细胞内NADPH转移到质外体中的分子氧来催化超氧化物产生的跨膜蛋白[52],可通过自发歧化或细胞壁超氧化物歧化酶(SOD)的催化活性将超氧化物进一步转化为H2O2。ROS产生的主要来源是NADPH氧化酶[53]。本研究显示柑橘能通过识别链格孢菌橘致病型的PPR导致CDPK活性升高,促使下游NADPH氧化酶同源基因rboh-Cs8g12000和Cs3g14240显著升高来调控ROS抑制该病原菌生长。过氧化物酶类如抗坏血酸过氧化物酶(APX)和谷胱甘肽过氧化物酶(GPX)被证实为植物受到病原菌侵染时产生的病程相关蛋白,是有效的活性氧清除剂,在植物调控细胞的氧化还原动态平衡、防御反应及胁迫响应中具有重要作用[54]。植物中的酶法ROS清除机制还包括SOD和过氧化氢酶(CAT),在链格孢菌橘致病型接种后的红橘中检测到Cs2g09310、Cs2g28680和Cs7g20700等20多个POD成员均受到真菌毒素刺激活性氧信号激发大量表达,其他抗氧化途径中包括超过30个谷胱甘肽转移酶(GST)受ROS诱导,抗坏血酸合成酶和APX都有不同程度上调,而在不同品种柑橘响应链格孢菌橘致病型的双向电泳中筛选出的差异蛋白同样发现GSTAPX[45],从转录层面和蛋白层面说明感病柑橘通过保护酶系统调整自身内部的ROS平衡来对抗该菌。
4 结论
链格孢菌橘致病型的侵染引起红橘大量基因差异表达。利用qRT-PCR验证了19个差异基因的表达量变化,均与RNA-seq分析结果一致。GO功能分类显示差异基因主要与分子功能、细胞成分和生物过程相关。KEGG和MapMan分析表明,受链格孢菌橘致病型诱导上调的差异表达基因可富集到病原菌识别、信号转导、活性氧消除、转录调控、次生代谢反应病程相关蛋白等生物胁迫相关基因类别。转录组数据分析显示,大量萜类合成酶在受链格孢菌橘致病型侵染后呈现下调表达,推测是红橘对该菌敏感的原因。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1094/Phyto-69-667URL [本文引用: 1]
DOI:10.1146/annurev.py.21.090183.000511URLPMID:25946338 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1094/PDIS-94-3-0375BURLPMID:30754225 [本文引用: 1]

Phalaris paradoxa (hood canarygrass) is one of the most abundant weeds in wheat fields of Iran. In a survey conducted from 2005 to 2007 in Ilam (Dehloran City) and Golestan (Gorgan City) provinces, leaf blotch symptoms were prevalent on P. paradoxa. Initial symptoms were pale brown and necrotic lesions that were 3 to 4 mm long on the leaves. Severity of the disease on the lower leaves was higher than on the upper leaves. Pycnidia were observed on the adaxial surface of infected leaves, scattered or sometimes in clusters, dark brown, globose, and 70 to 90 mum in diameter, with the ostiole approximately 10 mum in diameter. Conidia were filiform, hyaline, 0 to 3 septate (mostly 1 septate), and 17 to 40 x 1.5 to 2.0 mum. Conidiogenesis type was holoblastic. On the basis of the above morphological characters, this species was identified as Septoria phalaridis Cocc. & Morini (2,3). Sequencing the internal transcribed spacer (ITS) region of the fungus (GenBank Accession No. GU123926) showed 98% homology to Mycosphaerella graminicola strain 687 and 97% to S. passerinii strain ATCC26515 (GenBank Accession Nos. AB435068.1 and AF181696.1). To confirm pathogenicity of the fungus, 25 P. paradoxa seedlings were inoculated at the three-leaf stage with 20 ml of 1 x 10(7) spores/ml suspension with a hand sprayer. Plants were covered with a clear polyethylene bag to increase humidity and prevent cross contamination. After 72 h, bags were removed and plants were kept in a greenhouse at 21 +/- 2/16 +/- 2 degrees C (day/night) and a 16-h photoperiod. Control plants received sterilized distilled water only. Leaves of each plant were visually inspected every day and the appearance of disease symptoms was recorded. After 1 month, all inoculated leaves showed symptoms and signs of the disease such as chlorosis, necrosis, and pycnidia, whereas control plants showed no symptoms or signs of disease. The infected plant tissues were examined with a microscope, the pycnidia and pycnidiospores were measured, and S. phalaridis was reisolated from leaf lesions. The first description of S. phalaridis was on P. brachystachys (1); however, to our knowledge, this is the first report of this pathogen on P. paradoxa. In addition, this is a new fungal species for the mycobiota of Iran. Two voucher specimens (IRAN 14078 F and IRAN 14218 F) were deposited in the Fungus Collection of the Ministry of Jihad-e Agriculture, Tehran, Iran. References: (1) G. Cocconi and F. Morini. Mem. R. Accad. Sci. Ist. Bologna, Cl. Sci. Fis. Ser. 4, 6:371, 1884. (2) M. J. Priest. Fungi of Australia, Septoria. ABRS, Canberra. CSIRO Publishing, Melbourne, 2006. (3) D. N. Teterevnikova-Babayan. Fungi of the Genus Septoria in the USSR. Akademiya Nauk Armyanskoi SSR, Yerevan, 1987.
URL [本文引用: 1]
自2007 年起重庆万州的红橘(Citrus reticulata Blanco, CV. Hongjv)上爆发一种未曾见过的病害,损失严重,因病斑褐色,故被称之为褐斑病。褐斑病贯穿柑橘整个生长季节,主要为害叶片和果实,严重时也为害新梢,引起大量落叶和落果,甚至枝梢枯死。为明确其病原,本研究从病区多个果园采集发病的叶片、果实和枝梢,检查和分离培养发现病组织中广泛存在链格孢菌(Alternaria)和炭疽菌(Colletotrichum)。致病性试验证明只有链格孢菌才能引起类似褐斑症状,而炭疽菌和空白对照都不产生任何症状。通过比较研究这些链格孢菌菌株的形态学,培养性状和多聚半乳糖醛酸酶(endoPG)基因的部分序列,可以断定红橘褐斑病的病原为交链格孢菌[A. alternate (Fr. ) Keissler]。病原性质的确定将为红橘褐斑病发生规律的研究和控制奠定基础。
URL [本文引用: 1]
自2007 年起重庆万州的红橘(Citrus reticulata Blanco, CV. Hongjv)上爆发一种未曾见过的病害,损失严重,因病斑褐色,故被称之为褐斑病。褐斑病贯穿柑橘整个生长季节,主要为害叶片和果实,严重时也为害新梢,引起大量落叶和落果,甚至枝梢枯死。为明确其病原,本研究从病区多个果园采集发病的叶片、果实和枝梢,检查和分离培养发现病组织中广泛存在链格孢菌(Alternaria)和炭疽菌(Colletotrichum)。致病性试验证明只有链格孢菌才能引起类似褐斑症状,而炭疽菌和空白对照都不产生任何症状。通过比较研究这些链格孢菌菌株的形态学,培养性状和多聚半乳糖醛酸酶(endoPG)基因的部分序列,可以断定红橘褐斑病的病原为交链格孢菌[A. alternate (Fr. ) Keissler]。病原性质的确定将为红橘褐斑病发生规律的研究和控制奠定基础。
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/1462-2920.13125URLPMID:26567914 [本文引用: 2]
The ability to detoxify reactive oxygen species (ROS) is critical for pathogenicity in the necrotrophic fungus Alternaria alternata. We report a glutathione peroxidase 3 (AaGPx3) involved in the complex signalling network that is essential for the detoxification of cellular stresses induced by ROS and for A. alternata pathogenesis in citrus. AaGPx3 deletion mutants displayed increased sensitivity to H2 O2 and many ROS-generating compounds. AaGPx3 is required for correct fungal development as the AaGPx3 mutant strains showed a severe reduction in conidiation. AaGPx3 mutants accumulated higher chitin content than the wild-type and were less sensitive to the cell wall-targeting compounds calcofluor white and Congo red, as well as the fungicides fludioxonil and vinclozolin, suggesting a role of the glutathione systems in fungal cell wall construction. Virulence assays revealed that AaGPx3 is required for full virulence. The expression of AaGPx3 was downregulated in fungal strains carrying defective NADPH oxidase (Nox) or the oxidative stress responsive regulators YAP1 and HOG1, all implicated in ROS resistance. These results further support the important role of ROS detoxification during A. alternata pathogenesis in citrus. Overall, our study provides genetic evidence to define the central role of AaGPx3 in the biological and pathological functions of A. alternata.
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
DOI:10.1038/srep32437URLPMID:27582273 [本文引用: 1]
The tangerine pathotype of Alternaria alternata produces the A. citri toxin (ACT) and is the causal agent of citrus brown spot that results in significant yield losses worldwide. Both the production of ACT and the ability to detoxify reactive oxygen species (ROS) are required for A. alternata pathogenicity in citrus. In this study, we report the 34.41 Mb genome sequence of strain Z7 of the tangerine pathotype of A. alternata. The host selective ACT gene cluster in strain Z7 was identified, which included 25 genes with 19 of them not reported previously. Of these, 10 genes were present only in the tangerine pathotype, representing the most likely candidate genes for this pathotype specialization. A transcriptome analysis of the global effects of H2O2 on gene expression revealed 1108 up-regulated and 498 down-regulated genes. Expressions of those genes encoding catalase, peroxiredoxin, thioredoxin and glutathione were highly induced. Genes encoding several protein families including kinases, transcription factors, transporters, cytochrome P450, ubiquitin and heat shock proteins were found associated with adaptation to oxidative stress. Our data not only revealed the molecular basis of ACT biosynthesis but also provided new insights into the potential pathways that the phytopathogen A. alternata copes with oxidative stress.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1186/1471-2229-10-234URLPMID:21029438 [本文引用: 1]
BACKGROUND: Downy mildew (DM), caused by pathogen Plasmopara viticola (PV) is the single most damaging disease of grapes (Vitis L.) worldwide. However, the mechanisms of the disease development in grapes are poorly understood. A method for estimating gene expression levels using Solexa sequencing of Type I restriction-endonuclease-generated cDNA fragments was used for deep sequencing the transcriptomes resulting from PV infected leaves of Vitis amurensis Rupr. cv. Zuoshan-1. Our goal is to identify genes that are involved in resistance to grape DM disease. RESULTS: Approximately 8.5 million (M) 21-nt cDNA tags were sequenced in the cDNA library derived from PV pathogen-infected leaves, and about 7.5 M were sequenced from the cDNA library constructed from the control leaves. When annotated, a total of 15,249 putative genes were identified from the Solexa sequencing tags for the infection (INF) library and 14,549 for the control (CON) library. Comparative analysis between these two cDNA libraries showed about 0.9% of the unique tags increased by at least five-fold, and about 0.6% of the unique tags decreased more than five-fold in infected leaves, while 98.5% of the unique tags showed less than five-fold difference between the two samples. The expression levels of 12 differentially expressed genes were confirmed by Real-time RT-PCR and the trends observed agreed well with the Solexa expression profiles, although the degree of change was lower in amplitude. After pathway enrichment analysis, a set of significantly enriched pathways were identified for the differentially expressed genes (DEGs), which associated with ribosome structure, photosynthesis, amino acid and sugar metabolism. CONCLUSIONS: This study presented a series of candidate genes and pathways that may contribute to DM resistance in grapes, and illustrated that the Solexa-based tag-sequencing approach was a powerful tool for gene expression comparison between control and treated samples.
DOI:10.1093/jxb/err245URL [本文引用: 1]
The incompatible pathosystem between resistant cotton (Gossypium barbadense cv. 7124) and Verticillium dahliae strain V991 was used to study the cotton transcriptome changes after pathogen inoculation by RNA-Seq. Of 32 774 genes detected by mapping the tags to assembly cotton contigs, 3442 defence-responsive genes were identified. Gene cluster analyses and functional assignments of differentially expressed genes indicated a significant transcriptional complexity. Quantitative real-time PCR (qPCR) was performed on selected genes with different expression levels and functional assignments to demonstrate the utility of RNA-Seq for gene expression profiles during the cotton defence response. Detailed elucidation of responses of leucine-rich repeat receptor-like kinases (LRR-RLKs), phytohormone signalling-related genes, and transcription factors described the interplay of signals that allowed the plant to fine-tune defence responses. On the basis of global gene regulation of phenylpropanoid metabolism-related genes, phenylpropanoid metabolism was deduced to be involved in the cotton defence response. A closer look at the expression of these genes, enzyme activity, and lignin levels revealed differences between resistant and susceptible cotton plants. Both types of plants showed an increased level of expression of lignin synthesis-related genes and increased phenylalanine-ammonia lyase (PAL) and peroxidase (POD) enzyme activity after inoculation with V. dahliae, but the increase was greater and faster in the resistant line. Histochemical analysis of lignin revealed that the resistant cotton not only retains its vascular structure, but also accumulates high levels of lignin. Furthermore, quantitative analysis demonstrated increased lignification and cross-linking of lignin in resistant cotton stems. Overall, a critical role for lignin was believed to contribute to the resistance of cotton to disease.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/nmeth.3317URLPMID:25751142 [本文引用: 1]
HISAT (hierarchical indexing for spliced alignment of transcripts) is a highly efficient system for aligning reads from RNA sequencing experiments. HISAT uses an indexing scheme based on the Burrows-Wheeler transform and the Ferragina-Manzini (FM) index, employing two types of indexes for alignment: a whole-genome FM index to anchor each alignment and numerous local FM indexes for very rapid extensions of these alignments. HISAT's hierarchical index for the human genome contains 48,000 local FM indexes, each representing a genomic region of approximately 64,000 bp. Tests on real and simulated data sets showed that HISAT is the fastest system currently available, with equal or better accuracy than any other method. Despite its large number of indexes, HISAT requires only 4.3 gigabytes of memory. HISAT supports genomes of any size, including those larger than 4 billion bases.
[本文引用: 1]
DOI:10.1073/pnas.89.6.2389URLPMID:11607285 [本文引用: 1]
To deter pathogenic microorganisms and herbivores, plants have developed an inducible chemical defense system. It is known that the induced synthesis of low molecular weight compounds can be provoked by exposing cultured cells to fungal cell wall fragments. In this study we show that endogenous jasmonic acid and its methyl ester accumulate rapidly and transiently after treatment of plant cell suspension cultures of Rauvolfia canescens and Eschscholtzia californica with a yeast elicitor. Thirty-six plant species tested in cell suspension culture could be elicited with respect to the accumulation of secondary metabolites by exogenously supplied methyl jasmonate. Addition of methyl jasmonate initiates de novo transcription of genes, such as phenylalanine ammonia lyase, that are known to be involved in the chemical defense mechanisms of plants. These data demonstrate the integral role of jasmonic acid and its derivatives in the intracellular signal cascade that begins with interaction of an elicitor molecule with the plant cell surface and results, ultimately, in the accumulation of secondary compounds.
DOI:10.1104/pp.110.2.387URLPMID:12226190 [本文引用: 1]
It has been suggested that jasmonic acid (JA) could be an integral part of a general signal transduction system regulating inducible defense genes in plants. It was reported that treatment with an elicitor (N-acetylchitoheptaose) induced production of phytoalexin in suspension-cultured rice (Oryza sativa L.) cells. In this study, the role of JA in the induction of phytoalexin production by N-acetylchitoheptaose was investigated. Exogenously applied ([plus or minus])-JA (10-4 M) clearly induced the production of momilactone A, a major phytoalexin, in suspension-cultured rice cells. On the other hand, in rice cells treated with N-acetylchitoheptaose, endogenous JA was rapidly and transiently accumulated prior to accumulation of momilactone A. Treatment with ibuprofen, an inhibitor of JA biosynthesis, reduced production of momilactone A in the cells treated with N-acetylchitoheptaose, but the addition of ([plus or minus])-JA increased production of momilactone A to levels higher than those in the elicited rice cells. These results strongly suggest that JA functions as a signal transducer in the induction of biosynthesis of momilactone A by N-acetylchitoheptaose in suspension-cultured rice cells.
DOI:10.1104/pp.103.022186URLPMID:12805591 [本文引用: 1]
All tested accessions of Arabidopsis are resistant to the fungal pathogen Alternaria brassicicola. Resistance is compromised by pad3 or coi1 mutations, suggesting that it requires the Arabidopsis phytoalexin camalexin and jasmonic acid (JA)-dependent signaling, respectively. This contrasts with most well-studied Arabidopsis pathogens, which are controlled by salicylic acid-dependent responses and do not benefit from absence of camalexin or JA. Here, mutants with defects in camalexin synthesis (pad1, pad2, pad3, and pad5) or in JA signaling (pad1, coi1) were found to be more susceptible than wild type. Mutants with defects in salicylic acid (pad4 and sid2) or ethylene (ein2) signaling remained resistant. Plant responses to A. brassicicola were characterized using expression profiling. Plants showed dramatic gene expression changes within 12 h, persisting at 24 and 36 h. Wild-type and pad3 plants responded similarly, suggesting that pad3 does not have a major effect on signaling. The response of coi1 plants was quite different. Of the 645 genes induced by A. brassicicola in wild-type and pad3 plants, 265 required COI1 for full expression. It is likely that some of the COI1-dependent genes are important for resistance to A. brassicicola. Responses to A. brassicicola were compared with responses to Pseudomonas syringae infection. Despite the fact that these pathogens are limited by different defense responses, approximately 50% of the induced genes were induced in response to both pathogens. Among these, requirements for COI1 were consistent after infection by either pathogen, suggesting that the regulatory effect of COI1 is similar regardless of the initial stimulus.
[本文引用: 2]
DOI:10.1016/j.pmpp.2005.12.007URL [本文引用: 1]
DOI:10.1023/B:BIOP.0000041097.03177.2dURL [本文引用: 1]
The effect of application of jasmonic acid (JA) and salicylic acid (SA) on the induction of resistance in wheat to Stagonospora nodorum and on the induction of
 -1,3-glucanase and thaumatin-like proteins (TLPs) was studied. Western blot analysis revealed that two
-1,3-glucanase and thaumatin-like proteins (TLPs) was studied. Western blot analysis revealed that two  -1,3-glucanases with apparent molecular masses of 31 and 33 kDa that cross-reacted with a barley glucanase antiserum were induced in wheat leaves after treatment with JA and SA. When wheat plants were treated with SA and JA, a TLP with an apparent molecular mass of 25 kDa and several other isoforms of TLP were induced. Pre-treatment of wheat plants with SA and JA significantly reduced (up to 56 %) the incidence of leaf blotch disease incited by S. nodorum compared with untreated control plants.
-1,3-glucanases with apparent molecular masses of 31 and 33 kDa that cross-reacted with a barley glucanase antiserum were induced in wheat leaves after treatment with JA and SA. When wheat plants were treated with SA and JA, a TLP with an apparent molecular mass of 25 kDa and several other isoforms of TLP were induced. Pre-treatment of wheat plants with SA and JA significantly reduced (up to 56 %) the incidence of leaf blotch disease incited by S. nodorum compared with untreated control plants.DOI:10.1104/pp.109.138990URLPMID:19420325 [本文引用: 1]
DOI:10.1105/tpc.016980URLPMID:14742872 [本文引用: 1]
Cross talk between salicylic acid (SA)- and jasmonic acid (JA)-dependent defense signaling has been well documented in plants, but how this cross talk is executed and the components involved remain to be elucidated. We demonstrate that the plant-specific transcription factor WRKY70 is a common component in SA- and JA-mediated signal pathways. Expression of WRKY70 is activated by SA and repressed by JA. The early induction of WRKY70 by SA is NPR1-independent, but functional NPR1 is required for full-scale induction. Epistasis analysis suggested that WRKY70 is downstream of NPR1 in an SA-dependent signal pathway. Modulation of WRKY70 transcript levels by constitutive overexpression increases resistance to virulent pathogens and results in constitutive expression of SA-induced pathogenesis-related genes. Conversely, antisense suppression of WRKY70 activates JA-responsive/COI1-dependent genes. The effect of WRKY70 is not caused by subsequent changes in SA or JA levels. We suggest that WRKY70 acts as an activator of SA-induced genes and a repressor of JA-responsive genes, integrating signals from these mutually antagonistic pathways.
DOI:10.1111/j.1365-313X.2006.02849.xURLPMID:16925600 [本文引用: 1]
The expression profiles of Botrytis-inoculated Arabidopsis plants were studied to determine the nature of the defense transcriptome and to identify genes involved in host responses to the pathogen. Normally resistant Arabidopsis wild-type plants were compared with coi1, ein2, and nahG plants that are defective in various defense responses and/or show increased susceptibility to Botrytis. In wild-type plants, the expression of 621 genes representing approximately 0.48% of the Arabidopsis transcriptome was induced greater than or equal to twofold after infection. Of these 621 Botrytis-induced genes (BIGs), 462 were induced at or before 36 h post-inoculation, and may be involved in resistance to the pathogen. The expression of 181 BIGs was dependent on a functional COI1 gene required for jasmonate signaling, whereas the expression of 63 and 80 BIGs were dependent on ethylene (ET) signaling or salicylic acid accumulation, respectively, based on results from ein2 and nahG plants. BIGs encode diverse regulatory and structural proteins implicated in pathogen defense and abiotic and oxidative-stress responses. Thirty BIGs encode putative DNA-binding proteins that belong to ET response, zinc-finger, MYB, WRKY, and HD-ZIP family transcription-factor proteins. Fourteen BIGs were studied in detail to determine their role in resistance to Botrytis. T-DNA insertion alleles of ZFAR1 (At2G40140), the gene encoding a putative zinc-finger protein with ankyrin-repeat domains, showed increased local susceptibility to Botrytis and sensitivity to germination in the presence of abscisic acid (ABA), supporting the role of ABA in mediating responses to Botrytis infection. In addition, two independent T-DNA insertion alleles in the WRKY70 gene showed increased susceptibility to Botrytis. The transcriptional activation of genes involved in plant hormone signaling and synthesis, removal of reactive oxygen species, and defense and abiotic-stress responses, coupled with the susceptibility of the wrky70 and zfar1 mutants, highlights the complex genetic network underlying defense responses to Botrytis in Arabidopsis.
DOI:10.1111/j.1365-313X.2006.02901.xURLPMID:17059405 [本文引用: 1]
Plant WRKY transcription factors are key regulatory components of plant responses to microbial infection. In addition to regulating the expression of defense-related genes, WRKY transcription factors have also been shown to regulate cross-talk between jasmonate- and salicylate-regulated disease response pathways. The two pathways mediate resistance against different types of microbial pathogens, and there are numerous reports of antagonistic interactions between them. Here we show that mutations of the Arabidopsis WRKY33 gene encoding a WRKY transcription factor cause enhanced susceptibility to the necrotrophic fungal pathogens Botrytis cinerea and Alternaria brassicicola concomitant with reduced expression of the jasmonate-regulated plant defensin PDF1.2 gene. Ectopic over-expression of WRKY33, on the other hand, increases resistance to the two necrotrophic fungal pathogens. The wrky33 mutants do not show altered responses to a virulent strain of the bacterial pathogen Pseudomonas syringae, although the ectopic expression of WRKY33 results in enhanced susceptibility to this pathogen. The susceptibility of WRKY33-over-expressing plants to P. syringae is associated with reduced expression of the salicylate-regulated PR-1 gene. The WRKY33 transcript is induced in response to pathogen infection, or treatment with salicylate or the paraquat herbicide that generates activated oxygen species in exposed cells. WRKY33 is localized to the nucleus of plant cells and recognizes DNA molecules containing the TTGACC W-box sequence. Together, these results indicate that pathogen-induced WRKY33 is an important transcription factor that regulates the antagonistic relationship between defense pathways mediating responses to P. syringae and necrotrophic pathogens.
DOI:10.1038/sj.emboj.7600737URLPMID:15990873 [本文引用: 1]
Arabidopsis MAP kinase 4 (MPK4) functions as a regulator of pathogen defense responses, because it is required for both repression of salicylic acid (SA)-dependent resistance and for activation of jasmonate (JA)-dependent defense gene expression. To understand MPK4 signaling mechanisms, we used yeast two-hybrid screening to identify the MPK4 substrate MKS1. Analyses of transgenic plants and genome-wide transcript profiling indicated that MKS1 is required for full SA-dependent resistance in mpk4 mutants, and that overexpression of MKS1 in wild-type plants is sufficient to activate SA-dependent resistance, but does not interfere with induction of a defense gene by JA. Further yeast two-hybrid screening revealed that MKS1 interacts with the WRKY transcription factors WRKY25 and WRKY33. WRKY25 and WRKY33 were shown to be in vitro substrates of MPK4, and a wrky33 knockout mutant was found to exhibit increased expression of the SA-related defense gene PR1. MKS1 may therefore contribute to MPK4-regulated defense activation by coupling the kinase to specific WRKY transcription factors.
Arabidopsis map kinase 4 regulates gene expression through transcription factor release in the nucleus
DOI:10.1038/emboj.2008.147URLPMID:18650934 [本文引用: 1]
Plant and animal perception of microbes through pathogen surveillance proteins leads to MAP kinase signalling and the expression of defence genes. However, little is known about how plant MAP kinases regulate specific gene expression. We report that, in the absence of pathogens, Arabidopsis MAP kinase 4 (MPK4) exists in nuclear complexes with the WRKY33 transcription factor. This complex depends on the MPK4 substrate MKS1. Challenge with Pseudomonas syringae or flagellin leads to the activation of MPK4 and phosphorylation of MKS1. Subsequently, complexes with MKS1 and WRKY33 are released from MPK4, and WRKY33 targets the promoter of PHYTOALEXIN DEFICIENT3 (PAD3) encoding an enzyme required for the synthesis of antimicrobial camalexin. Hence, wrky33 mutants are impaired in the accumulation of PAD3 mRNA and camalexin production upon infection. That WRKY33 is an effector of MPK4 is further supported by the suppression of PAD3 expression in mpk4-wrky33 double mutant backgrounds. Our data establish direct links between MPK4 and innate immunity and provide an example of how a plant MAP kinase can regulate gene expression by releasing transcription factors in the nucleus upon activation.
DOI:10.1038/s41438-020-0245-0URLPMID:32140232 [本文引用: 1]
Chrysanthemum (Chrysanthemum morifolium) black spot disease (CBS) poses a major threat to Chrysanthemum cultivation owing to suitable climate conditions and current lack of resistant cultivars for greenhouse cultivation. In this study, we identified a number of genes that respond to Alternaria alternata infection in resistant and susceptible Chrysanthemum cultivars. Based on RNA sequencing technology and a weighted gene coexpression network analysis (WGCNA), we constructed a model to elucidate the response of Chrysanthemum leaves to A. alternata infection at different stages and compared the mapped response of the resistant cultivar 'Jinba' to that of the susceptible cultivar 'Zaoyihong'. In the early stage of infection, when lesions had not yet formed, abscisic acid (ABA), salicylic acid (SA) and EDS1-mediated resistance played important roles in the Chrysanthemum defense system. With the formation of necrotic lesions, ethylene (ET) metabolism and the Ca(2+) signal transduction pathway strongly responded to A. alternata infection. During the late stage, when necrotic lesions continued to expand, members of the multidrug and toxic compound extrusion (MATE) gene family were highly expressed, and their products may be involved in defense against A. alternata invasion by exporting toxins produced by the pathogen, which plays important roles in the pathogenicity of A. alternata. Furthermore, the function of hub genes was verified by qPCR and transgenic assays. The identification of hub genes at different stages, the comparison of hub genes between the two cultivars and the highly expressed genes in the resistant cultivar 'Jinba' provide a theoretical basis for breeding cultivars resistant to CBS.
[本文引用: 1]
DOI:10.1104/pp.108.117523URLPMID:18467450 [本文引用: 2]
Plant defense against pathogens depends on the action of several endogenously produced hormones, including jasmonic acid (JA) and ethylene. In certain defense responses, JA and ethylene signaling pathways synergize to activate a specific set of defense genes. Here, we describe the role of the Arabidopsis (Arabidopsis thaliana) APETALA2/ETHYLENE RESPONSE FACTOR (AP2/ERF) domain transcription factor ORA59 in JA and ethylene signaling and in defense. JA- and ethylene-responsive expression of several defense genes, including PLANT DEFENSIN1.2 (PDF1.2), depended on ORA59. As a result, overexpression of ORA59 caused increased resistance against the fungus Botrytis cinerea, whereas ORA59-silenced plants were more susceptible. Several AP2/ERF domain transcription factors have been suggested to be positive regulators of PDF1.2 gene expression based on overexpression in stably transformed plants. Using two different transient overexpression approaches, we found that only ORA59 and ERF1 were able to activate PDF1.2 gene expression, in contrast to the related proteins AtERF1 and AtERF2. Our results demonstrate that ORA59 is an essential integrator of the JA and ethylene signal transduction pathways and thereby provide new insight into the nature of the molecular components involved in the cross talk between these two hormones.
DOI:10.1038/nchembio.164URLPMID:19377457 [本文引用: 1]
Plants live in complex environments in which they intimately interact with a broad range of microbial pathogens with different lifestyles and infection strategies. The evolutionary arms race between plants and their attackers provided plants with a highly sophisticated defense system that, like the animal innate immune system, recognizes pathogen molecules and responds by activating specific defenses that are directed against the invader. Recent advances in plant immunity research have provided exciting new insights into the underlying defense signaling network. Diverse small-molecule hormones play pivotal roles in the regulation of this network. Their signaling pathways cross-communicate in an antagonistic or synergistic manner, providing the plant with a powerful capacity to finely regulate its immune response. Pathogens, on the other hand, can manipulate the plant's defense signaling network for their own benefit by affecting phytohormone homeostasis to antagonize the host immune response.
DOI:10.1016/j.pbi.2004.04.007URL [本文引用: 1]
Abstract
The AP2 transcription factor family, found only in plants, includes several genes that encode proteins involved in the regulation of disease resistance pathways. These genes are members of the ethylene response factor (ERF) subfamily of AP2 transcription factor genes, which have only a single DNA-binding domain and are distinct from members of the dehydration-responsive element binding (DREB) subfamily. Some ERF subgroups are enriched in such genes, suggesting that they have conserved functions that are required for the regulation of disease resistance pathways. The expression of several ERF genes is regulated by plant hormones, such as jasmonic acid, salicylic acid and ethylene, as well as by pathogen challenge. A phylogenetic overview of these genes, with a focus on Arabidopsis, rice and tomato, suggests that despite broad conservation of their function in monocots and dicots, some structural elements are specialized within each of these two lineages.[本文引用: 1]
DOI:10.1094/MPMI.2004.17.7.763URLPMID:15242170 [本文引用: 1]
Ethylene response factor 1 (ERF1) is a transcriptional factor from Arabidopsis thaliana that regulates plant resistance to the necrotrophic fungi Botrytis cinerea and Plectosphaerella cucumerina and whose overexpression enhances resistance to these fungi. Here, we show that ERF1 also mediates Arabidopsis resistance to the soilborne fungi Fusarium oxysporum sp. conglutinans and F. oxysporum f. sp. lycopersici, because its constitutive expression in Arabidopsis confers enhanced resistance to these pathogens. Expression of ERF1 was upregulated after inoculation with F. oxysporum f. sp. conglutinans, and this response was blocked in ein2-5 and coi1-1 mutants, impaired in the ethylene (ET) and jasmonic acid (JA) signal pathways, respectively, which further indicates that ERF1 is a downstream component of ET and JA defense responses. The signal transduction network controlling resistance to F. oxysporum fungi was explored using signaling-defective mutants in ET (ein2-5), JA (jar1-1), and salicylic acid (SA) (NahG, sid2-1, eds5-1, npr1-1, pad4-1, eds1-1, and pad2-1) transduction pathways. This analysis revealed that Arabidopsis resistance to F. oxysporum requires the ET, JA, and SA signaling pathways and the NPR1 gene, although it is independent of the PAD4 and EDS1 functions.
DOI:10.1046/j.1365-313x.2002.01191.xURLPMID:12060224 [本文引用: 1]
Infection of a plant by a pathogen induces a variety of defense responses that imply the action of several signaling molecules, including salicylic acid (SA), jasmonic acid (JA) and ethylene (E). Here we describe the role of ETHYLENE-RESPONSE-FACTOR1 (ERF1) as a regulator of ethylene responses after pathogen attack in Arabidopsis. The ERF1 transcript is induced on infection by Botrytis cinerea, and overexpression of ERF1 in Arabidopsis is sufficient to confer resistance to necrotrophic fungi such as B. cinerea and Plectosphaerella cucumerina. A positive co-operation between E and SA pathways was observed in the plant response to P. cucumerina. Infection by Pseudomonas syringae tomato DC3000, however, does not affect ERF1 expression, and activation of ethylene responses by ERF1 overexpression in Arabidopsis plants reduces tolerance against this pathogen, suggesting negative crosstalk between E and SA signaling pathways, and demonstrating that positive and negative interactions between both pathways can be established depending on the type of pathogen.
DOI:10.1094/MPMI-23-2-0187URLPMID:20064062 [本文引用: 1]
Cross-talk between jasmonate (JA), ethylene (ET), and Salicylic acid (SA) signaling is thought to operate as a mechanism to fine-tune induced defenses that are activated in response to multiple attackers. Here, 43 Arabidopsis genotypes impaired in hormone signaling or defense-related processes were screened for their ability to express SA-mediated suppression of JA-responsive gene expression. Mutant cev1, which displays constitutive expression of JA and ET responses, appeared to be insensitive to SA-mediated suppression of the JA-responsive marker genes PDF1.2 and VSP2. Accordingly, strong activation of JA and ET responses by the necrotrophic pathogens Botrytis cinerea and Alternaria brassicicola prior to SA treatment counteracted the ability of SA to suppress the JA response. Pharmacological assays, mutant analysis, and studies with the ET-signaling inhibitor 1-methylcyclopropene revealed that ET signaling renders the JA response insensitive to subsequent suppression by SA. The APETALA2/ETHYLENE RESPONSE FACTOR transcription factor ORA59, which regulates JA/ET-responsive genes such as PDF1.2, emerged as a potential mediator in this process. Collectively, our results point to a model in which simultaneous induction of the JA and ET pathway renders the plant insensitive to future SA-mediated suppression of JA-dependent defenses, which may prioritize the JA/ET pathway over the SA pathway during multi-attacker interactions.
DOI:10.1016/j.plaphy.2007.12.009URL [本文引用: 1]
Abstract
As a major component of plant specialized metabolism, phenylpropanoid biosynthetic pathways provide anthocyanins for pigmentation, flavonoids such as flavones for protection against UV photodamage, various flavonoid and isoflavonoid inducers of Rhizobium nodulation genes, polymeric lignin for structural support and assorted antimicrobial phytoalexins. As constituents of plant-rich diets and an assortment of herbal medicinal agents, the phenylpropanoids exhibit measurable cancer chemopreventive, antimitotic, estrogenic, antimalarial, antioxidant and antiasthmatic activities. The health benefits of consuming red wine, which contains significant amounts of 3,4′,5-trihydroxystilbene (resveratrol) and other phenylpropanoids, highlight the increasing awareness in the medical community and the public at large as to the potential dietary importance of these plant derived compounds. As recently as a decade ago, little was known about the three-dimensional structure of the enzymes involved in these highly branched biosynthetic pathways. Ten years ago, we initiated X-ray crystallographic analyses of key enzymes of this pathway, complemented by biochemical and enzyme engineering studies. We first investigated chalcone synthase (CHS), the entry point of the flavonoid pathway, and its close relative stilbene synthase (STS). Work soon followed on the O-methyl transferases (OMTs) involved in modifications of chalcone, isoflavonoids and metabolic precursors of lignin. More recently, our groups and others have extended the range of phenylpropanoid pathway structural investigations to include the upstream enzymes responsible for the initial recruitment of phenylalanine and tyrosine, as well as a number of reductases, acyltransferases and ancillary tailoring enzymes of phenylpropanoid-derived metabolites. These structure–function studies collectively provide a comprehensive view of an important aspect of phenylpropanoid metabolism. More specifically, these atomic resolution insights into the architecture and mechanistic underpinnings of phenylpropanoid metabolizing enzymes contribute to our understanding of the emergence and on-going evolution of specialized phenylpropanoid products, and underscore the molecular basis of metabolic biodiversity at the chemical level. Finally, the detailed knowledge of the structure, function and evolution of these enzymes of specialized metabolism provide a set of experimental templates for the enzyme and metabolic engineering of production platforms for diverse novel compounds with desirable dietary and medicinal properties.[本文引用: 1]
DOI:10.1016/j.ijbiomac.2019.06.069URLPMID:31199975 [本文引用: 2]
Alternaria brown spot (ABS) is a disease caused by the necrotrophic fungus Alternaria alternata, which induces necrotic lesions on fruits and young leaves due to the production of the host-specific ACT toxin by the fungus. To better understand the citrus-A. alternata interaction and to identify putative resistance proteins, as well as the receptor of the ACT toxin, citrus plants susceptible ('Minneola' mandarin) and resistant ('Clemenules' tangor) to A. alternata, infected or not (control) with the pathogen were analyzed by proteomics. Protein changes were observed between citrus genotypes after infection, and 150 candidate proteins were obtained. A general scheme of the metabolic processes involved in susceptible and resistant citrus-A. alternata interactions was designed. Susceptible plants presented a high level of proteins involved in stress response at the final stages of the infection, whereas resistant plants presented high level of ROS proteins, metabolic proteins, and proteins involved in the immune system process. Proteins like ferredoxin and cyclophilin are specific to the susceptible variety and may be good candidates as fungal effector-interacting proteins. This is the first citrus-A. alternata proteomics analysis, which has allowed a better understanding of the molecular bases of the citrus response to ABS disease.
DOI:10.1093/jxb/eru203URL [本文引用: 1]
Alternaria alternata (tobacco pathotype) is a necrotrophic fungus causing severe losses in Nicotiana species by infection of mature leaves. Similar to what has been observed in cultivated tobacco, N. tabacum, young leaves of wild tobacco, N. attenuata, were more resistant to A. alternata than mature leaves, and this was correlated with stronger blue fluorescence induced after infection. However, the nature of the fluorescence-emitting compound, its role in defence, and its regulation were not clear. Silencing feruloyl-CoA 6'-hydroxylase 1 (F6'H1), the gene encoding the key enzyme for scopoletin biosynthesis, by virus-induced gene silencing (VIGS) revealed that the blue fluorescence was mainly emitted by scopoletin and its beta-glycoside form, scopolin. Further analysis showed that scopoletin exhibited strong antifungal activity against A. alternata in vitro and in vivo. Importantly, jasmonic acid (JA) levels were highly elicited in young leaves but much less in mature leaves after infection; and fungus-elicited scopoletin was absent in JA-deficient plants, but was largely restored with methyl jasmonate treatments. Consistent with this, plants strongly impaired in JA biosynthesis and perception were highly susceptible to A. alternata in the same way scopoletin/scopolin-depleted VIGS F6'H1 plants. Furthermore, silencing MYC2, a master regulator of most JA responses, reduced A. alternata-induced NaF6'H1 transcripts and scopoletin. Thus, it is concluded that JA signalling is activated in N. attenuata leaves after infection, which subsequently regulates scopoletin biosynthesis for the defence against A. alternata partly through MYC2, and higher levels of scopoletin accumulated in young leaves account for their strong resistance.
DOI:10.1007/10_2014_295URLPMID:25583224 [本文引用: 1]
Terpenoids (isoprenoids) represent the largest and most diverse class of chemicals among the myriad compounds produced by plants. Plants employ terpenoid metabolites for a variety of basic functions in growth and development but use the majority of terpenoids for more specialized chemical interactions and protection in the abiotic and biotic environment. Traditionally, plant-based terpenoids have been used by humans in the food, pharmaceutical, and chemical industries, and more recently have been exploited in the development of biofuel products. Genomic resources and emerging tools in synthetic biology facilitate the metabolic engineering of high-value terpenoid products in plants and microbes. Moreover, the ecological importance of terpenoids has gained increased attention to develop strategies for sustainable pest control and abiotic stress protection. Together, these efforts require a continuous growth in knowledge of the complex metabolic and molecular regulatory networks in terpenoid biosynthesis. This chapter gives an overview and highlights recent advances in our understanding of the organization, regulation, and diversification of core and specialized terpenoid metabolic pathways, and addresses the most important functions of volatile and nonvolatile terpenoid specialized metabolites in plants.
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/pce.13602URLPMID:31222757 [本文引用: 1]
Plants emit a large variety of volatile organic compounds during infection by pathogenic microbes, including terpenes, aromatics, nitrogen-containing compounds, and fatty acid derivatives, as well as the volatile plant hormones, methyl jasmonate, and methyl salicylate. Given the general antimicrobial activity of plant volatiles and the timing of emission following infection, these compounds have often been assumed to function in defence against pathogens without much solid evidence. In this review, we critically evaluate current knowledge on the toxicity of volatiles to fungi, bacteria, and viruses and their role in plant resistance as well as how they act to induce systemic resistance in uninfected parts of the plant and in neighbouring plants. We also discuss how microbes can detoxify plant volatiles and exploit them as nutrients, attractants for insect vectors, and inducers of volatile emissions, which stimulate immune responses that make plants more susceptible to infection. Although much more is known about plant volatile-herbivore interactions, knowledge of volatile-microbe interactions is growing and it may eventually be possible to harness plant volatiles to reduce disease in agriculture and forestry. Future research in this field can be facilitated by making use of the analytical and molecular tools generated by the prolific research on plant-herbivore interactions.
[本文引用: 1]
DOI:10.1016/j.tplants.2011.10.001URL [本文引用: 1]
Reactive oxygen species (ROS) are highly reactive molecules able to damage cellular components but they also act as cell signalling elements. ROS are produced by many different enzymatic systems. Plant NADPH oxidases, also known as respiratory burst oxidase homologues (RBOHs), are the most thoroughly studied enzymatic ROS-generating systems and our understanding of their involvement in various plant processes has increased considerably in recent years. In this review we discuss their roles as ROS producers during cell growth, plant development and plant response to abiotic environmental constraints and biotic interactions, both pathogenic and symbiotic. This broad range of functions suggests that RBOHs may serve as important molecular 'hubs' during ROS-mediated signalling in plants.
DOI:10.1111/j.1399-3054.2009.01326.xURLPMID:20002601 [本文引用: 1]
Production of reactive oxygen species (ROS) is a hallmark of successful recognition of infection and activation of plant defenses. ROS play multifaceted signaling functions mediating the establishment of multiple responses and can act as local toxins. Controversy surrounds the origin of these ROS. Several enzymatic mechanisms, among them a plasma membrane NADPH oxidase and cell wall peroxidases, can be responsible for the ROS detected in the apoplast. However, high levels of ROS from metabolic origins and/or from downregulation of ROS-scavenging systems can also accumulate in different compartments of the plant cell. This compartmentalization could contribute to the specific functions attributed to ROS. Additionally, ROS interact with other signals and phytohormones, which could explain the variety of different scenarios where ROS signaling plays an important part. Interestingly, pathogens have developed ways to alter ROS accumulation or signaling to modify plant defenses. Although ROS have been mainly associated with pathogen attack, ROS are also detected in other biotic interactions including beneficial symbiotic interactions with bacteria or mycorrhiza, suggesting that ROS production is a common feature of different biotic interactions. Here, we present a comprehensive review describing the newer views in ROS signaling and function during biotic stress.
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}