 ,, 朱雅楠, 李秋方, 苏松坤,, 聂红毅,福建农林大学动物科学学院(蜂学学院),福州 350002
,, 朱雅楠, 李秋方, 苏松坤,, 聂红毅,福建农林大学动物科学学院(蜂学学院),福州 350002Transcriptomic Analysis of Genes Related to Nursing Behavior in the Brains of Apis mellifera ligustica
GAO Yan,, ZHU YaNan, LI QiuFang, SU SongKun,, NIE HongYi,College of Animal Sciences (College of Bee Science), Fujian Agriculture and Forestry University, Fuzhou 350002通讯作者:
责任编辑: 岳梅
收稿日期:2020-01-17接受日期:2020-03-10网络出版日期:2020-10-01
| 基金资助: |
Received:2020-01-17Accepted:2020-03-10Online:2020-10-01
作者简介 About authors
高艳,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (982KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
高艳, 朱雅楠, 李秋方, 苏松坤, 聂红毅. 转录组学分析意大利蜜蜂脑部哺育行为相关基因[J]. 中国农业科学, 2020, 53(19): 4092-4102 doi:10.3864/j.issn.0578-1752.2020.19.021
GAO Yan, ZHU YaNan, LI QiuFang, SU SongKun, NIE HongYi.
0 引言
【研究意义】蜜蜂脑是调节其生物学各项行为的中心器官,脑部的神经系统能够调控蜜蜂表现出的各种行为响应,其中包括从哺育行为到采集行为的生命过渡过程,如分泌蜂王浆哺育幼虫、采集、识别颜色和嗅觉线索、舞蹈交流、群体防御等[1,2,3]。工蜂的哺育行为在蜜蜂幼虫生长发育、维护蜂群稳定等方面发挥重要作用,同时工蜂的哺育行为也受到幼虫信息素的调节[4]。蜂王浆的分泌由哺育蜂哺育行为所介导,这一哺育行为已被人工用于生产蜂王浆供人类食用。一般认为,蜂王浆由适龄工蜂的咽下腺、上颚腺共同分泌,同时哺育行为介导的蜂王浆分泌过程离不开脑部复杂的网络机制调控[5,6]。因此,研究哺育蜂脑部调控哺育行为相关的分子网络机制可为深入解析意大利蜜蜂(Apis mellifera ligustica)蜂王浆分泌的分子机理提供新的思路和线索。【前人研究进展】随着日龄的发育,工蜂从巢内工作向巢外采集活动转变,包括环境调节的行为变化[7,8,9],这一过程涉及大脑的结构、基因表达和蛋白质合成的变化[10];同样也涉及到脑中激素和神经化学物质的生理过程以及成千上万基因表达的变化[11],例如,其在功能和形态方面的转变会受保幼激素的控制[12]。此外,脑部中枢神经系统通过调控咽下腺和上颚腺相关基因的表达,促进这些腺体的发育并且增强它们分泌蜂王浆的功能[13]。研究表明,哺育蜂的大脑对蜂王浆分泌产生了一种独特的神经肽,哺育蜂分泌蜂王浆能力的增强与调节行为过程中高度增强的神经肽有关,并通过调节水稳态、对幼虫信息素的识别、采集能力和收集花粉来增加蜂群群体的营养供应进而提高蜂王浆的分泌,从而在提高蜂王浆产量方面发挥了作用[13]。哺育蜂脑中高量表达的基因可能参与轴突形成和细胞黏附,这些基因可能涉及到哺育蜂在转向采集活动之前的脑结构变化[7]。哺育蜂脑部磷脂酰肌醇信号转导和花生四烯酸代谢水平的提高有助于增强嗅觉对幼虫信息素刺激的反应[14]。【本研究切入点】关于脑部调控蜂王浆分泌机制的研究主要集中于不同日龄工蜂脑部磷酸化蛋白质组学和膜蛋白质组学方面,以及不同日龄普通意大利蜜蜂与高产王浆意大利蜜蜂之间的脑部蛋白质组学[2,6,10,13]。此外,借助微阵列分析技术展开不同日龄工蜂脑部基因表达变化也有相关的报道[15]。目前,尚未有排除日龄因素影响而全面开展的脑部调控蜂王浆分泌网络机制相关的研究。【拟解决的关键问题】为了排除日龄因素的干扰,人工组建蜂群,收集3日龄工蜂、10日龄哺育蜂、10日龄采集蜂、21日龄哺育蜂、21日龄采集蜂,利用RNA-seq技术全面筛选哺育蜂脑部哺育行为相关的关键差异表达基因(differentially expressed gene,DEG),为深入研究哺育蜂脑部调控蜂王浆分泌的分子机制提供参考。1 材料与方法
1.1 供试蜂种
供试蜂群为‘蜂强1号’意大利蜜蜂蜂种,样品于2017年10月至2018年8月采自福建农林大学动物科学学院(蜂学学院)教学蜂场。1.2 主要仪器及试剂
体视显微镜1台、冷光源仪器1台、DEPC水(上海生工生物工程股份有限公司)、SYBR? Premix Ex TaqTM II(Tli RNaseH Plus)(TaKaRa公司)、75%酒精、干冰、碎冰。1.3 标记蜜蜂及收集样品
试验所用蜂群为正常健康的5群强群,每群蜂中至少含有2—3张即将出房的封盖子脾。在组建新的蜂群之前,去除封盖子脾上的蜜蜂,放于恒温恒湿培养箱(34.5℃,相对湿度60%)中,每隔24 h用不同颜色的记号笔在刚出房的蜜蜂胸部或腹部做好标记,连续标记5 d,每天标记刚出房工蜂数目约为2 000— 3 000头。标记完后投入由一只蜂王、一张蜜粉脾、一张幼虫脾组建的人工蜂群。标记的工蜂发育到第3天时,直接收取工蜂作为3日龄工蜂;第10、21天时,收集头部伸到有幼虫巢房且持续时间超过10 s的工蜂作为哺育蜂;第10、21天时,在巢门口收集后足花粉筐中载有花粉的外勤蜂,将其作为采集蜂。按照这个要求,收集3日龄工蜂(3 d)、10日龄哺育蜂(10 dN)、10日龄采集蜂(10 dF)、21日龄哺育蜂(21 dN)、21日龄采集蜂(21 dF)。
1.4 样品测序
参考赵元洪等[16]解剖蜜蜂大脑的方法,解剖5组样品(3 d、10 dN、10 dF、21 dN、21 dF)的脑部,每组解剖10—15只,每组均设置3个生物学重复。3 d的3个生物学重复分别为3 d_1、3 d_2、3 d_3;10 dN的3个生物学重复分别为10 dN_1、10 dN_2、10 dN_3;10 dF的3个生物学重复分别为10 dF_1、10 dF_2、10 dF_3;21 dN的3个生物学重复分别为21 dN_1、21 dN_2、21 dN_3;21 dF的3个生物学重复分别为21 dF_1、21 dF_2、21 dF_3。委托北京诺禾致源生物有限公司开展总RNA质量控制、cDNA文库构建和Illumina测序。1.5 哺育蜂脑部哺育行为相关DEG的筛选及表达水平分析
根据FPKM(Fragments Per Kilobase of exon model per Million mapped reads)值法计算每个基因在5组样本中的表达量。利用DESeq2软件分析差异表达基因,将校正后的P-adjust<0.05作为筛选DEG的标准[17],其中log2 fold change (FC)>0和log2 FC<0分别作为筛选上调和下调DEG的标准。筛选与哺育行为密切相关的DEG的标准如下:DEG在10 dN脑中的表达量显著高于3 d和21 dF;DEG在10 dN脑中的表达量显著高于10 dF;DEG在21 dN脑中的表达量显著高于21 dF;DEG在10 dN与21 dN脑中的表达量无显著差异。将10 dN与3 d之间产生的DEG记为10 dN vs 3 d组,10 dN与21 dF之间产生的DEG记为10 dN vs 21 dF组,10 dN与10 dF之间产生的DEG记为10 dN vs 10 dF组,21 dN与21 dF之间产生的DEG记为21 dN vs 21dF组,10 dN与21 dN产生的DEG为10 dN vs 21 dN组。
通过韦恩图分析,10 dN vs 3 d与10 dN vs 21 dF、10 dN vs 10 dF、21 dN vs 21 dF做交集会产生共有的DEG,这一共有DEG包括调控哺育行为及日龄发育等相关,最后为减少日龄发育对筛选的影响,从共有DEG中去除10 dN vs 21 dN与其交集产生的DEG,最后得到的即为严格筛选条件下与哺育行为密切相关的DEG。
通过clusterProfiler R软件包和基因本体(GO)将关键DEG映射到GO数据库中。通过BLAST软件将筛选得到的DEG与KEGG数据库比对。利用诺禾Novogene售后工具平台(https://magic.novogene.com/ public/customer/main#/tool_rna/add_tool)对DEG进行KEGG pathway富集分析。
1.6 DEG的实时荧光定量PCR(qPCR)验证
在DEG中随机选取4个基因。利用Primer Premier 6设计特异性引物(表1),其中以Actin(NM_001185146.1)作为内参基因,委托上海生工生物工程有限公司进行引物合成。反应体系:SYBR Premix Ex Taq Ⅱ(TaKaRa公司,日本)5 μL,引物(2 μmol·L-1)2 μL,模板(500 ng·μL-1)2 μL,ddH2O补充至10 μL。PCR程序分为3步:第一步为95℃预变性30 s;第二步为95℃变性5 s,60℃退火30 s,共 40个循环;最后一步为熔解曲线分析:65℃开始,每5 s上升0.5℃,直至上升到95℃。整个反应程序在荧光定量PCR仪(Bio-Rad公司,美国)上进行,按照说明书进行操作。以2-ΔΔCt法[18]计算哺育蜂脑部DEG的相对表达量。Table 1
表1
表1qPCR引物信息
Table 1
| 基因 Gene | 登录号 Accession number | 引物序列 Primer sequence (5′-3′) | 产物大小 Amplicon length (bp) |
|---|---|---|---|
| LOC409709 | 409709 | F: TTGGTCAGTTGGATGGGTAGA | 236 |
| R: CCGACGGTGTAAGAAAGGC | |||
| LOC551813 | 551813 | F: CAAAATTCAACAGCTCCTGC | 210 |
| R: TCATCGCTACCGAAATCATAA | |||
| LOC409708 | 409708 | F: TTGTTGATCGCAATCCTGTC | 480 |
| R: CGTCGCATCGTCATCGTAA | |||
| LOC551165 | 551165 | F:TGCCTGTTGTATTCTCGTAT | 304 |
| R:TCTGACCTTGCCCTCCTC | |||
| Actin | NM_001185146.1 | F: CCTAGCACCATCCACCATGAA | 87 |
| R: GAAGCAAGAATTGACCCACCAA |
新窗口打开|下载CSV
2 结果
2.1 不同发育时期工蜂脑部测序文库的测序数据质控与评估
15个工蜂脑样品经建库和测序,有效读段数介于56 348 822—82 964 416,样本的单一匹配率均在72%以上,Q30值也都在88%以上,说明RNA-Seq数据质量较好,测序数据可靠性高(表2)。Table 2
表2
表2RNA-seq数据总览
Table 2
| 样品 Sample | 原始读数 Number of raw reads | 有效读数 Number of clean reads | 有效匹配数(匹配率) Clean map (Map rate) | 单一匹配数(匹配率)Unique map (Map rate) | 多匹配数(匹配率)Multiple map (Map rate) | 99.9%的碱基正确率99.9% base accuracy (%) |
|---|---|---|---|---|---|---|
| 3 d_1 | 67515798 | 66708824 | 56766708 (85.10%) | 55951632 (83.87%) | 815076 (1.22%) | 88.25 |
| 3 d_2 | 83920302 | 81832028 | 70899666 (86.64%) | 69821133 (85.32%) | 1078533 (1.32%) | 90.59 |
| 3 d_3 | 81546254 | 80442702 | 69502321 (86.40%) | 68471536 (85.12%) | 1030785 (1.28%) | 89.66 |
| 10 dN_1 | 57288572 | 56348822 | 49321933 (87.53%) | 48015717 (85.21%) | 1306216 (2.32%) | 90.51 |
| 10 dN_2 | 83787622 | 82964416 | 63408429 (76.43%) | 60124557 (72.47%) | 3283872 (3.96%) | 90.66 |
| 10 dN_3 | 72837334 | 71663646 | 56922446 (79.43%) | 56028890 (78.18%) | 893556 (1.25%) | 91.00 |
| 10 dF_1 | 67610626 | 66671128 | 55057317 (82.58%) | 54208585 (81.31%) | 848732 (1.27%) | 90.37 |
| 10 dF_2 | 81258562 | 80246018 | 63725456 (79.41%) | 62620480 (78.04%) | 1104976 (1.38%) | 90.06 |
| 10 dF_3 | 66021926 | 65365544 | 52522944 (80.35%) | 51744723 (79.16%) | 778221 (1.19%) | 91.00 |
| 21 dN_1 | 71882296 | 71020506 | 56181047 (79.11%) | 54846870 (77.23%) | 8223024 (13.18%) | 92.19 |
| 21 dN_2 | 72018192 | 70803290 | 60295752 (85.16%) | 58523770 (82.66%) | 11236986 (15.51%) | 92.33 |
| 21 dN_3 | 72458810 | 71460376 | 59684702 (83.52%) | 57902404 (81.03%) | 8742852 (13.63%) | 92.50 |
| 21 dF_1 | 72168956 | 70890254 | 59385891 (83.77%) | 58485427 (82.50%) | 900464 (1.27%) | 91.94 |
| 21 dF_2 | 69418794 | 68335020 | 55559809 (81.31%) | 54623413 (79.93%) | 936396 (1.37%) | 88.98 |
| 21 dF_3 | 81670548 | 80692702 | 67432186 (83.57%) | 66368993 (82.25%) | 1063193 (1.32%) | 91.86 |
新窗口打开|下载CSV
2.2 哺育蜂哺育行为相关DEG的筛选及趋势分析
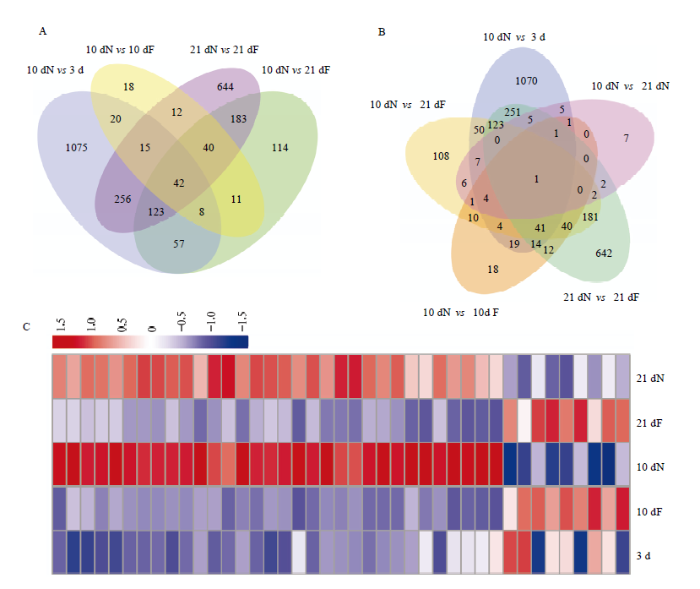
韦恩图分析发现,10 dN vs 3 d有1 596个DEG;10 dN vs 21 dF有578个DEG;10 dN vs 10 dF有166个DEG;21 dN vs 21 dF有1 315个DEG,将这4组进行交集分析得到42个DEG(图1-A),这42个DEG包括调控哺育行为及日龄发育等相关。为减少日龄对筛选与哺育行为相关DEG的干扰,去除10 dN vs 21 dN与42个DEG交集产生的1个DEG,最后得到41个DEG(图1-B)。图1
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1哺育蜂脑部哺育行为相关DEG表达趋势分析
A、B:筛选哺育蜂脑部哺育行为相关DEG韦恩图 Venn diagram of selected-DEGs related to nursing behavior of nurses;C:41个DEG表达量聚类热
Fig. 1Analysis on the expression trend of DEGs related to nurses’ nursing behavior
41个DEG的表达量热图聚类分析发现,这些DEG主要在21 dN和10 dN组表达量较高(图1-C);其中,在哺育蜂阶段均上调表达的DEG有32个,均下调表达的有6个,剩余的3个DEG除在10 dN vs 3 d中呈上调表达外,其余都是在哺育蜂阶段呈下调表达(表3)。
Table 3
表3
表3脑部41个DEG基于日龄依赖性在5组样本中的显著性趋势
Table 3
| 基因数目 Gene number | 10 dN vs 3 d显著性比较Significant comparison of 10 dN vs 3 d | 10 dN vs 21 dF显著性比较Significant comparison of 10 dN vs 21 dF | 10 dN vs 10 dF显著性比较Significant comparison of 10 dN vs 10 dF | 21 dN vs 21 dF显著性比较Significant comparison of 21 dN vs 21 dF | 10 dN vs 21 dN显著性比较 Significant comparison of 10 dN vs 21 dN |
|---|---|---|---|---|---|
| 32 | ↑ | ↑ | ↑ | ↑ | ns |
| 6 | ↓ | ↓ | ↓ | ↓ | ns |
| 3 | ↑ | ↓ | ↓ | ↓ | ns |
新窗口打开|下载CSV
32个在哺育蜂阶段均上调表达的DEG为脑中与哺育行为密切相关的DEG。将32个DEG在10 dN vs 10 dF和21 dN vs 21 dF中按照log2 FC值从高到低的顺序排列,其中差异倍数最大的为编码王浆主蛋白1(MRJP1)的基因LOC551813(log2 FC (10 dN vs 10 dF)=9.44,log2 FC (21 dN vs 21 dF)=5.71),该基因在哺育蜂阶段高量表达(10 dN、21 dN脑中FPKM值分别为1 564.78和510.71),在3日龄工蜂和采集蜂阶段呈低量表达(3 d、10 dF和21 dF脑中FPKM值分别为0.40、2.27和9.64);其次为LOC725215(编码毒酸磷酸酶Acph-1样)、LOC411186(编码突触小泡糖蛋白2C)、LOC410149(编码羧肽酶Q样)、LOC409709(编码葡萄糖基神经酰胺酶4),其中LOC409709在哺育蜂阶段呈高量表达(10 dN和21 dN脑中FPKM值分别为75.03、50.90),在3日龄工蜂和采集蜂阶段呈低量表达(3 d、10 dF和21 dF脑中FPKM值分别为2.83、7.81和8.63);还在上调DEG中发现编码气味结合蛋白(odorant binding protein,OBP)的3个基因Obp17、Obp21、Obp14,以及编码细胞色素P450家族之一的基因CYP6AQ1(表4)。
Table 4
表4
表4脑部前11个上调DEG在5组样本中的表达信息
Table 4
| 基因登录号 Gene_ID | 基因 Gene | 基因描述 Gene description | 平均FPKM值 Average FPKM value | log2 FC (10 dN vs 10 dF) | log2 FC (21 dN vs 21 dF) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 3 d | 10 dN | 10 dF | 21 dN | 21 dF | |||||
| 551813 | LOC551813 | 王浆主蛋白1 Major royal jelly protein 1 | 0.40 | 1564.78 | 2.27 | 510.71 | 9.64 | 9.44 | 5.71 |
| 725215 | LOC725215 | 毒酸磷酸酶Acph-1样 Venom acid phosphatase Acph-1-like | 0.31 | 29.97 | 0.66 | 23.80 | 0.73 | 5.52 | 5.01 |
| 411186 | LOC411186 | 突触小泡糖蛋白2C Synaptic vesicle glycoprotein 2C | 0.22 | 12.81 | 0.47 | 11.96 | 0.60 | 4.78 | 4.29 |
| 410149 | LOC410149 | 羧肽酶Q样 Carboxypeptidase Q-like | 1.59 | 31.09 | 1.82 | 26.29 | 2.81 | 4.10 | 3.21 |
| 409709 | LOC409709 | 假定的葡萄糖基神经酰胺酶4 Putative glucosylceramidase 4 | 2.83 | 75.03 | 7.81 | 50.90 | 8.63 | 3.27 | 2.54 |
| 552478 | Obp17 | 气味结合蛋白17 Odorant binding protein 17 | 1.77 | 6.43 | 0.73 | 4.05 | 0.80 | 3.14 | 2.33 |
| 551935 | Obp21 | 气味结合蛋白21 Odorant binding protein 21 | 4.50 | 34.70 | 4.43 | 22.20 | 7.71 | 2.97 | 1.51 |
| 677673 | Obp14 | 气味结合蛋白14 Odorant binding protein 14 | 1.00 | 7.70 | 1.01 | 3.32 | 0.69 | 2.92 | 2.24 |
| 409708 | LOC409708 | 葡糖基神经酰胺酶样 Glucosylceramidase-like | 13.84 | 132.92 | 22.42 | 113.84 | 27.93 | 2.57 | 2.01 |
| 726798 | LOC726798 | Sn1特异性二酰基甘油脂肪酶β Sn1-specific diacylglycerol lipase beta | 0.53 | 2.67 | 0.49 | 3.46 | 0.85 | 2.45 | 2.01 |
| 408383 | CYP6AQ1 | 细胞色素P450 6AQ1 Cytochrome P450 6AQ1 | 0.49 | 1.97 | 0.42 | 1.41 | 0.39 | 2.22 | 1.84 |
新窗口打开|下载CSV
2.3 哺育蜂哺育行为相关DEG的调控网络分析
GO富集分析(表5)发现32个上调DEG主要参与氧化还原酶活性(4个DEG)、气味结合(3个DEG)、跨膜运输(2个DEG),GO富集中未发现有显著性富集的基因。Table 5
表5
表5上调DEG富集的GO条目
Table 5
| GO条目 GO term | 基因 Gene |
|---|---|
| 氧化还原酶活性 Oxidoreductase activity | CYP6AQ1/LOC726418/ LOC408734/LOC413590 |
| 气味结合 Odorant binding | Obp21/Obp14/Obp17 |
| 跨膜运输 Transmembrane transport | LOC411186/LOC725462 |
| 蛋白质水解 Proteolysis | LOC725215/LOC725154 |
| 翻译 Translation | LOC550715 |
| 脂质代谢 Lipid metabolism | LOC726798 |
| DNA复制 DNA replication | LOC725238 |
| 细胞器组分Organelle part | LOC408444 |
新窗口打开|下载CSV
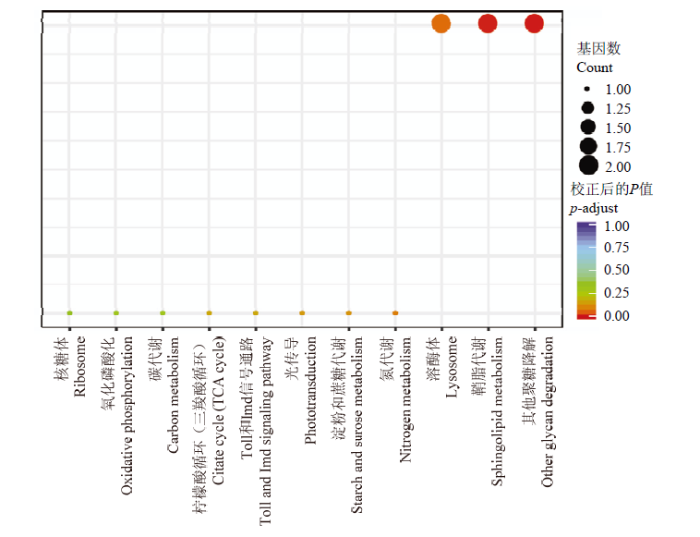
通过KEGG富集分析,发现上调表达的DEG(图2)主要参与蛋白质代谢(核糖体)、能量代谢(氧化磷酸化、碳代谢、三羧酸循环、淀粉和蔗糖代谢、氮代谢、其他聚糖降解通路)、信号转导(Toll和Imd信号通路、光传导、鞘脂代谢)、消化作用(溶酶体),其中显著性富集在鞘脂代谢和其他聚糖降解通路上。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2哺育蜂哺育行为相关DEG的KEGG功能富集分析
Fig. 2KEGG functional enrichment analysis of DEGs related to nurses’ nursing behavior
2.4 转录组中DEG的qPCR验证
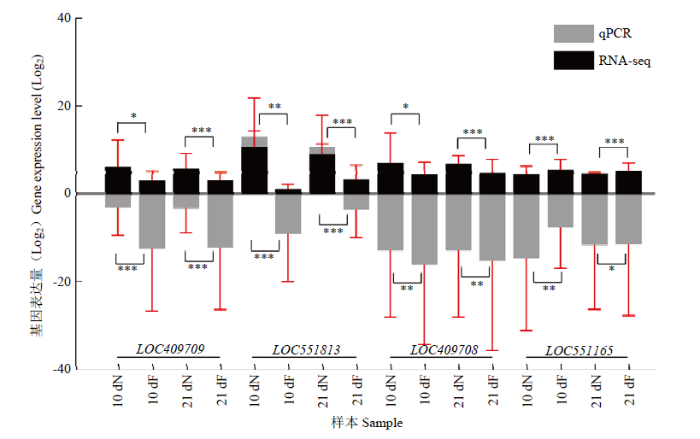
为验证测序数据的准确性,从41个DEG中选取3个上调基因(LOC409709、LOC551813和LOC409708)和1个下调基因(LOC551165),结果显示这些基因表达水平的趋势变化与RNA-seq数据中的变化趋势基本一致(图3),证实了测序结果的可信性。图3
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3转录组数据的qPCR验证
黑色柱状图表示RNA-seq数据中基因的FPKM值,灰色表示qPCR数据中基因的相对表达量,纵坐标均为相应取过以2为底的对数值The black bar graph represents the FPKM value of the gene in the RNA-seq data, and the gray represents the relative expression of genes in the qPCR data; the ordinates are the corresponding logarithmic values with a base of 2。图中数据为平均值±标准误,采用单因素方差分析 Data in the figure are the mean±SE, One-way ANOVA. *P<0.05; **P<0.01; ***P<0.001
Fig. 3qPCR validation of transcriptome data
3 讨论
蜜蜂脑部的研究大多基于脑细胞的化学功能、结构、内分泌活动以及脑部基因和蛋白质表达的时态变化,如保幼激素、生物胺、多巴胺、5-羟色胺和章鱼胺在脑中对蜜蜂行为发育的调节起着关键作用[19]。此外,神经分子(例如神经肽)被称为中枢神经系统回路中的神经递质和神经调节剂[20],对蜜蜂脑中神经功能的肽能调节产生重大影响[21],也有研究表明脑部神经肽组的重构用以适应蜂王浆高产的需要[14]。哺育蜂对幼虫的哺育行为是建立在其大脑对幼虫信息素信号的接收和反馈基础上的[4,14]。哺育蜂哺育行为介导的蜂王浆分泌机制的研究大多集中在咽下腺和上颚腺[6,22-23],有关脑部调控哺育行为介导蜂王浆分泌的分子机制方面的研究相对较少。本研究人工组建蜂群收集3日龄工蜂,10日龄哺育蜂、采集蜂,21日龄哺育蜂、采集蜂,利用RNA-seq技术,在减小日龄的干扰下严格筛选出41个DEG,其中32个与哺育蜂哺育行为密切相关。WHITFIELD等通过基因芯片技术检测到哺育蜂和采集蜂脑之间约有1 800个DEG [7],而本研究只筛选出41个DEG,主要有以下原因:首先本研究的试验样本来自人工构建相同日龄的哺育蜂和采集蜂;构建两个时间点(10日龄和21日龄)的同日龄哺育蜂和采集蜂,然后取共有的DEG。这两个严格条件可以排除日龄的影响,导致筛选的DEG数目较少,进一步保证结果更加可靠。GO富集分析显示上调DEG主要参与氧化还原酶活性、气味结合、跨膜运输;KEGG富集通路显示这些上调DEG主要参与蛋白质代谢、能量代谢、信号转导等(图2),其中显著性富集在鞘脂代谢和其他聚糖降解通路上。
3.1 哺育蜂脑中信号转导相关的上调DEG
突触囊泡糖蛋白2(SV2)具有许多功能,包括囊泡运输、稳定神经递质囊泡负载、锚定囊泡蛋白、协助囊泡运输、调节钙敏感性以及与细胞外基质相互作用;在脊椎动物中,SV2家族由3个副产物组成,包括SV2A、SV2B和SV2C,其中SV2C的表达是最受限制的,仅局限于进化古老的大脑区域,在纹状体、中脑和腹侧苍白球均有较强的表达,而在新皮层的表达很少[24]。SV2C参与中枢系统中多巴胺的释放,其缺失可以引起小鼠Mures运动障碍、增加α-突触核蛋白单体的聚集和降低纹状体多巴胺的释放[25]。本研究在上调DEG中发现编码SV2C的基因LOC411186,该基因在3日龄工蜂和采集蜂阶段基本不表达(3 d、10 dF和21 dF中该基因的FPKM值分别为0.22、0.47和0.60),而在哺育蜂阶段的表达量相对较高(10 dN和21 dN脑中FPKM值分别为12.81和11.96)。化学神经传递对神经元之间的交流至关重要,在这个交流过程中,Ca2+涌入突触前末端,触发神经递质释放进入突触,作用于突触后受体[24]。此外,GO富集结果也显示LOC411186参与跨膜运输功能,推测工蜂脑部的LOC411186在调节Ca2+敏感性,促进神经递质作用于突触后受体,从而引发哺育行为方面发挥重要作用。葡萄糖神经酰胺是300多种结构不同的鞘糖脂(包括神经节苷脂和硫苷脂)的骨架,对于哺乳动物的发育至关重要。葡萄糖神经酰胺是一种膜鞘磷脂,是许多糖脂的前体[26]。葡糖苷神经酰胺合成酶催化神经酰胺产生葡萄糖神经酰胺,它是神经节苷脂类的前体物质[27]。在上调DEG中发现编码葡萄糖基神经酰胺酶的基因LOC409709和LOC409708,KEGG富集结果显示这两个基因显著性富集在鞘脂代谢路径上。鞘脂及其代谢产物不仅是构成细胞膜的重要结构分子,而且参与调节细胞的生长、分化、衰老和细胞程序性死亡等许多重要的信号转导过程,使细胞产生各种不同的生物学功能[28]。
药理学研究表明,在发育的大脑中,sn-1特异性二酰基甘油脂肪酶活性是轴突生长和引导所必需的[29,30]。在哺育蜂阶段富集的光传导途径可能表明该通路在哺育蜂从事巢外工作接受光信号中发挥重要作用[31]。本研究在上调DEG中发现编码sn-1特异性二酰基甘油脂肪酶β的基因LOC726798富集在光传导途径中。
在昆虫中,气味结合蛋白(OBP)和化学感受蛋白(chemosensory protein,CSP)在运载疏水性气味分子和信息素穿过淋巴液的信号转导中发挥重要作用[32,33]。OBP14在蜂群的上颚腺中含量丰富,与单萜类结构有较好的亲和力;OBP21在老蜂体内含量丰富,并与法尼素结合,而法尼素是一种吸引蜂群的信息素[34]。OBP和CSP可以与多种配体结合,但是与蜜蜂的幼虫信息素有更高的结合力[33,35]。本研究在上调DEG中发现了Obp17、Obp21、Obp14;NIE等在意蜂的触角转录组中发现Obp17、Obp21、Obp14,其中Obp17、Obp21在触角中的表达量从刚出房工蜂到哺育蜂阶段呈增加趋势,而在哺育蜂阶段到采集蜂阶段呈减小趋势[36],这与本研究在上调DEG中发现的这两个基因的表达趋势基本一致,推测这两个基因可能在脑部化学信息的传递和调控方面发挥重要作用。
哺育蜂通过调节对幼虫信息素的识别和采集蜂的采集能力等来增加蜂群群体的营养供应进而提高蜂王浆的分泌。细胞的一切行为都需要信号分子和受体结合,通过信号转导实现。这些与信号转导相关的基因(LOC409709、LOC409708和LOC726798)在哺育蜂脑中呈上调表达,暗示哺育蜂可能通过这些基因增强信号转导,进而在感知挥发物信息素后启动高效蛋白合成的机制。
3.2 哺育蜂脑中与能量代谢相关的上调DEG
氧化磷酸化是真核生物体内有机物包括糖、脂、氨基酸等在分解过程中发生氧化并驱动ATP合成的过程,生物体内95%的ATP来自这种方式[37]。脑部为满足其较高的代谢速率需要消耗很多的ATP,因此有大量的线粒体位于大脑细胞中。除氧化磷酸化外,三羧酸循环(TCA循环)和碳代谢也是生物体能量代谢产生ATP的重要途径。在细胞和器官中,三羧酸循环(TCA循环)是产生能量的关键过程,本研究在上调DEG中发现编码琥珀酸脱氢酶[泛醌]黄素蛋白亚基的基因LOC408734,表明哺育蜂脑部可能通过有氧氧化(TCA循环)葡萄糖产生大量ATP。此外,该基因还显示富集在氧化磷酸化、碳代谢、三羧酸循环通路,这些结果表明哺育蜂脑部可能通过多种途径为哺育行为提供充足的能量。昆虫细胞色素P450酶参与多种代谢活动,包括外源性物质降解、保幼激素和蜕皮类固醇生物合成、信息素代谢[9]。在相同日龄哺育蜂和采集蜂脑中,发现CYP6AQ1在10日龄哺育蜂和21日龄哺育蜂脑中的表达量均显著高于相同日龄采集蜂中表达量;GO富集分析表明该基因参与氧化还原酶活性。唐晓伟根据细胞色素P450与血红素结合区域设计的简并引物克隆出一段蜜蜂细胞色素P450基因片段,进而克隆得到该基因全长,并由细胞色素P450命名委员会命名为CYP6AQ1[38]。该基因属于CYP6家族成员,但是关于该基因的功能尚未有深入研究。在果蝇中,CYP6家族的成员CYP6A8羟化了月桂酸的ω-1位置,这表明CYP6家族的成员参与了昆虫的脂肪酸羟化反应[23,39]。BOUTIN等通过在清理行为和非清理行为工蜂脑部的差异表达基因中发现在清理行为工蜂脑部高表达的细胞色素P450基因,推测可能是清理行为工蜂对外源性物质(如幼虫发出的信息素等气味)的高敏感性引发细胞色素P450基因的高量表达[40]。在哺育蜂脑中发现CYP6AQ1上调表达,推测该基因可能在哺育蜂脑部调控脂肪酸羟基化或降解幼虫释放的请求饲喂的信息素过程中发挥作用。哺育蜂能够感知幼虫信息素并通过外围化学感受器官传输信号到大脑;通过高级中枢神经器官-大脑调控上颚腺和咽下腺合成蛋白质和脂肪酸等重要大分子物质,引发脑部产生哺育行为,进而促进咽下腺和上颚腺等腺体产生蜂王浆分泌的行为。上述富集的通路说明大脑在这一时期的基础能量供应和蛋白、脂肪酸合成都得到加强,这样更有利于大脑进行信息处理和行为调控,从而促进哺育蜂的哺育行为。
值得注意的是,在上调DEG中发现编码王浆主蛋白1(MRJP1)的LOC551813,目前该基因尚未有相关功能的报道。该基因在NCBI意大利蜜蜂基因组新版本Amel_HAv3.1中尚未注释,但以前版本Amel_4.5中有注释。针对该基因特异序列设计引物,通过荧光定量检测该基因在脑中表达,且在哺育蜂脑中显著高量表达(图3),该基因有可能是蜂王浆主蛋白家族的新成员,后续可以通过克隆基因全长进行验证。前人在蜜蜂脑中也检测到蜂王浆主蛋白家族基因的表达,但在脑中发挥的作用尚不明确。RNA-seq测序将具有卫生行为和不具有卫生行为工蜂脑进行比较[40],也发现LOC551813在这两种行为工蜂脑部高量表达。蛋白质组学发现MRJP1、MRJP2和MRJP7蛋白在哺育蜂脑中的含量显著高于采集蜂脑中含量,推测这些基因可能为脑中蛋白质的合成过程储存氨基酸[41]。在本研究构建的相同日龄哺育蜂和采集蜂脑的转录组数据中,检测到MRJP1(LOC551813)在哺育蜂脑中显著高量表达,暗示该基因可能在哺育行为调控蜂王浆合成和分泌过程中发挥重要作用。
4 结论
在mRNA组学水平对3日龄工蜂、10日龄哺育蜂、10日龄采集蜂、21日龄哺育蜂、21日龄采集蜂5组样本脑组织进行全面表征分析,获得了相同日龄哺育蜂和采集蜂脑部基因的转录组图谱,分析得到了哺育行为密切相关的32个上调差异表达基因。哺育蜂脑部的差异表达基因主要通过信号转导和能量代谢等途径调控哺育蜂的哺育行为,研究结果可为阐明哺育蜂哺育行为的分子机制提供理论参考。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1038/s41598-017-02192-3URLPMID:28512345 [本文引用: 1]

The honeybee brain is a central organ in regulating wide ranges of honeybee biology, including life transition from nurse to forager bees. Knowledge is still lacking on how protein phosphorylation governs the neural activity to drive the age-specific labor division. The cerebral phosphoproteome of nurse and forager honeybees was characterized using Ti(4+)-IMAC phosphopeptide enrichment mass-spectrometry-based proteomics and protein kinases (PKs) were predicted. There were 3,077 phosphosites residing on 3,234 phosphopeptides from 1004 phosphoproteins in the nurse bees. For foragers the numbers were 3,056, 3,110, and 958, respectively. Notably, among the total 231 PKs in honeybee proteome, 179 novel PKs were predicted in the honeybee brain, of which 88 were experimentally identified. Proteins involved in wide scenarios of pathways were phosphorylated depending on age: glycolysis/gluconeogenesis, AGE/RAGE and phosphorylation in nurse bees and metal ion transport, ATP metabolic process and phototransduction in forager bees. These observations suggest that phosphorylation is vital to the tuning of protein activity to regulate cerebral function according to the biological duties as nursing and foraging bees. The data provides valuable information on phosphorylation signaling in the honeybee brain and potentially useful resource to understand the signaling mechanism in honeybee neurobiology and in other social insects as well.
DOI:10.1021/acs.jproteome.7b00371URLPMID:28879772 [本文引用: 2]
The brain is a vital organ in regulating complex social behaviors of honeybees including learning and memory. Knowledge of how brain membrane proteins and their phosphorylation underlie the age-related behavioral polyethism is still lacking. A hitherto age-resolved brain membrane proteome and phosphoproteome were reported in adult worker bees from two strains of honeybee (Apis mellifera ligustica): Italian bee (ITB) and Royal Jelly bee (RJB), a line selected from ITB for increased RJ outputs over four decades. There were 1079 membrane protein groups identified, and 417 unique phosphosites were located in 179 membrane protein groups mainly phosphorylated by kinase families of MAPKs, CDKs, and CK2. Age-resolved dynamics of brain membrane proteome and phosphoproteome are indicative of their correlation with the neurobiological requirements during the adult life of honeybee workers. To stimulate immature brain cell development in newly emerged bees (NEBs), the enriched functional classes associated with metabolism of carbohydrates, nucleosides, and lipids by the up-regulated proteins suggest their enhanced role in driving cell maturity of the brain. In nurse bees (NBs) and forager bees (FBs), a higher number of membrane proteins and phosphoproteins were expressed as compared with in the young stages, and the enriched signal-transduction-related pathways by the up-regulated proteins suggest their significances in sustaining the intensive information processing during nursing and foraging activities. Notably, RJB has shaped unique membrane proteome and phosphoproteome settings to consolidate nursing and foraging behaviors in response to decades of selection underpinning the elevated RJ yields. In RJB NBs, the enriched pathways of phosphatidylinositol signaling and arachidonic acid metabolism indicate a stronger olfaction sensation in response to larval pheromone stimulation. In RJB FBs, the enriched pathways related to signal processing such as SNARE interactions in vesicular transport, wnt signaling, TGF-beta signaling, and taurine and hypotaurine metabolism suggest an enhanced nerve sensitivity to prime the stronger tendency to pollen collection. Our data gain a novel insight into membrane proteome and phosphoproteome-driven cerebral regulation of honeybee behaviors, which is potentially useful for further neurobiological investigation in both honeybees and other social insects.
DOI:10.1002/pmic.201900094URLPMID:31115157 [本文引用: 1]
The olfactory conditioning of the bee proboscis extension reflex (PER) is extensively used as a paradigm in associative learning of invertebrates but with limited molecular investigations. To investigate which protein changes are linked to olfactory conditioning, a non-sophisticated conditioning model is applied using the PER in the honeybee (Apis mellifera). Foraging honeybees are assigned into three groups based on the reflex behavior and training: conditioned using 2-octanone (PER-conditioned), and sucrose and water controls. Thereafter, the brain synaptosomal proteins are isolated and analyzed by quantitative proteomics using stable isotope labeling (TMT). Additionally, the complex proteome dataset of the bee brain is generated with a total number of 5411 protein groups, including key players in neurotransmitter signaling. The most significant categories affected during olfactory conditioning are associated with
DOI:10.1098/rspb.2000.1345URLPMID:11209886 [本文引用: 2]
Primer pheromones are thought to act in a variety of vertebrates and invertebrates but only a few have been chemically identified. We report that a blend of ten fatty-acid esters found on the cuticles of honeybee larvae, already known as a kairomone, releaser pheromone and primer pheromone, also act as a primer pheromone in the regulation of division of labour among adult workers. Bees in colonies receiving brood pheromone initiated foraging at significantly older ages than did bees in control colonies in five out of five trials. Laboratory and additional field tests also showed that exposure to brood pheromone significantly depressed blood titres of juvenile hormone. Brood pheromone exerted more consistent effects on age at first foraging than on juvenile hormone, suggesting that the primer effects of this pheromone may occur via other, unknown, mechanisms besides juvenile hormone. These results bring the number of social factors known to influence honeybee division of labour to three: worker-worker interactions, queen mandibular pheromone and brood pheromone.
DOI:10.1021/pr300700eURLPMID:23157659 [本文引用: 1]
To identify candidate royal jelly (RJ) proteins that might affect the physiologic status of honeybee colony members, we used shotgun proteomics to comprehensively identify the RJ proteome as well as proteomes of the hypopharyngeal gland (HpG), postcerebral gland (PcG), and thoracic gland (TG), from which RJ proteins are assumed to be derived. We identified a total of 38 nonredundant RJ proteins, including 22 putative secretory proteins and Insulin-like growth factor-binding protein complex acid labile subunit. Among them, 9 proteins were newly identified from RJ. Comparison of the RJ proteome with the HpG, PcG, and TG proteomes revealed that 17 of the 22 putative secretory RJ proteins were derived from some of these glands, suggesting that the RJ proteome is a cocktail of proteins from these three glands. Furthermore, pathway analysis suggested that the HpG proteome represents the molecular basis of the extremely high protein-synthesizing ability, whereas the PcG proteome suggests that the PcG functions as a reservoir for the volatile compounds and a primer pheromone. Finally, to further characterize the possible total RJ proteome, we identified putative secretory proteins in the proteomes of these three glands. This will be useful for predicting novel RJ protein components in future studies.
DOI:10.1007/s13592-019-00656-1URL [本文引用: 3]
DOI:10.1126/science.1086807URLPMID:14551438 [本文引用: 3]
We show that the age-related transition by adult honey bees from hive work to foraging is associated with changes in messenger RNA abundance in the brain for 39% of approximately 5500 genes tested. This result, discovered using a highly replicated experimental design involving 72 microarrays, demonstrates more extensive genomic plasticity in the adult brain than has yet been shown. Experimental manipulations that uncouple behavior and age revealed that messenger RNA changes were primarily associated with behavior. Individual brain messenger RNA profiles correctly predicted the behavior of 57 out of 60 bees, indicating a robust association between brain gene expression in the individual and naturally occurring behavioral plasticity.
DOI:10.1186/gb-2002-3-2-research0007URLPMID:11864369 [本文引用: 1]
BACKGROUND: The honeybee (Apis mellifera) has been used with great success in a variety of behavioral studies. The lack of genomic tools in this species has, however, hampered efforts to provide genome-based explanations for behavioral data. We have combined the power of DNA arrays and the availability of distinct behavioral stages in honeybees to explore the dynamics of gene expression during adult development in this insect. In addition, we used caffeine treatment, a procedure that accelerates learning abilities in honeybees, to examine changes in gene expression underlying drug-induced behavioral modifications. RESULTS: Spotted microarrays containing several thousand cDNAs were interrogated with RNAs extracted from newly emerged worker bees, experienced foragers and caffeine-treated bees. Thirty-six differentially expressed cDNAs were verified by northern blot hybridization and characterized in silico by sequencing and database searches. Experienced foragers overexpressed royal jelly proteins, a putative imaginal disc growth factor, a transcriptional regulator (Stck) and several enzymes, including alpha-glucosidases, aminopeptidases and glucose dehydrogenase. Naive workers showed increased expression of members of the SPARC and lectin families, heat-shock cognate proteins and several proteins related to RNA translation and mitochondrial function. A number of novel genes overexpressed in both naive and experienced bees, and genes induced by caffeine, have also been identified. CONCLUSIONS: We have shown the usefulness of this transcriptome-based approach for gene discovery, in particular in the context of the efficacy of drug treatment, in a model organism in which routine genetic techniques cannot be applied easily.
DOI:10.1177/0748730411431404URLPMID:22306970 [本文引用: 2]
Honey bee workers care for (
DOI:10.1021/pr2007818URLPMID:22181811 [本文引用: 2]
A large-scale mapping of the worker honeybee brain proteome was achieved by MudPIT. We identified 2742 proteins from forager and nurse honeybee brain samples; 17% of the total proteins were found to be differentially expressed by spectral count sampling statistics and a G-test. Sequences were compared with the EuKaryotic Orthologous Groups (KOG) catalog set using BLASTX and then categorized into the major KOG categories of most similar sequences. According to this categorization, nurse brain showed increased expression of proteins implicated in translation, ribosomal structure, and biogenesis (14.5%) compared with forager (1.8%). Experienced foragers overexpressed proteins involved in energy production and conversion, showing an extensive difference in this set of proteins (17%) in relation to the nurse subcaste (0.6%). Examples of proteins selectively expressed in each subcaste were analyzed. A comparison between these MudPIT experiments and previous 2-DE experiments revealed nine coincident proteins differentially expressed in both methodologies.
DOI:10.1371/journal.pone.0142917URLPMID:26588091 [本文引用: 1]
We investigated the association between the expression of a gene encoding gustatory receptor (G10) and division of labor in the honey bee, Apis mellifera. Among 10 GR genes encoding proteins 15% ~ 99% amino acid identity in the honey bee, we found that AmGR10 with 99% identity is involved in nursing or brood care. Expression of AmGR10 was restricted to organs of the hypopharyngeal gland, brain, and ovary in the nurse bee phase. Members of an extended nursing caste under natural conditions continued to express this gene. RNAi knockdown of AmGR10 accelerated the transition to foraging. Our findings demonstrate that this one gene has profound effects on the division of labor associated with the development and physiology of honeybee society.
DOI:10.1051/apido:19900507URL [本文引用: 1]
URLPMID:26310634 [本文引用: 3]
[D].
[本文引用: 3]
[D].
[本文引用: 3]
URLPMID:14745100 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1186/gb-2010-11-10-r106URLPMID:20979621 [本文引用: 1]
High-throughput sequencing assays such as RNA-Seq, ChIP-Seq or barcode counting provide quantitative readouts in the form of count data. To infer differential signal in such data correctly and with good statistical power, estimation of data variability throughout the dynamic range and a suitable error model are required. We propose a method based on the negative binomial distribution, with variance and mean linked by local regression and present an implementation, DESeq, as an R/Bioconductor package.
DOI:10.1006/meth.2001.1262URLPMID:11846609 [本文引用: 1]
The two most commonly used methods to analyze data from real-time, quantitative PCR experiments are absolute quantification and relative quantification. Absolute quantification determines the input copy number, usually by relating the PCR signal to a standard curve. Relative quantification relates the PCR signal of the target transcript in a treatment group to that of another sample such as an untreated control. The 2(-Delta Delta C(T)) method is a convenient way to analyze the relative changes in gene expression from real-time quantitative PCR experiments. The purpose of this report is to present the derivation, assumptions, and applications of the 2(-Delta Delta C(T)) method. In addition, we present the derivation and applications of two variations of the 2(-Delta Delta C(T)) method that may be useful in the analysis of real-time, quantitative PCR data.
DOI:10.1159/000067788URLPMID:12563167 [本文引用: 1]
Efficient division of labor is one of the main reasons for the success of the social insects. In honey bees the division of labor is principally achieved by workers changing tasks as they age. Typically, young adult bees perform a series of tasks within the colony before ultimately making the transition to foraging outside the hive for resources. This lifelong behavioral development is a well-characterized example of naturally occurring behavioral plasticity, but its neural bases are not well understood. Two techniques were used to assess the role of biogenic amines in the transition from in-hive work to foraging, which is the most dramatic and obvious transition in honey bee behavioral development. First, associations between amines and tasks were determined by measuring the levels of amines in dissected regions of individual bee brains using HPLC analysis. Second, colonies were orally treated with biogenic amines and effects on the onset of foraging were observed. Octopamine concentration in the antennal lobes of the bee brain was most reliably associated with task: high in foragers and low in nurses regardless of age. In contrast, octopamine in the mushroom bodies, a neighboring neuropil, was associated with age and not behavior, indicating independent modulation of octopamine in these two brain regions. Treating colonies with octopamine resulted in an earlier onset of foraging in young bees. In addition, octopamine levels were not elevated by non-foraging flight, but were already high on return from the first successful foraging trip and subsequently remained high, showing no further change with foraging experience. This observation suggests that octopamine becomes elevated in the antennal lobes in anticipation of foraging and is involved in the release and maintenance of the foraging state. Foraging itself, however, does not modulate octopamine levels. Behaviorally related changes in octopamine are modulated by juvenile hormone, which has also been implicated in the control of honey bee division of labor. Treatment with the juvenile hormone analog methoprene elevated octopamine and octopamine treatment 'rescued' the delay in behavioral development caused by experimentally depleting juvenile hormone in bees. Although the pathways linking juvenile hormone and octopamine are presently unknown, it is clear that octopamine acts 'downstream' of juvenile hormone to influence behavior and that juvenile hormone modulates brain octopamine levels. A working hypothesis is that octopamine acts as an activator of foraging by modulating responsiveness to foraging-related stimuli. This is supported by the finding that octopamine treatment increased the response of bees to brood pheromone, a stimulator of foraging activity. Establishing a role for octopamine in honey bee behavioral development is a first step in understanding the neural bases of this example of naturally occurring, socially mediated, behavioral plasticity. The next level of analysis will be to determine precisely where and how octopamine acts in the nervous system to coordinate this complex social behavior.
URLPMID:31565815 [本文引用: 1]
DOI:10.1021/acs.jproteome.8b00098URLPMID:29775065 [本文引用: 1]
Aggressiveness in honeybees seems to be regulated by multiple genes, under the influence of different factors, such as polyethism of workers, environmental factors, and response to alarm pheromones, creating a series of behavioral responses. It is suspected that neuropeptides seem to be involved with the regulation of the aggressive behavior. The role of allatostatin and tachykinin-related neuropeptides in honeybee brain during the aggressive behavior is unknown, and thus worker honeybees were stimulated to attack and to sting leather targets hung in front of the colonies. The aggressive individuals were collected and immediately frozen in liquid nitrogen; the heads were removed and sliced at sagittal plan. The brain slices were submitted to MALDI spectral imaging analysis, and the results of the present study reported the processing of the precursors proteins into mature forms of the neuropeptides AmAST A (59-76) (AYTYVSEYKRLPVYNFGL-NH2), AmAST A (69-76) (LPVYNFGL-NH2), AmTRP (88-96) (APMGFQGMR-NH2), and AmTRP (254-262) (ARMGFHGMR-NH2), which apparently acted in different neuropils of the honeybee brain during the aggressive behavior, possibly taking part in the neuromodulation of different aspects of this complex behavior. These results were biologically validated by performing aggressiveness-related behavioral assays using young honeybee workers that received 1 ng of AmAST A (69-76) or AmTRP (88-96) via hemocele. The young workers that were not expected to be aggressive individuals presented a complete series of aggressive behaviors in the presence of the neuropeptides, corroborating the hypothesis that correlates the presence of mature AmASTs A and AmTRPs in the honeybee brain with the aggressiveness of this insect.
DOI:10.1371/journal.pone.0081001URLPMID:24339892 [本文引用: 1]
Transcriptome sequencing has become the main methodology for analyzing the relationship between genes and characteristics of interests, particularly those associated with diseases and economic traits. Because of its role of functional food for humans, commercial royal jelly (RJ) and its production are major research focuses in the field of apiculture. Multiple lines of evidence have demonstrated that many factors affect RJ output by activating or inhibiting various target genes and signaling pathways. Available coding sequences from the Honey Bee Genome Sequencing Consortium have permitted a pathway-based approach for investigating the development of the hypopharyngeal glands (HGs). In the present study, 3573941, 3562730, 3551541, 3524453, and 3615558 clean reads were obtained from the HGs of five full-sister honey bee samples using Solexa RNA sequencing technology. These reads were then assembled into 18378, 17785, 17065, 17105, and 17995 unigenes, respectively, and aligned to the DFCI Honey Bee Gene Index database. The differentially expressed genes (DEGs) data were also correlated with detailed morphological data for HGs acini.
DOI:10.1038/s41598-017-04879-zURLPMID:28674395 [本文引用: 2]
Secretions from mandibular glands (MGs) have important caste-specific functions that are associated with the social evolution of honey bees. To gain insights into the molecular architecture underlying these caste differences, we compared the gene expression patterns of MGs from queens, queenright workers (WQRs) and queenless workers (WQLs) using high-throughput RNA-sequencing technology. In total, we identified 46 candidate genes associated with caste-specific biosynthesis of fatty acid pheromones in the MG, including members of cytochrome P450 (CYP450) family and genes involved in fatty acid beta-oxidation and omega-oxidation. For further identification of the CYP450s genes involved in the biosynthesis of MG secretions, we analyzed by means of qPCR, the expression levels of six of the CYP450 genes most abundantly expressed in the transcriptome analysis across different castes, ages, tasks and tissues. Our analysis revealed that CYP6AS8 and CYP6AS11, the most abundantly expressed CYP450 genes in worker and queen MGs, respectively, are selectively expressed in the MGs of workers and queens compared to other tissues. These results suggest that these genes might be responsible for the critical bifurcated hydroxylation process in the biosynthesis pathway. Our study contributes to the description of the molecular basis for the biosynthesis of fatty acid-derived pheromones in the MGs.
URLPMID:31394034 [本文引用: 2]
DOI:10.1007/PL00000722URLPMID:11130460 [本文引用: 1]
Ras-related guanosine triphosphatases (GTPases) couple receptor activity to a number of intracellular signalling events culminating in the control of cell morphology and gene transcription. In culture cells, the best-understood Ras-dependent signalling pathway involves the mitogen-activated protein kinase/extracellular-regulated kinase (MAPK/ERK) cascade. A growing body of evidence has recently been accumulating to suggest a crucial role of Ras and MAPK signalling in neuronal functions connected to synaptic plasticity. In the present review article we discuss the experimental basis supporting the notion that the Ras/MAPK pathway interacts with other synaptic mechanisms to regulate invertebrate and vertebrate behavioural responses such as those implicated in learning and memory processes.
DOI:10.1016/j.biochi.2015.11.009URLPMID:26582417 [本文引用: 1]
Sphingolipids represent a major class of lipids that are essential constituents of eukaryotic cells. They are predominantly located in plasma membrane microdomains, and play an important structural role in regulating membrane fluidity. They are also bioactive effectors involved in diverse key cellular functions such as apoptosis and proliferation. The implication of some sphingolipids in cancer is well established whereas that of some others is still a matter of intense investigation. Glucosylceramide is the backbone of more than 300 structurally different glycosphingolipids including gangliosides and sulfatides, and is essential for mammalian development. Therefore, glucosylceramidases (also named GBA1, GBA2 and GBA3 beta-glucosidases), the enzymes that hydrolyse beta-glucosylceramide, play important functions. GBA1 is a lysosomal hydrolase whose deficiency causes Gaucher disease, the most prevalent inherited lysosomal storage disorder. GBA2 is a ubiquitous non-lysosomal glucosylceramidase whose mutations have been associated with some forms of hereditary spastic paraplegia. GBA3 is a cytosolic beta-glucosidase, mostly present in the kidney, liver, spleen, intestine and lymphocytes of mammals, the function of which is still unclear. Whereas glucosylceramide synthase is implicated in multidrug resistance, the role of glucosylceramide breakdown in cancer is not yet fully appreciated. Defective GBA1 enzyme activity in humans, i.e., Gaucher disease, is associated with an increased risk of multiple myeloma and other malignancies. Putative molecular links between Gaucher disease and cancer, which might implicate the malignant cell and/or its microenvironment, are reviewed. The functions of GBA2 and GBA3 in cancer progression are also discussed.
[本文引用: 1]
[本文引用: 1]
DOI:10.1042/BST0391341URLPMID:21936812 [本文引用: 1]
LEV (levetiracetam), an antiepileptic drug which possesses a unique profile in animal models of seizure and epilepsy, has as its unique binding site in brain, SV2A (synaptic vesicle protein 2A). Previous studies have used a chimaeric and site-specific mutagenesis approach to identify three residues in the putative tenth transmembrane helix of SV2A that, when mutated, alter binding of LEV and related racetam derivatives to SV2A. In the present paper, we report a combined modelling and mutagenesis study that successfully identifies another 11 residues in SV2A that appear to be involved in ligand binding. Sequence analysis and modelling of SV2A suggested residues equivalent to critical functional residues of other MFS (major facilitator superfamily) transporters. Alanine scanning of these and other SV2A residues resulted in the identification of residues affecting racetam binding, including Ile273 which differentiated between racetam analogues, when mutated to alanine. Integrating mutagenesis results with docking analysis led to the construction of a mutant in which six SV2A residues were replaced with corresponding SV2B residues. This mutant showed racetam ligand-binding affinity intermediate to the affinities observed for SV2A and SV2B.
URLPMID:14610053 [本文引用: 1]
DOI:10.1006/mcne.1996.0051URLPMID:8954627 [本文引用: 1]
During the early stages of development various cell adhesion molecules (CAMs) and fibroblast growth factor receptors (FGFR) are expressed throughout the retinal neuroepithelium. The ability of retinal ganglion cells to project their axons to the optic fissure depends, in part, on cell-cell interactions mediated by cell adhesion molecules. In the present study we show that the ability of the firstborn rat retinal ganglion cells to extend axons in vitro can be stimulated by NCAM and L1, but not N-cadherin. Both CAM responses can be fully inhibited by antibodies that block neuronal fibroblast growth factor receptor function and by agents that block defined steps in the FGFR signal transduction cascade. When added to living E13.5 rat retinal whole-mount preparations the same agents induced errors in the orderly establishment of young axon patterns in the retinal periphery and caused axons in the retinal center to defasciculate. These results suggest that the activation of the fibroblast growth factor receptor signal cascade not only promotes survival and proliferation of various cell types but can also mediate intraretinal axon guidance.
DOI:10.1016/S0968-0004(03)00172-5URLPMID:13678959 [本文引用: 1]
Vertebrate visual phototransduction represents one of the best-characterized G-protein-coupled receptor-mediated signaling pathways. Structural analyses of rhodopsin, G protein, arrestin and several other phototransduction components have revealed common folds and motifs that are important for function. Static and dynamic information has been acquired through the application of X-ray diffraction, solution and solid-state nuclear magnetic resonance spectroscopy's, electron and atomic force microscopy's, and a host of indirect structural methods. A comprehensive understanding of phototransduction requires further structural work on individual components and their relevant complexes in solution and the native disk membrane. Given the accelerated pace of structure determination, it is anticipated that this will be the first G-protein-coupled pathway for which a complete molecular description is ultimately available.
DOI:10.1111/nyas.1998.855.issue-1URL [本文引用: 1]
[本文引用: 2]
DOI:10.1021/pr2000754URLPMID:21707107 [本文引用: 1]
Odorant-binding proteins (OBPs) and chemosensory proteins (CSPs) mediate both perception and release of chemical stimuli in insects. The genome of the honey bee contains 21 genes encoding OBPs and 6 encoding CSPs. Using a proteomic approach, we have investigated the expression of OBPs and CSPs in the mandibular glands of adult honey bees in relation to caste and age. OBP13 is mostly expressed in young individuals and in virgin queens, while OBP21 is abundant in older bees and is prevalent in mated queens. OBP14, which had been found in larvae, is produced in hive workers' glands. Quite unexpectedly, the mandibular glands of drones also contain OBPs, mainly OBP18 and OBP21. We have expressed three of the most represented OBPs and studied their binding properties. OBP13 binds with good specificity oleic acid and some structurally related compounds, OBP14 is better tuned to monoterpenoid structures, while OBP21 binds the main components of queen mandibular pheromone as well as farnesol, a compound used as a trail pheromone in the honey bee and other hymenopterans. The high expression of different OBPs in the mandibular glands suggests that such proteins could be involved in solubilization and release of semiochemicals.
DOI:10.1016/j.ijbiomac.2019.09.041URLPMID:31499112 [本文引用: 1]
Honeybees communicate with members of their intra-species via pheromones. The volatile pheromones, beta-ocimene and allo-ocimene, are the primary signals of larvae to beg for the care from the nurses. Of the odorant binding proteins (OBPs)/chemosensory proteins (CSPs), CSP4 has the best affinity with beta-ocimene and allo-ocimene. To reveal the binding mechanism of CPS4 with them, fluorescent quenching, UV absorption spectra, circular dichroism (CD) spectra, isothermal titration calorimetry (ITC), molecular docking, molecular dynamic (MD) simulation, and site-directed mutagenesis were applied. The quenching constant Ksv decreased with temperature increase, and the interaction distance was 2.73nm and 2.43nm (<10nm), indicating that beta-ocimene and allo-ocimene could form stable complexes with CSP4. The observed big up tri, openH<0 and big up tri, openS>0 of thermodynamics suggest the main driving forces are electrostatic or hydrophobic force. All above thermodynamics findings are in line with the results of ITC experiments. Furthermore, molecular docking, MD simulation and site-directed mutagenesis indicate the binding cavities are located at cavity 1 in C-terminal of CSP4, where Tyr98 and Asp67 are vital amino acids in maintaining the stable form of protein and larval pheromones, and electrostatic energies are the main driving forces. Our findings gain novel insight into the binding mechanism of chemosensory protein with volatile larval pheromones and are important for understanding olfactory interaction of honeybees.
DOI:10.1007/s00438-017-1382-5URLPMID:29043489 [本文引用: 1]
Honey bee is a social insect. Its colony is mainly coordinated by the chemical signals such as pheromones produced by queen or brood. Correspondingly, the worker bee developed numerous complicated olfactory sensilla in antennae for detection of these colony chemical signals and nectar/pollen signals in foraging. With the normal development of new emerged workers, young adults (nurse bee) worked in colony at the first 2-3 weeks and then followed by the foraging activity outside of the hive, which give rise to great change of the surrounding chemical signals. However, the olfactory adaption mechanism of worker bee in these processes of behavioral development is still unclear. In this study, we conducted a comprehensive and quantitative analysis of gene expression in Apis mellifera antenna of newly emerged workers, nurses and foragers using transcriptome analysis. Meanwhile, we constructed experimental colonies to collect age-matched samples, which were used to determine whether task is the principal determinant of differential expression. RNA sequencing and quantitative real-time polymerase chain reaction revealed that 6 and 14 genes were closely associated with nurse and forager behaviors, respectively. Furthermore, a broad dynamic range of chemosensory gene families and candidate odorant degrading enzymes were analyzed at different behavior statuses. We firstly reported genes associated with nursing/foraging behavior from antennae and the variations of expression of genes belonging to various olfactory gene families at different development stages. These results not only could contribute to elucidating the relationship between olfactory and behavior-related changes, but also provide a new perspective into the molecular mechanism underlying honey bee division of labor.
DOI:10.1016/j.arr.2006.04.001URLPMID:16831573 [本文引用: 1]
This review addresses the data that support the presence and contribution of decreased mitochondrial oxidative phosphorylation during aging to impaired cellular metabolism. Aging impairs substrate oxidation, decreases cellular energy production and increases the production of reactive intermediates that are toxic to the cell. First, the basic principles of mitochondrial oxidative physiology are briefly reviewed. Second, the focus on the relationship of altered mitochondrial respiration to the increased production of reactive oxygen species that are employed by the
[D].
[本文引用: 1]
[D].
[本文引用: 1]
DOI:10.1016/j.bbrc.2004.10.194URLPMID:15555597 [本文引用: 1]
Only a handful of P450 genes have been functionally characterized from the approximately 90 recently identified in the genome of Drosophila melanogaster. Cyp6a8 encodes a 506-amino acid protein with 53.6% amino acid identity with CYP6A2. CYP6A2 has been shown to catalyze the metabolism of several insecticides including aldrin and heptachlor. CYP6A8 is expressed at many developmental stages as well as in adult life. CYP6A8 was produced in Saccharomyces cerevisiae and enzymatically characterized after catalytic activity was reconstituted with D. melanogaster P450 reductase and NADPH. Although several saturated or non-saturated fatty acids were not metabolized by CYP6A8, lauric acid (C12:0), a short-chain unsaturated fatty acid, was oxidized by CYP6A8 to produce 11-hydroxylauric acid with an apparent V(max) of 25 nmol/min/nmol P450. This is the first report showing that a member of the CYP6 family catalyzes the hydroxylation of lauric acid. Our data open new prospects for the CYP6 P450 enzymes, which could be involved in important physiological functions through fatty acid metabolism.
DOI:10.1186/s12864-015-1714-yURL [本文引用: 2]
URLPMID:19203288 [本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}