 ,1,2,3, 陈万权,1,2,4, 曹世勤4,5, 孙振宇4,5, 贾秋珍4,5, 高利2,4, 刘博2,4, 刘太国,2,3,4
,1,2,3, 陈万权,1,2,4, 曹世勤4,5, 孙振宇4,5, 贾秋珍4,5, 高利2,4, 刘博2,4, 刘太国,2,3,4Surveillance and Genetic Diversity Analysis of Puccinia striiformis f. sp. tritici in Gansu and Qinghai Provinces
HUANG MiaoMiao,1,2,3, CHEN WanQuan,1,2,4, CAO ShiQin4,5, SUN ZhenYu4,5, JIA QiuZhen4,5, GAO Li2,4, LIU Bo2,4, LIU TaiGuo,2,3,4通讯作者:
责任编辑: 岳梅
收稿日期:2020-01-20接受日期:2020-03-20网络出版日期:2020-09-16
| 基金资助: |
Received:2020-01-20Accepted:2020-03-20Online:2020-09-16
作者简介 About authors
黄苗苗,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (1435KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄苗苗, 陈万权, 曹世勤, 孙振宇, 贾秋珍, 高利, 刘博, 刘太国. 甘肃、青海地区小麦条锈菌监测及群体遗传多样性分析[J]. 中国农业科学, 2020, 53(18): 3693-3706 doi:10.3864/j.issn.0578-1752.2020.18.006
HUANG MiaoMiao, CHEN WanQuan, CAO ShiQin, SUN ZhenYu, JIA QiuZhen, GAO Li, LIU Bo, LIU TaiGuo.
0 引言
【研究意义】小麦条锈病是一种气传病害,是危害我国小麦生产的主要流行病害之一,在世界各主要麦区也时有发生,其病原菌为条形柄锈菌小麦专化型(Puccinia striiformis f. sp. tritici,Pst)。小麦条锈菌主要在纬度较高或高海拔地区越夏,具有喜凉怕热、远距离气流传播、分布范围广、流行频率高、暴发性强、流行速度快、危害程度重等特点[1]。该病害流行时,可造成10%—70%的产量损失,严重时可至绝收[2,3],长期威胁着我国小麦主产区,包括西北、西南、黄淮海和长江中下游地区。新中国成立以来,小麦条锈病在我国年均发生400万公顷左右,特别是1950、1964、1990、2002和2017年的5次大流行,小麦产量损失达1 380万吨[4]。我国小麦条锈病的病菌来源分为秋季菌源基地和春季菌源基地,秋季菌源基地为甘肃陇南陇东、川西北、青海东部等,向条锈病菌冬繁区秋苗提供菌源[5]。前人在甘肃小麦条锈菌流行关系研究中,对与青海菌源的关系少有涉及,没有将甘肃和青海的菌源作为研究主体,存在样本量较小、条锈菌的标样未采集于病害流行初期等缺陷。因此,明确甘肃、青海两省春季流行期间的菌源传播以及生殖方式对条锈菌大区流行预测及确定越夏初始菌源来源等具有重要意义。【前人研究进展】我国西北、华北、西南主要流行区的条锈菌群体遗传多样性和遗传结构已进行了广泛而深入的研究,确定了我国小麦条锈菌具有较高的遗传多样性水平,尤其是甘肃陇南地区,不同流行区群体间存在着一定的基因交流[6,7,8,9,10,11]。西北地区的甘肃和青海是我国小麦条锈菌最重要的越夏区,条锈菌新小种的首次发现几乎均在这一区域。该区域秋季条锈菌菌源可向东部广大冬麦区传播,对周边地区有重要影响[1]。关于青海、甘肃、新疆、西藏4省(自治区)的小麦条锈菌毒性及遗传多样性的研究表明,青海和甘肃的条锈菌群体相似性较高,存在一定的菌源交换关系[12]。青海与甘肃条锈菌群体的菌源交流主要存在于陇南地区,基因流较强;与临夏群体的菌源交流则受到一定程度的限制,基因流相对较弱[10]。因此,****们多认为青海春季、夏季菌源全部由甘肃陇南传播到青海[13]。【本研究切入点】虽然前人对甘肃、青海地区小麦条锈病做了相关研究,但关于小麦条锈菌在甘肃、青海地区周年循环的研究,包括越夏、越冬以及春夏季菌源传播,特别是春季流行期间甘肃、青海菌源传播以及两地可能存在的生殖模式等问题,尚缺乏一些直接的证据。【拟解决的关键问题】以采自自然发病条件下甘肃、青海地区小麦变异观察圃材料上的小麦条锈菌群体为对象,利用微卫星(simple sequence repeat,SSR)分子标记的方法研究甘肃和青海两省6个市(县)小麦条锈菌春季流行传播路线及群体遗传多样性和群体的生殖模式,阐明甘肃和青海两省小麦条锈菌的群体遗传关系,明确两省小麦条锈菌之间的传播流行关系及菌源交换规律,进而为两省小麦条锈病的预测预报、确定越夏初始菌源来源和有效治理提供理论依据。1 材料与方法
1.1 试验点及标样采集
选择历年种植小麦、条锈病常发生的地区作为调查和研究区域。甘肃省共4个试验点:陇南市文县、陇东平凉市崆峒区、中部麦区定西市临洮县、临夏州临夏县;青海省2个试验点:西宁市城北区、海东市互助县。供试6套82份小麦品种组成的抗病性变异观察圃材料由中国农业科学院植物保护研究所麦类真菌病害研究组提供,根据各地小麦播种适期,按照编号顺序依次种植在试验田中。每个小麦材料种植1行,行长2 m,行距0.33 m,每隔一段距离(一般18行)设置当地抗、感病品种各1行作为对照。在试验区周围播种2—3行铭贤169(高度感染条锈病品种)作为诱发品种。种植管理按照当地大田常规栽培管理方法进行,同时注意以下几点:(1)保持水肥适中,防止土壤干旱和植株倒伏;(2)小麦生育期内禁用任何杀菌剂;(3)不可进行人工接种,并尽可能远离接种试验地;(4)观察记载自然发病结果(始发期、盛发期)及各品种的小麦生长状况。
标样采集:从6个试验点82份变异观察圃上共收集到551份始发期小麦条锈病标样(表1)。每个试验点调查病害的普遍率、严重度和反应型[14]并采集条形柄锈菌夏孢子堆之间分界明显,无其他病原菌污染的叶片作为发病标样,用单张吸水纸包裹叶片,并单个放入干燥的信封中,对每个信封进行编号,并标明地名(市、县、乡镇)、小麦品种及采集时间,带回实验室后4℃干燥器中保存备用。
Table 1
表1
表1甘肃、青海两省6个观察圃内小麦条锈病自然发病调查结果及采集的551份标样信息
Table 1
| 试验点 Test site | 文县 Wenxian | 平凉(崆峒区) Pingliang (Kongtong) | 临夏 Linxia | 临洮 Lintao | 互助 Huzhu | 西宁(城北区) Xining (Chengbei) | |
|---|---|---|---|---|---|---|---|
| 播种时间(年-月-日) Day of seeding time (Year-month-day) | 2017-10-08 | 2017-09-25 | 2017-09-28 | 2017-10-08 | 2018-04-04 | 2018-04-25 | |
| 采集时间(年-月-日) Day of collection (Year-month-day) | 2018-04-14 | 2018-05-24 | 2018-06-16 | 2018-06-18 | 2018-08-08 | 2018-08-09 | |
| 经度 Longitude | 104°53′17″ | 106°40′11″ | 103°11′13″ | 103°31′18″ | 101°53′24″ | 101°44′56″ | |
| 纬度 Latitude | 32°50′43″ | 35°33′06″ | 35°36′49″ | 35°13′24″ | 36°42′41″ | 36°43′15″ | |
| 海拔 Elevation (m) | 786 | 1190 | 2010 | 1893 | 2360 | 2350 | |
| 样本数量 Number of samples | 27 | 132 | 156 | 178 | 47 | 11 | |
| 编号 Code | 品种名称 Cultivar | IT/S/I (N) | |||||
| 1 | 水源11 Suwon 11 | 3/5/T (1) | 3/5/T (7) | 4/60/80 (5) | 4/10/10 (3) | 0 | 0 |
| 2 | 洛夫林10 Lovrin 10 | 3/5/T (1) | 3/5/10 (8) | 4/40/60 (6) | 4/10/T (4) | 0 | 0 |
| 3 | 洛夫林13 Lovrin 13 | 0 | 3/20/40 (4) | 4/20/60 (6) | 4/20/T (4) | 0 | 0 |
| 4 | 抗引655 Kangyin 655 | 0 | 3/10/20 (5) | 4/20/40 (5) | 0 | 0 | 0 |
| 5 | 中四(无芒)Zhong 4 (un-awn) | 0 | 0 | 4/40/60 (5) | 0 | 0 | 0 |
| 6 | 贵农22 Guinong 22 | 0 | 4/10/10 (2) | 0 | 4/T/T (2) | 0 | 0 |
| 7 | Triticum spelta album | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | Hybrid 46 | 0 | 0 | 0 | 4/10/10 (3) | 0 | 0 |
| 9 | Reichersberg 42 | 0 | 0 | 4/10/10 (3) | 4/10/T (3) | 0 | 0 |
| 10 | Heines Peko | 0 | 3/5/10 (6) | 4/60/80 (5) | 4/10/20 (4) | 0 | 0 |
| 11 | Nord Desprez | 0 | 0 | 4/80/80 (6) | 4/10/T (3) | 4/10/T (2) | 0 |
| 12 | Compair | 0 | 3/5/10 (2) | 4/40/40 (5) | 4/10/10 (3) | 2/T/T (1) | 3/T/T (1) |
| 13 | Carsten V | 0 | 3/5/T (3) | 4/20/20 (6) | 0 | 3/T/T (2) | 0 |
| 14 | Spaldings Prolific | 0 | 3/10/T (4) | 4/10/20 (4) | 4/10/T (2) | 0 | 0 |
| 15 | Heines Ⅶ | 0 | 0 | 4/10/20 (4) | 0 | 0 | 0 |
| 16 | Joss Cambier | 0 | 0 | 0 | 0 | 0 | 0 |
| 17 | Mega | 0 | 0 | 0 | 3/5/T (2) | 0 | 0 |
| 18 | Hobbit | 0 | 3/T/T (4) | 4/20/40 (5) | 3/10/T (4) | 3/40/20 (4) | 0 |
| 19 | 铭贤169 Mingxian 169 | 4/60/40 (1) | 4/40/20 (7) | 4/60/60 (4) | 4/20/10 (3) | 4/40/20 (2) | 4/40/40 (3) |
| 20 | 铭贤169*6/Yr5 Mingxian 169*6/Yr5 | 0 | 3/10/T (2) | 4/60/60 (6) | 0 | 0 | 0 |
| 21 | 铭贤169*6/Yr10 Mingxian 169*6/Yr10 | 0 | 4/10/10 (5) | 4/80/80 (4) | 4/10/10 (6) | 4/60/60 (7) | 0 |
| 22 | 周麦22 Zhoumai 22 | 0 | 0 | 0 | 0 | 4/10/10 (4) | 0 |
| 23 | 烟农15 Yannong 15 | 0 | 3/5/T (5) | 4/80/80 (2) | 4/20/20 (4) | 0 | 0 |
| 24 | 鲁麦23 Lumai 23 | 3/10/10 (4) | 4/20/10 (2) | 4/80/80 (4) | 4/60/40 (6) | 0 | 0 |
| 25 | 冀麦38 Jimai 38 | 0 | 4/10/20 (6) | 4/20/80 (6) | 4/20/10 (6) | 0 | 0 |
| 26 | 石4185 Shi 4185 | 0 | 3/10/10 (3) | 4/20/20 (5) | 4/60/30 (3) | 4/T/T (3) | 0 |
| 27 | 晋太170 Jintai 170 | 0 | 4/20/20 (1) | 4/60/40 (3) | 4/20/40 (4) | 4/10/20 (3) | 0 |
| 28 | 杨麦158 Yangmai 158 | 0 | 3/5/T (2) | 4/20/20 (5) | 0 | 0 | 0 |
| 29 | 兰天19 Lantain 19 | 3/5/T (4) | 0 | 0 | 0 | 0 | 0 |
| 30 | 兰天31 Lantian 31 | 3/5/T (1) | 0 | 4/T/T (2) | 0 | 0 | 0 |
| 31 | 兰天26 Lantian 26 | 3/10/T (4) | 0 | 0 | 0 | 0 | 0 |
| 32 | 天选50 Tianxuan 50 | 3/5/T (1) | 0 | 0 | 0 | 0 | 0 |
| 33 | 陇鉴386 Longjian 386 | 0 | 0 | 4/T/T (2) | 0 | 4/40/60 (7) | 0 |
| 34 | Chancellor | 0 | 4/20/20 (1) | 0 | 4/20/20 (4) | 4/30/40 (7) | 0 |
| 35 | Ulka/8cc | 0 | 4/20/30 (5) | 0 | 3/5/T (1) | — | 0 |
| 36 | Maris Huntsman | 0 | — | 4/10/10 (2) | — | 0 | 0 |
| 37 | 小白冬麦 Xiaobaidongmai | 0 | 3/10/T (1) | 0 | 4/40/10 (2) | 0 | 0 |
| 38 | Khaplic/8cc | 0 | 3/10/10 (1) | 0 | 4/10/10 (5) | 0 | 0 |
| 39 | Armada | 0 | 0 | 0 | 0 | 0 | 0 |
| 40 | 81-7241 | 0 | 3/20/20 (4) | 4/10/T (4) | 4/80/40 (3) | — | 0 |
| 41 | Aquila | 0 | — | 4/5/10 (2) | — | 4/20/20 (3) | — |
| 42 | 赤牙糙 Chiyacao | 0 | 4/10/30 (4) | 4/10/10 (2) | 4/80/60 (4) | 0 | 0 |
| 43 | 蚂蚱麦 Mazhamai | 0 | 3/10/T (3) | 4/10/T (3) | 4/5/10 (3) | 0 | — |
| 44 | 92R137 | 0 | 0 | 4/10/10 (5) | 4/T/T (2) | 0 | 0 |
| 45 | 保丰104 Baofen 104 | 0 | 3/10/T (3) | 4/10/T (1) | 4/40/40 (5) | 0 | 4/40/20 (5) |
| 46 | 高加索 Gaojiasuo | 0 | 0 | 4/20/10 (4) | 0 | 0 | 0 |
| 47 | 京双16 Jingshuang 16 | 0 | 3/20/10 (1) | 4/10/10 (1) | 0 | 0 | 0 |
| 48 | 白兔3号 Baitu No. 3 | 0 | 3/10/20 (5) | 4/10/10 (3) | 4/10/10 (2) | 0 | — |
| 49 | 绵麦37 Mianmai 37 | 0 | 0 | 4/10/10 (3) | 0 | 0 | 0 |
| 50 | 繁6 Fan 6 | 0 | 4/5/T (3) | 4/T/T (3) | 4/10/T (3) | 0 | 0 |
| 51 | 绵阳11 Mianyang 11 | 0 | 3/40/40 (5) | 4/T/T (3) | 4/10/T (3) | 0 | 0 |
| 52 | 川麦107 Chuanmai 107 | 3/10/T(2) | 2/5/T (2) | 4/10/T (1) | 4/T/T (1) | 0 | 0 |
| 53 | 川农19 Chuannong 19 | 3/5/T(4) | 0 | 4/20/T (1) | 0 | 0 | 0 |
| 54 | 川农27 Chuannong 27 | 3/5/T(2) | 0 | 4/20/10 (4) | 0 | 0 | 0 |
| 55 | 川麦58 Chuanmai 58 | 3/5/T(1) | 0 | 0 | 4/T/T (2) | 0 | 0 |
| 56 | 川育23 Chuanyu 23 | 0 | 0 | 0 | 0 | 0 | 0 |
| 57 | Avocet S*6/Yr9 | 0 | 0 | 4/5/10 (2) | 0 | 0 | 0 |
| 58 | Avocet S*6/Yr15 | 0 | 0 | 4/10/10 (2) | — | 0 | 0 |
| 59 | Avocet S*6/Yr24 | 0 | 0 | 0 | 0 | 0 | 0 |
| 60 | Avocet S*6/Yr26 | 0 | 0 | 0 | 0 | 0 | 0 |
| 61 | Jupateco R Yr18 | 0 | 0 | 0 | 0 | 0 | 0 |
| 62 | C591 | 0 | 0 | 4/10/10 (3) | 0 | 0 | 0 |
| 63 | TcLr9 | 0 | 0 | 0 | 4/10/T (4) | 0 | 0 |
| 64 | TcLr16 | 0 | 0 | 4/T/T (2) | 4/40/10 (3) | 0 | 0 |
| 65 | TcLr19 | 0 | 4/40/10 (1) | 4/10/10 (3) | 0 | 0 | 0 |
| 66 | TcLr24 | 0 | 3/10/10 (4) | 4/10/10 (2) | 0 | 0 | 0 |
| 67 | TcLr26 | 0 | 3/5/T (4) | 4/10/T (2) | 0 | 0 | 0 |
| 68 | TcLr38 | 0 | 0 | 0 | 0 | 0 | 0 |
| 69 | TcLr46 | 0 | 0 | 4/60/40 (3) | 0 | 0 | 0 |
| 70 | ISr5 Ra | 0 | 0 | 4/60/80 (4) | 0 | 0 | 0 |
| 71 | ISr6 Ra | 0 | 0 | 0 | 0 | 0 | 0 |
| 72 | Verstein Sr9e | 0 | 3/40/20 (6) | 0 | — | 0 | 0 |
| 73 | W2691Sr10 | 0 | 4/10/10 (6) | 4/10/20 (2) | 4/20/40 (5) | 0 | 0 |
| 74 | ISr11 Ra | 0 | 0 | 0 | 0 | 0 | 0 |
| 75 | CnS-T-mono-deri=Sr21 | 0 | 3/T/T (3) | 0 | 0 | 0 | 0 |
| 76 | LcSr24Ag | 0 | 3/20/T (5) | 4/10/T (1) | 4/10/10 (3) | 0 | 0 |
| 77 | Ealge Sr26 | 0 | 2/5/T (1) | 4/10/10 (1) | 4/5/T (1) | 0 | 0 |
| 78 | BtSr30 Wst | 0 | 3/10/T (2) | 0 | 0 | 0 | 0 |
| 79 | Sr31/6*LMPG | 3/5/T (1) | 2/T/T (2) | 4/5/T (1) | 4/5/T (3) | 0 | 0 |
| 80 | W2691 SrTt-1=Sr36 | 0 | 3/10/T (5) | 4/5/T (1) | 4/5/T (2) | 0 | 0 |
| 81 | Trident Sr38 | 0 | 0 | 0 | 0 | 0 | 0 |
| 82 | Little Club | 0 | 4/20/30 (1) | 0 | 0 | 0 | 4/T/T (2) |
新窗口打开|下载CSV
1.2 小麦条锈菌基因组DNA的提取及微卫星位点的PCR扩增
对采集到的小麦条锈菌标样,小心将单一病斑剪下装入2 mL离心管中,按采集地点和品种进行编号,试验过程要严防交叉污染。基因组DNA的提取参照天根植物DNA提取试剂盒的方法进行,使用Nano Drop(ND-1000,USA)测量DNA的浓度,并将DNA浓度稀释至50 ng·μL-1后置于-20℃冰箱保存备用。用于SSR分子标记的15对引物[2,15](表2)均由上海生工合成。PCR反应体系及程序参照文献[2,15],对扩增产物进行2%的琼脂糖凝胶电泳后筛选符合要求的反应产物,使用3500XL Genetic Analyzer(Applied Biosystems,USA)对扩增子进行检测。
Table 2
表2
表2本试验使用的15对SSR小麦条锈菌引物信息
Table 2
| 引物名称 Primer name | 重复单元 Repeat motif | 引物序列 Sequence (5′-3′) | 染料 Dye | 退火温度 Tm (℃) | 参考文献 Reference | |
|---|---|---|---|---|---|---|
| RJ03 | (TGG)8 | F: | GCAGCACTGGCAGGTGG | FAM | 61 | 文献[15] Reference [15] |
| R: | GATGAATCAGGATGGCTCC | |||||
| RJ04 | (TGG)8 | F: | GTGGGTTGGGCTGGAGTC | HEX | 58 | 文献[15] Reference [15] |
| R: | GCTAATCCATTCCACGCACC | |||||
| RJ12 | (AC)7 | F: | ATC ATT CCG ATT TCT TTC TCA CC | FAM | 56 | 文献[15] Reference [15] |
| R: | TCA CAC TGA TCC CAA TAG ATC AG | |||||
| RJ15 | (TG)7 | F: | ATC GAG CAC GTC CAA ATC G | HEX | 56 | 文献[15] Reference [15] |
| R: | CAC TGG ACA GAC GAC GGT TG | |||||
| RJ18 | (TGT)5 | F: | CTG CCC ATG CTC TTC GTC | ROX | 59 | 文献[15] Reference [15] |
| R: | GAT GAA GTG GGT GCT GCT G | |||||
| RJ20 | (CAG)4 | F: | AGA AGA TCG ACG CAC CCG | ROX | 56 | 文献[15] Reference [15] |
| R: | CCT CCG ATT GGC TTA GGC | |||||
| RJ21 | (GTT)6 | F: | TTC CTG GAT TGA ATT CGT CG | FAM | 55 | 文献[15] Reference [15] |
| R: | CAG TTC TCA CTC GGA CCC AG | |||||
| RJ24 | (GTT)5+9 | F: | TTG CTG AGT AGT TTG CGG TGA G | HEX | 58 | 文献[15] Reference [15] |
| R: | CTC AAG CCC ATC CTC CAA CC | |||||
| RJ27 | (TC)10 | F: | CGTCCCGACTAATCTGGTCC | ROX | 52 | 文献[15] Reference [15] |
| R: | ATGAGTTAGTTTAGATCAGGTCGAC | |||||
| CPS08 | (CAG)14 | F: | GAT AAG AAA CAA GGG ACA GC | FAX | 56 | 文献[2] Reference [2] |
| R: | CAG TGA ACC CAA TTA CTC AG | |||||
| CPS09 | (GTT)9 | F: | CGG GAG AAG ACC TGA GC | ROX | 55 | 文献[2] Reference [2] |
| R: | AGA AAA CGG AAT GTA ATG TG | |||||
| CPS10 | (TAG)8 | F: | TCT ACT GGG CAG ACT GGT C | HEX | 56 | 文献[2] Reference [2] |
| R: | CGG TTT GTT TTG TCG TTT C | |||||
| CPS15 | (ATG)5 | F: | GAT GGG GAA AAG TAA GAA GT | FAM | 56 | 文献[2] |
| R: | GGT GGG GGA TGT AAG TAT GTA | Reference [2] | ||||
| CPS34 | (TTTGG)4 | F: | GTT GGC TAC GAG TGG TCA TC | HEX | 56 | 文献[2] |
| R: | TAA CAC TAC ACA AAA GGG GTC | Reference [2] | ||||
| CPS36 | (CTCTAG)3 | F: | TCC AGG CAG TAA ATC AGA CGC | ROX | 58 | 文献[2] |
| R: | ATC AGC AGG TGT AGC CCC ATC | Reference [2] |
新窗口打开|下载CSV
3500XL Genetic Analyzer上机前的准备:按照1﹕50的比例对PCR产物进行稀释,吸取1 μL稀释液加入9 μL的Hi-Di+Liz混合液(Hi-Di﹕Liz = 1 000﹕15),混匀后全部加入3500XL Genetic Analyzer配套的96孔板中,贴胶贴,在振荡仪上充分混匀后,2 000 r/min离心1 min收集待测反应液于管底,95℃变性5 min,冰浴5 min,随后上机。
1.3 数据处理
扩增子长度由3500XL Genetic Analyzer(Applied Biosystems,USA)检测分析后,用GeneMaker(V2.7.0)(Soft Genetics)读取数据。然后将数据按照二倍体的格式录入Excel表,利用GenAlEx 6.5[16]将数据转化为可供R软件POPPR v2.5.0分析的CSV数据格式,超过一半位点有缺失数据的样品去除。利用GenAlEx计算以下遗传参数:(1)分子方差分析(analysis of molecular variation,AMOVA);(2)群体遗传分化系数(FST);(3)基因流(gene flow value,Nm)。
利用POPPR v2.5.0[17]计算以下参数:(1)观察到的等位基因数量(Na);(2)均匀度(Evenness);(3)位点缺失比率(locus missing ratio);(4)每个采样试验点的样本个数(N);(5)特定个体在特定位点上携带的等位基因的组合,携带相同等位基因组合的个体称为具有相同的多位点基因型(multi locus genotype,MLG);(6)基因型多样性(G);(7)标准化关联指数(rbarD)[18];(8)基因型累积曲线(genotype accumulating curve):反映位点是否足以进行群体分析,基因型累积曲线达到100%,代表所选择的位点数足够进行群体分析[19];(9)采用Bruvo’s distance绘制不同个体间的最小时空网络图(minimum spanning networks,MSN)[20];(10)主成分判别分析(discriminant analysis of principal components,DAPC)[21]。
2 结果
2.1 甘肃、青海地区6个试验点小麦条锈病监测
田间调查结果显示,82份全国变异观察圃材料在甘肃地区发病比青海地区严重,其中在临夏地区发病材料最多(54份),其次是平凉(44份)、临洮(40份)和文县(13份);有11份材料(Triticum spelta album、Joss Cambier、Armada、川育23、Avocet S*/Yr24、Avocet S*/Yr26、Jupateco R Yr18、TcLr38、ISr6 Ra、ISr11 Ra、Trident Sr38)在6个地区均未发病,占供试材料的13.41%,其余71份材料至少在1个试验点发病;青海互助14份材料发病,西宁仅4份材料发病;青海地区发病的18份材料仅1份材料(铭贤169)在甘肃4个地区都发病,其他17份材料均未在甘肃文县发病,但在平凉和临夏地区部分材料发病(表1),显示文县和青海群体小麦条锈菌毒性有一定差异。2.2 小麦条锈菌遗传多样性水平
15对引物组合共扩增出81个位点,每对引物组合产生的多态性位点为2—12个。均匀度值在0.2719—0.8648,表明等位基因频率在样本之间分布不均,其中引物RJ21的均匀度接近0.5,即该引物的等位基因在样本中均等分布。各个位点的缺失比率在0—9.62%(表3)。Table 3
表3
表315对引物的遗传多样性信息
Table 3
| 引物 Primer name | 等位基因 Allele | 均匀度 Evenness | 缺失比率 Missing ratio (%) |
|---|---|---|---|
| RJ12 | 6 | 0.3695 | 2.72 |
| RJ15 | 6 | 0.3383 | 5.63 |
| RJ18 | 12 | 0.7109 | 8.89 |
| RJ27 | 4 | 0.2719 | 4.72 |
| RJ20 | 5 | 0.8056 | 8.71 |
| RJ21 | 6 | 0.5285 | 2.36 |
| RJ24 | 7 | 0.8571 | 3.63 |
| CPS08 | 6 | 0.7591 | 0.36 |
| CPS09 | 4 | 0.8648 | 9.62 |
| CPS10 | 2 | 0.2997 | 0 |
| CPS15 | 7 | 0.8171 | 1.09 |
| CPS34 | 2 | 0.5949 | 0 |
| CPS36 | 3 | 0.6670 | 0.36 |
| RJ03 | 7 | 0.8327 | 5.63 |
| RJ04 | 4 | 0.2982 | 6.17 |
| 平均Mean | 5.4 | 0.6010 | 3.99 |
新窗口打开|下载CSV
551份样本克隆矫正后,共鉴定出505个MLG,甘肃和青海群体总的基因型多样性(0.917)高,群体的基因型多样性在0.841—0.974,其中平凉群体的基因型多样性最高,互助群体次之,甘肃临洮群体的最低(表4),可以看出,6个群体的基因型多样性均比较高。
Table 4
表4
表4重采基因型及克隆校正群体的基因型多样性统计
Table 4
| 群体 Population | 样本数 N | 多基因座基因型 MLG | 重采基因型编号及数量 Resampled MLG codes and numbers | 基因型多样性 G | 标准化关联指数rbarD | P值 P value |
|---|---|---|---|---|---|---|
| 文县 Wenxian | 27 | 23 | 61 (2), 427 (3), 429 (2) | 0.852 | 0.0139 | 0.186 |
| 临洮 Lintao | 132 | 111 | 29 (3), 74 (2), 169 (2), 184 (6), 185 (4), 187 (2), 192 (2), 193 (2), 194 (3), 197 (3), 210 (2), 228 (2) | 0.841 | 0.1163 | 0.001 |
| 平凉 Pingliang | 156 | 152 | 94 (2), 251 (2), 260 (2), 433 (2) | 0.974 | 0.0286 | 0.001 |
| 临夏 Linxia | 178 | 164 | 141 (5), 143 (2), 146 (2), 254 (2), 289 (2), 308 (2), 314 (2), 320 (2), 394 (2), 489 (2) | 0.921 | 0.0110 | 0.010 |
| 互助 Huzhu | 47 | 45 | 358 (2), 358 (2) | 0.957 | 0.0416 | 0.002 |
| 西宁 Xining | 11 | 10 | 272 (2) | 0.909 | 0.0718 | 0.025 |
| 总数 Total | 551 | 505 | 32 | 0.917 | 0.0260 | 0.001 |
新窗口打开|下载CSV
2.3 甘肃与青海小麦条锈菌群体菌源关系
在505个MLG中(表4),仅有32个MLG被克隆并进行了2—6次重新采样,各群体内部均检测到重新采样的MLG。甘肃、青海群体分别有29和3个MLG被重新采样,其中MLG-184在临洮的群体中重新采样6次,MLG-141在临夏的群体中重新采样5次,没有重新采样的MLG有473个,表明甘肃和青海小麦条锈菌群体中存在明显的遗传差异,MLG多样性与小麦条锈菌流行期的主要无性繁殖模式形成了明显的对比,即无性繁殖在群体中所占的比例不高。青海西宁群体与甘肃平凉群体之间的基因流最大,临夏次之,表明西宁群体与平凉群体菌源交流最频繁,与临夏群体菌源交流较频繁;其与文县和临洮群体的基因流小且群体遗传分化系数大(0.234和0.246),说明西宁群体与文县、临洮群体之间有较大的遗传分化,菌源之间交流不频繁。青海互助群体与临夏群体之间基因流最大,菌源交流最频繁;与文县群体之间基因流最小且群体遗传分化系数最大,说明互助群体与文县群体之间遗传分化大,菌源交流不频繁。可推断,青海地区小麦条锈菌的菌源大多数来源于甘肃平凉和临夏地区的传播。甘肃的4个群体之间,平凉群体与临夏群体之间的基因流最大,菌源交流最频繁(表5)。
Table 5
表5
表5甘肃和青海两省6个小麦条锈菌群体成对的群体遗传分化系数(对角线下部)和基因流(对角线上部)
Table 5
| 文县Wenxian | 临洮Lintao | 平凉Pingliang | 临夏Linxia | 互助Huzhu | 西宁Xining | |
|---|---|---|---|---|---|---|
| 文县 Wenxian | 2.408 | 2.337 | 3.408 | 2.916 | 1.074 | |
| 临洮 Lintao | 0.143*** | 3.567 | 3.541 | 4.426 | 1.036 | |
| 平凉 Pingliang | 0.156*** | 0.098*** | 6.238 | 5.217 | 2.601 | |
| 临夏 Linxia | 0.114*** | 0.103*** | 0.056*** | 8.055 | 2.072 | |
| 互助 Huzhu | 0.112*** | 0.084*** | 0.069*** | 0.043*** | 1.373 | |
| 西宁 Xining | 0.234*** | 0.246*** | 0.124*** | 0.138*** | 0.178*** |
新窗口打开|下载CSV
最小时空网络图(MSN)结果表明,两省6个群体相互之间未发现共享MLG。临洮群体的MLG与其他5个群体之间的遗传关系较小,从而形成了一个独特的MLG基因簇。甘肃省4个小麦条锈菌群体之间存在不同程度的遗传变异;西宁群体的MLG紧密聚集在一起,与甘肃临夏和平凉群体的部分个体菌源遗传距离相近,支持菌源从甘肃的临夏或平凉传播到西宁,随后在西宁群体产生了克隆分支。这说明尽管种群内部存在多样性,但个体亲缘关系仍然密切。文县群体同样紧密聚集在一起,并与青海西宁群体之间的遗传距离较远,来自甘肃临夏、平凉和青海互助的许多MLG非常接近,表明这些群体之间遗传关系更密切(图1)。
图1
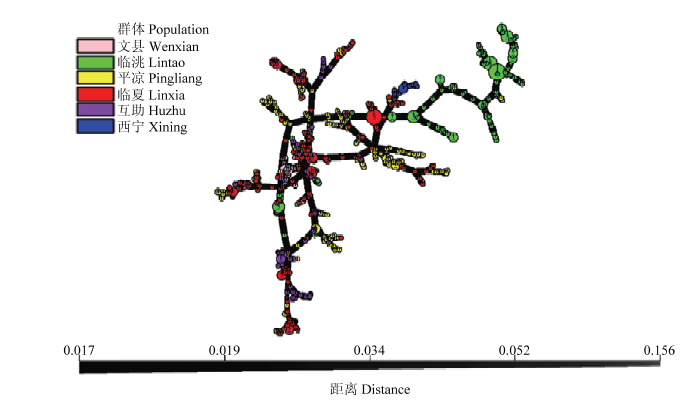 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1根据Bruvo’s距离进行的所有小麦条锈菌分离株的最小网络图
Fig. 1Minimum spanning network (MSN) of all isolates of Pst based on Bruvo’s distance
每个节点代表一个MLG,其中相同颜色的节点表示来自于同一地区的群体。每个个体之间的遗传距离越大,线条的颜色越细和越亮。节点越大,具有相同微卫星MLG的样本数量就越多Each node represents a MLG, the nodes of the same color indicate the population from the same area. The greater the genetic distance, the thinner and lighter the color of the line. The larger the node size, the greater the number of samples with the same microsatellite profile同样,非参数主成分分析(DAPC)验证了MSN的结果,青海互助和西宁的群体与来自于甘肃平凉和临夏的群体之间菌源关系最密切,差异最小;与临洮群体遗传距离相对较远且临洮群体为相对独立;文县群体则是一个完全独立的群体,与其他5个群体之间的差异最大(图2)。DAPC的结果以及群体间较大的FST(0.131)(表6)再次说明小麦条锈菌各群体之间存在遗传变异。
图2
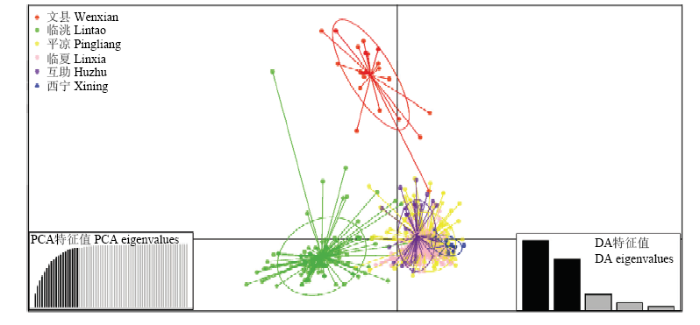 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2不同小麦条锈菌群体主成分判别分析图
不同颜色点代表相应小麦条锈菌群体内个体
Fig. 2The discriminant analysis of principal components (DAPC) of different Pst populations
Different colors represent individuals within the corresponding Pst population
Table 6
表6
表6甘肃、青海两省6个小麦条锈菌群体分子方差分析
Table 6
| 变异来源 Source of variance | 自由度 Degree of freedom | 变异组分 Variance component | 变异百分数 Percentage of variation (%) | 遗传分化系数 FST | P值 P value (rand≥data) |
|---|---|---|---|---|---|
| 群体间Among populations | 5 | 0.393 | 13 | 0.131 | 0.001 |
| 样本间Among individual | 545 | 0.620 | 21 | ||
| 样本内Within individual | 551 | 1.983 | 66 | ||
| 总数Total | 1101 | 2.996 | 100 |
新窗口打开|下载CSV
分子方差分析结果表明,小麦条锈菌群体间和个体间存在着一定的遗传分化,大多数变异(87%,P= 0.001)来自群体样本内部,而群体之间的遗传变异仅贡献了13%,说明有部分变异来自于地区之间(表6)。
2.4 甘肃、青海小麦条锈菌群体繁殖方式分析
结果显示,等位基因与个体以及群体之间的基因座相关联这一假说得到了重要支持,整体上甘肃和青海小麦条锈菌群体表现出无性繁殖特征(表4)。而来自于甘肃文县、临夏的群体以及青海西宁的群体具有与其他地区群体不同的特征,这3个群体存在不显著的rbarD值(表4、图3)表示连锁平衡,可能是有性生殖群体,尤其文县群体(rbarD=0.0139,P=0.186)显示出明显的有性重组特征,其他3个群体均为无性繁殖模式。图3
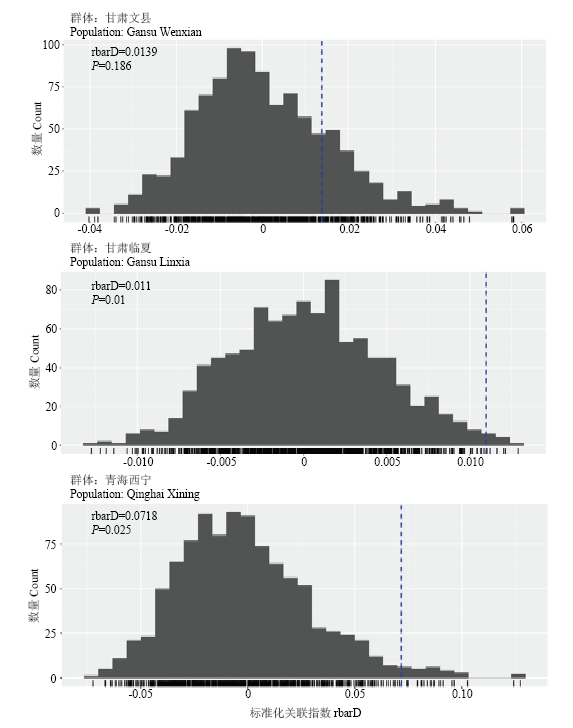 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3甘肃、青海3个小麦条锈菌有性群体连锁不平衡分析
Fig. 3Linkage disequilibrium analysis of three Pst populations from Gansu and Qinghai
3 讨论
甘肃、青海地处西北内陆,地域辽阔,地形地貌复杂多样,区域间气候差异大,形成了小麦种植呈垂直分布,冬、春小麦成片交错种植的农田生态景观,是小麦条锈病周年流行的适宜场所,使其成为我国小麦条锈菌重要的越夏区。小麦条锈菌除了在甘肃、青海区域内往返传播循环外,还通过孢子的远程传播危及到我国东部广大麦区[1]。条锈菌的远距离传播一直是植物病害流行学中的热点研究领域之一,为大区病害流行规律的研究提供了极具价值的材料,也极大地丰富了流行学理论,可从根本上提供地域性病害防治与品种抗病性布局的科学依据。因此通过分子标记方法明确甘肃、青海小麦条锈菌传播及遗传多样性水平,对该地区小麦条锈病的准确预测和指导防治具有重要作用。本研究中,春季流行时小麦条锈菌在甘肃和青海之间存在广泛的基因交流。根据田间实地调查条锈病初始发病时间,甘肃4个地区比青海2个地区发病早,且发病程度严重。最小时空网络图和主成分判别分析结果显示甘肃省4个群体之间,文县和临洮群体是2个相对独立的群体,临夏和平凉群体菌源关系最近,交流最频繁,且平凉地区发病时间早于临夏,两群体之间存在很大的基因流,可推断菌源从平凉传播到临夏,然后进行繁殖存活。文县群体和其他群体不同的原因主要是甘肃省春季发病初始菌源不同,甘肃省春季菌源结构复杂,有来自于甘肃省本地的越冬菌源,也有其他地方的菌源。2018年4月中旬,笔者在甘肃省春季病害实地调查中发现与陇南文县相邻的陕西汉中宁强、略阳的小麦生长期为灌浆期,其条锈病已进入暴发期;而甘肃天水、定西地区的小麦仅处于苗期,其条锈病呈始发或未发状态,平均病田率、病点率和病叶率均轻于陕西宁强和略阳。陕西黄陵以南是条锈菌在西北的主要越冬场所,也是条锈病的春季流行区[1]。另外,前人研究也表明陇南地区菌源可能来自于四川、云南的越冬菌源或陕西的春季流行菌源,而不是当地冬前菌源[22]。同时,陇中、陇东地区小麦种植以甘肃育成小麦品种为主,而陇南以四川及20世纪70年代引进的抗病品种为主[23,24],差异较大的寄主造成甘肃条锈菌群体出现一定差异。陇南地区在小麦条锈病综合治理中引进了较多抗病品种,使得小麦群体的抗病基因趋于多样,并对抗病品种进行合理布局和轮换,所以多元化的寄主抗病基因和较低的选择压力也是陇南地区条锈菌群体结构不同的主要成因。
本研究中群体差异主要来自各群体内部个体之间,这与之前研究各流行区群体遗传分化结果一致[6-8,10]。基因流反应群体的遗传分化水平,基因流大表示群体间的遗传分化小。本研究中,群体间成对的基因流均>1(表5),表明小麦条锈菌在各群体间有一定的基因交流,与LU等[10]研究不同,未发现6个群体之间共享任何基因型,仅在各个群体内部存在少数基因型重新采样,青海群体的基因交流不如甘肃群体频繁,较高的基因型多样性与小麦条锈菌流行周期的主要克隆性形成了明显的对比。
青海互助和西宁群体与甘肃陇东平凉和陇中临夏群体菌源之间的基因流要大于其与陇南文县群体之间的基因流,本研究表明青海的群体与甘肃临夏、平凉群体之间的遗传距离比陇南文县的近,文县群体是一个相对独立的群体。青海地区发病的变异观察圃材料,除了铭贤169,其他材料均未在陇南文县地区发病,也是群体有差异的证据之一。而LU等研究认为青海春季菌源主要自陇南地区传播而来,两地群体间基因流较强,菌源交流广泛;与临夏群体的菌源交流则受到一定程度的限制,基因流相对较弱[10],这与本研究结果不同。造成本研究结果的原因,一是与各地区的地形地貌特征和区域间距离相关,文县地区高山林立,山川纵横,生态环境较为封闭,病菌与外地交流需借助较强的上升气流和适当的风向才可实现,且气候温和,冬季比较温暖,夏季凉爽,有利于病原菌完成周年循环;二是环境的改变,随着近年来温度的升高,菌株发生变异,加之种植制度的变化等原因;三是有性繁殖群体存在的影响。总的来说,导致陇南文县地区小麦条锈菌群体遗传多样性与其他地区不同的主要因素是地理气候特征,其次,有性生殖的存在也有一定的影响[10,25-26]。
小麦条锈病为气传病害,条锈菌夏孢子在风力作用下可大范围传播。其中青海互助和西宁群体之间基因交流不频繁,原因可能是这些地区有一部分菌源来源于青海已发病地区的菌源传播。在早春病情调查中发现,各地区小麦品种不尽一致,而青海尖扎、贵德地区的小麦条锈病发病时间略早于甘肃临夏等相邻地区,青海尖扎地区5月21日初始发病,发病严重度和普遍率较低(3/10/10),甘肃临夏地区6月16日发病程度(4/40/60) 比尖扎严重,青海贵德6月14日发病(4/20/40),青海贵德和尖扎两地群体菌源来源也不相同。有研究表明,青海一些地区小麦条锈菌可以少量越冬[27]。所以青海春季流行的初始菌源不一定都来自于甘肃省,两省菌源有一定的交流。由此推断,青海小麦条锈菌越冬菌源在春季病害流行具有一定的作用。
本研究中,两省小麦条锈菌群体遗传多样性均较高,这与样本来源于地理跨度较大的不同县(市)有一定关系,是病菌受复杂多样生态环境自然选择的结果[28]。同时,样本来自不同的变异观察圃品种,小麦品种拥有多样化的抗性背景,寄主对病原菌群体结构产生了重新配置的作用[29,30]。另外,甘肃和青海地区是地形相似的自然区域,或为高寒冬春麦区,或为山川交错、冬春麦垂直分布的麦区,为条锈菌的生长和越夏提供了适宜的气候条件和生态环境,条锈菌在该地有较长的进化史。因此,这些地区的条锈菌群体有较高的遗传多样性和重组事件。6个群体间没有共享基因型存在,也说明条锈菌可通过遗传重组、突变、寄主选择及对当地生态环境的适应,在各地区进行了相对独立的进化。
根据哈迪-温伯格定律,无性繁殖的群体由于基因不是完全独立的遗传会出现显著的连锁不平衡现象,有性生殖的群体因配子的随机交配使得基因座间在遗传过程中没有相关性,处于连锁平衡状态。甘肃、青海省6个地区的群体中,甘肃文县、临夏的群体以及青海西宁的群体存在连锁平衡现象(图3),说明这些地区存在有性重组的可能。ZHAO等[31]在甘肃省从锈菌侵染的小檗上分离纯化的2个锈子器可以产生条锈菌夏孢子,为甘肃小麦条锈菌群体存在有性生殖提供了最直接的证据,前人也报道了甘肃地区存在有性重组现象[9,25-26]。但是关于青海地区存在有性繁殖现象少有报道。青海地区存在大量的转主寄主小檗[32],且种类丰富。在5—7月份,青海大多数小檗可在自然条件下受到锈菌侵染,能产生锈子器,并大量分布在冬麦和春麦田边。青海东部地区广泛分布的鲜黄小檗、直穗小檗、置疑小檗、甘肃小檗、近似小檗、细叶小檗、匙叶小檗、松潘小檗、堆花小檗和秦岭小檗10 种小檗可以作为小麦条锈菌的转主寄主[31,33-34],这是青海群体可以发生有性生殖的必要条件之一。随着全球气候的变暖,耕作制度的变革,青海省东部地区冬小麦种植面积不断扩大,使得小麦条锈病的发生流行规律发生显著变化。甘肃和青海小檗种类繁多,为小麦条锈菌有性生殖发生提供了基础条件。本研究发现青海西宁地区可能存在有性生殖,群体具有很高的遗传多样性,赵杰等研究表明青海地区小檗分布较为广泛,春季小檗锈病发生非常严重[35],但目前尚未确定青海其他地区是否存在小麦条锈菌的有性生殖,需要结合更大的样本量和更多的采样点进一步深入研究证实。
4 结论
小麦条锈病春季流行期,甘肃和青海群体均具有较高的基因型多样性,甘肃地区与青海东部地区的传播路线以甘肃平凉、临夏到青海的传播为主,甘肃文县到青海的传播为辅。甘肃文县、临夏和青海西宁3个群体存在有性生殖现象,对该地区条锈菌丰富的遗传多样性的形成具有一定作用。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 4]
[本文引用: 4]
DOI:10.1111/j.1755-0998.2008.02423.xURLPMID:21564613 [本文引用: 8]

We described twenty polymorphic microsatellite loci derived from the expressed sequence tags of Puccinia striiformis f. sp. tritici, which causes yellow rust disease on wheat. The numbers of alleles range from two to six and eight microsatellite loci show significant similarities to known genes. Observed and expected heterozygosities ranged from 0.12 to 0.78 and from 0.24 to 0.87, respectively.
DOI:10.1080/07060660509507230URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.3864/j.issn.0578-1752.2013.20.008URL [本文引用: 1]
小麦条锈病是影响小麦安全生产的重要生物灾害。文中介绍了全国小麦锈病工作者通过60多年通力协作,对小麦条锈病综合治理理论和技术研究取得的显著成绩,系统揭示了中国小麦条锈病的越冬、越夏规律、菌源传播规律、病菌致病性变异途径以及品种抗病性“丧失”的规律与原因,发现中国小麦条锈病存在秋季菌源和春季菌源2大菌源基地。提出了“综合治理越夏异变区、持续控制冬季繁殖区和全面预防春季流行区”的病害源头治理策略,研发出小麦条锈病分子诊断、异地测报以及抗锈良种、药剂拌种、退麦改种、适期晚种和带药侦查、打点保面等一系列病害监测预警和关键防治技术,构建了以生物多样性利用为核心的中国小麦条锈病菌源基地综合治理技术体系,在生产上大规模推广应用,防病保产效果极其显著。文中并对病菌致病性变异机制、早期预警和越夏易变区生态治理等问题进行了讨论。
DOI:10.3864/j.issn.0578-1752.2013.20.008URL [本文引用: 1]
小麦条锈病是影响小麦安全生产的重要生物灾害。文中介绍了全国小麦锈病工作者通过60多年通力协作,对小麦条锈病综合治理理论和技术研究取得的显著成绩,系统揭示了中国小麦条锈病的越冬、越夏规律、菌源传播规律、病菌致病性变异途径以及品种抗病性“丧失”的规律与原因,发现中国小麦条锈病存在秋季菌源和春季菌源2大菌源基地。提出了“综合治理越夏异变区、持续控制冬季繁殖区和全面预防春季流行区”的病害源头治理策略,研发出小麦条锈病分子诊断、异地测报以及抗锈良种、药剂拌种、退麦改种、适期晚种和带药侦查、打点保面等一系列病害监测预警和关键防治技术,构建了以生物多样性利用为核心的中国小麦条锈病菌源基地综合治理技术体系,在生产上大规模推广应用,防病保产效果极其显著。文中并对病菌致病性变异机制、早期预警和越夏易变区生态治理等问题进行了讨论。
DOI:10.1094/PDIS-02-16-0190-REURLPMID:30681929 [本文引用: 2]
Wheat stripe rust, caused by Puccinia striiformis f. sp. tritici (Pst), is an important disease on wheat, seriously threatening wheat production worldwide. China is one of the largest stripe rust epidemic regions in the world. The pathogen sexual reproduction and migration routes between Tibet and the other regions in China are still unknown. In this study, we obtained 961 Pst isolates from 1,391 wheat leaf samples from Gansu (277), Shaanxi (253), Sichuan (172), and Tibet (259), comprising 13 natural populations, and genotyped them with simple sequence repeat (SSR) markers. The isolates can be divided into two distinct clusters based on DAPC and STRUCTURE analyses. The genetic diversity of Longnan (in Gansu) and Yibin (in Sichuan) populations was the highest and lowest among the 13 populations, respectively. The hypothesis of multilocus linkage disequilibrium was rejected for the populations from Linzhi in the Himalayan, Longnan, Hanzhong, Guangyuan, Mianyang, Liangshan, and Chendu in the south Qinling Mountains at the level of P = 0.01, which indicated significant linkage among markers in these populations. Populations in the other regions had extensive gene exchange (Nm > 4); little gene exchange was found between Tibet and the other regions (Nm < 1). The results suggest that the Tibet epidemic region of Pst is highly differentiated from the other epidemic regions in China.
DOI:10.1094/PHYTO-03-15-0081-RURLPMID:26506459 [本文引用: 1]
Puccinia striiformis f. sp. tritici is the causal pathogen of interregional epidemics of wheat stripe rust in China via long-distance migration. Gansu Province serves as putative inoculum center providing oversummering inoculum, while Sichuan Basin area serves as a region providing huge amounts of overwintering inoculum. Thus, the relationship between these two regions in population exchange and migration become important in prediction of interregional epidemics. In this study, we compared the population genetic structure and race composition between Gansu and Sichuan Basin populations to infer their migration relationships. A total of 526 isolates, spanning 3 years, were genotyped using eight pairs of amplified fragment length polymorphism markers, and a subset of 98 isolates were inoculated onto 19 Chinese differentials to perform the race analysis. Twenty-three common races and 26 shared genotypes supplied molecular evidence for migration between Gansu and Sichuan Basin populations. Bayesian assignment and principal component analysis revealed that the genetic group assignment of the Sichuan Basin populations (10SB and 11SB) changed in the spring to align with the fall Gansu populations in the prior seasons (09GS and 10GS), which indicated an asymmetric migration from Gansu Province to the Sichuan Basin area. The linkage disequilibrium and the parsimony tree length permutation test revealed a strong annual recombination signal in the Gansu populations and an inconsistent signal in the Sichuan Basin populations.
DOI:10.1094/PDIS-01-12-0072-REURL [本文引用: 2]
Liang, J.-M., Wan, Q., Luo, Y., and Ma, Z.-H. 2013. Population genetic structures of Puccinia striiformis in Ningxia and Gansu provinces of China. Plant Dis. 97:501-509. Wheat stripe rust, caused by the fungal pathogen Puccinia striiformis f. sp. tritici, is one of the most destructive plant diseases in China. Gansu and Ningxia Provinces are considered to be the key areas for oversummering of this pathogen in China. In this study, 283 P striiformis f. sp. tritici isolates were collected in these two provinces. Amplified fragment length polymorphism (AFLP) was used to analyze the population genetics and to infer the chance of population exchanges between different geographic locations and seasons. The genotypic diversity of the Gansu population (0.514) was slightly higher than that of the Ningxia population (0.489). The occurrence of frequent population exchanges between these two regions was observed, showing that 40 AFLP genotypes were shared by the populations of the two provinces. Gene flow between these two regions in autumn and spring subpopulations was also detected. The genotype distribution in three populations of Ningxia from opposite sides of the Liupan Mountains revealed possible significant effects of the mountains on limiting gene flow and population exchange. Phylogenetic analysis confirmed the possibility of recombination in some of the studied subpopulations in both provinces.
DOI:10.1007/s10658-010-9688-8URL [本文引用: 2]
Wheat stripe rust, caused by Puccinia striiformis f. sp. tritici, is one of the most destructive wheat diseases in China. Yunnan Province, located in south-western China, possesses unique features of geography, climate, wheat growth and stripe rust epidemics, different from main epidemic regions in China. The isolates of this pathogen were collected from nine counties in Yunnan Province during February to May of 2008. Used as a comparison, isolates were also collected from five counties of Gansu Province, the province important in inter-regional stripe rust epidemics in China. Amplified fragment length polymorphism (AFLP) method was applied to study the population genetics of the pathogen among different populations in these two provinces. Forty one AFLP genotypes were obtained from 150 isolates and the genotype qj3 showed the highest frequency in Yunnan Province. While 22 genotypes were detected from 40 isolates, no genotype showing as predominant was identified in Gansu Province. Genotypic diversity in Gansu Province was higher than that in Yunnan Province. A free recombination signature was detected in Gansu Province but not in Yunnan Province. We concluded that the population of P. striiformis in Yunnan Province can be considered as a clonal population.
DOI:10.1007/s10658-011-9842-yURL [本文引用: 6]
In China, wheat stripe rust, caused by Puccinia striiformis f. sp. tritici, is one of the most destructive diseases of wheat. The Longnan and Linxia regions in Gansu Province and Qinghai Province are the major over-summering regions for the pathogen and key epidemiological zones in Northwest China. Population genetic diversity and interregional long-distance spread of the wheat stripe rust pathogen in Northwest China were studied using SSR markers. The genetic diversity in the Longnan population was much higher than those in the Linxia and Qinghai populations. Therefore, the molecular data confirmed that the Longnan region is a center of genetic diversity for P. striiformis f. sp. tritici in Northwest China. The low genetic differentiation (Gst=0.15) and the extensive gene flow (Nm=1.37) were found among the three regions in Northwest China. The most important conclusion of this study is that the stripe rust inoculum in Qinghai can come from both Longnan and Linxia, but mainly from Longnan directly in the spring.
URL [本文引用: 1]
【目的】甘肃陇南地区是小麦条锈菌最主要和最大的越夏区,本研究的目的是分析该地区的小麦条锈菌自然群体的遗传结构,探索其分子遗传变异规律。【方法】采用TP-M13-SSR 荧光标记技术,对甘肃省陇南地区8个种群409个小麦条锈菌分离株基因组DNA进行SSR标记分析。【结果】陇南地区小麦条锈菌的观察等位基因数(Na)为1.95,有效等位基因数目(Ne)为1.43,Nei's (1973)基因多样性指数(H)为0.27,Shannon 信息指数(I)为0.41。武都、文县和秦城种群遗传多样性较高,徽县、成县和西和种群相对较低。AMOVA分析结果表明,小麦条锈菌群体间和群体内都存在着一定的遗传分化,群体间的遗传变异占总变异的12.5%,群体内遗传变异占87.5%。地区间的基因流Nm =1.83。【结论】陇南地区小麦条锈菌群体遗传多样性很丰富,但地区之间有一定的差异;群体遗传变异主要存在于群体内部,不同地区间存在基因的交流和病原菌的移动。
URL [本文引用: 1]
【目的】甘肃陇南地区是小麦条锈菌最主要和最大的越夏区,本研究的目的是分析该地区的小麦条锈菌自然群体的遗传结构,探索其分子遗传变异规律。【方法】采用TP-M13-SSR 荧光标记技术,对甘肃省陇南地区8个种群409个小麦条锈菌分离株基因组DNA进行SSR标记分析。【结果】陇南地区小麦条锈菌的观察等位基因数(Na)为1.95,有效等位基因数目(Ne)为1.43,Nei's (1973)基因多样性指数(H)为0.27,Shannon 信息指数(I)为0.41。武都、文县和秦城种群遗传多样性较高,徽县、成县和西和种群相对较低。AMOVA分析结果表明,小麦条锈菌群体间和群体内都存在着一定的遗传分化,群体间的遗传变异占总变异的12.5%,群体内遗传变异占87.5%。地区间的基因流Nm =1.83。【结论】陇南地区小麦条锈菌群体遗传多样性很丰富,但地区之间有一定的差异;群体遗传变异主要存在于群体内部,不同地区间存在基因的交流和病原菌的移动。
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 10]
DOI:10.1093/bioinformatics/bts460URL [本文引用: 1]
GenAlEx: Genetic Analysis in Excel is a cross-platform package for population genetic analyses that runs within Microsoft Excel. GenAlEx offers analysis of diploid codominant, haploid and binary genetic loci and DNA sequences. Both frequency-based (F-statistics, heterozygosity, HWE, population assignment, relatedness) and distance-based (AMOVA, PCoA, Mantel tests, multivariate spatial autocorrelation) analyses are provided. New features include calculation of new estimators of population structure: G'(ST), G ''(ST), Jost's Dest and F'(ST) through AMOVA, Shannon Information analysis, linkage disequilibrium analysis for biallelic data and novel heterogeneity tests for spatial autocorrelation analysis. Export to more than 30 other data formats is provided. Teaching tutorials and expanded step-by-step output options are included. The comprehensive guide has been fully revised.
DOI:10.3389/fgene.2015.00348URLPMID:26734060 [本文引用: 1]
The study of microorganisms that pervade each and every part of this planet has encountered many challenges through time such as the discovery of unknown organisms and the understanding of how they interact with their environment. The aim of this review is to take the reader along the timeline and major milestones that led us to modern metagenomics. This new and thriving area is likely to be an important contributor to solve different problems. The transition from classical microbiology to modern metagenomics studies has required the development of new branches of knowledge and specialization. Here, we will review how the availability of high-throughput sequencing technologies has transformed microbiology and bioinformatics and how to tackle the inherent computational challenges that arise from the DNA sequencing revolution. New computational methods are constantly developed to collect, process, and extract useful biological information from a variety of samples and complex datasets, but metagenomics needs the integration of several of these computational methods. Despite the level of specialization needed in bioinformatics, it is important that life-scientists have a good understanding of it for a correct experimental design, which allows them to reveal the information in a metagenome.
URLPMID:17249067 [本文引用: 1]
The association of alleles among different loci was studied in natural populations of Hordeum spontaneum, the evolutionary progenitor of cultivated barley. The variance of the number of heterozygous loci in two randomly chosen gametes affords a useful measure of such association. The behavior of this statistic in several particular models is described. Generally, linkage (gametic phase) disequilibrium tends to increase the variance above the value expected under complete independence. This increase is greatest when disequilibria are such as to maximize the sum of squares of the two-locus gametic frequencies.-When data on several loci per individual are available, the observed variance may be tested for its agreement with that expected under the hypothesis of complete interlocus independence, using the sampling theory of this model. When applied to allozyme data from 26 polymorphic populations of wild barley, this test demonstrated the presence of geographically widespread multilocus organization. On average, the variance was 80% higher than expected under random association. Gametic frequencies for four esterase loci in both of these populations of wild barley and two composite crosses of cultivated barley were analyzed. Most generations of the composites showed less multilocus structure, as measured by the indices of association, than the wild populations.
DOI:10.1094/PHYTO-12-16-0425-RVWURLPMID:28513284 [本文引用: 1]
Population genetic analysis is a powerful tool to understand how pathogens emerge and adapt. However, determining the genetic structure of populations requires complex knowledge on a range of subtle skills that are often not explicitly stated in book chapters or review articles on population genetics. What is a good sampling strategy? How many isolates should I sample? How do I include positive and negative controls in my molecular assays? What marker system should I use? This review will attempt to address many of these practical questions that are often not readily answered from reading books or reviews on the topic, but emerge from discussions with colleagues and from practical experience. A further complication for microbial or pathogen populations is the frequent observation of clonality or partial clonality. Clonality invariably makes analyses of population data difficult because many assumptions underlying the theory from which analysis methods were derived are often violated. This review provides practical guidance on how to navigate through the complex web of data analyses of pathogens that may violate typical population genetics assumptions. We also provide resources and examples for analysis in the R programming environment.
DOI:10.1111/j.1365-294X.2004.02209.xURLPMID:15189230 [本文引用: 1]
Microsatellites are powerful molecular markers, used commonly to estimate intraspecific genetic distances. With the exception of band sharing similarity index, available distance measures were developed specifically for diploid organisms and are unsuited for comparisons of polyploids. Here, we present a simple method for calculation of microsatellite genotype distances, which takes into account mutation processes and permits comparison of individuals with different ploidy levels. This method should provide a valuable tool for intraspecific analyses of polyploid organisms, which are widespread among plants and some animal taxa. An illustration is given using data from the planarian flatworm Schmidtea polychroa (Platyhelminthes).
DOI:10.1186/1471-2156-11-94URLPMID:20950446 [本文引用: 1]
BACKGROUND: The dramatic progress in sequencing technologies offers unprecedented prospects for deciphering the organization of natural populations in space and time. However, the size of the datasets generated also poses some daunting challenges. In particular, Bayesian clustering algorithms based on pre-defined population genetics models such as the STRUCTURE or BAPS software may not be able to cope with this unprecedented amount of data. Thus, there is a need for less computer-intensive approaches. Multivariate analyses seem particularly appealing as they are specifically devoted to extracting information from large datasets. Unfortunately, currently available multivariate methods still lack some essential features needed to study the genetic structure of natural populations. RESULTS: We introduce the Discriminant Analysis of Principal Components (DAPC), a multivariate method designed to identify and describe clusters of genetically related individuals. When group priors are lacking, DAPC uses sequential K-means and model selection to infer genetic clusters. Our approach allows extracting rich information from genetic data, providing assignment of individuals to groups, a visual assessment of between-population differentiation, and contribution of individual alleles to population structuring. We evaluate the performance of our method using simulated data, which were also analyzed using STRUCTURE as a benchmark. Additionally, we illustrate the method by analyzing microsatellite polymorphism in worldwide human populations and hemagglutinin gene sequence variation in seasonal influenza. CONCLUSIONS: Analysis of simulated data revealed that our approach performs generally better than STRUCTURE at characterizing population subdivision. The tools implemented in DAPC for the identification of clusters and graphical representation of between-group structures allow to unravel complex population structures. Our approach is also faster than Bayesian clustering algorithms by several orders of magnitude, and may be applicable to a wider range of datasets.
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.7606/j.issn.1009-1041.2006.01.028URL [本文引用: 1]
在我国小麦条锈病常发易变区陇南地区,从1973年起对里勃留拉、N.斯特拉姆潘列、成农4号、清农1号、中梁5号和清农3号6个品种抗杀锈病的持久性进行了鉴定与分析。结果表明,在陇南小麦生产品种太多仅经3~8年即丧失抗务锈性的情况下,里勃留拉和N.斯特拉姆潘列在近30年间表现了突出的持久抗性。里勃留拉一年表现免疫,其余年份均发病,但病情指数仅为0.01%~5%。N.斯特拉姆潘列则以表现免疫为主,少数年份发病,病情指数0.1%~2.5%。成农4号也已大面积种植20余年,表现感病的反应型,少数年份病情指数较高.但多年来均能保持正常落黄和较为稳定的产量。此外,清农1号抗性保持16年,但以后却成为高感品种。本文在上述试验的基础上对小麦蒂锈病抗病性的持久性等问题进行了讨论。
DOI:10.7606/j.issn.1009-1041.2006.01.028URL [本文引用: 1]
在我国小麦条锈病常发易变区陇南地区,从1973年起对里勃留拉、N.斯特拉姆潘列、成农4号、清农1号、中梁5号和清农3号6个品种抗杀锈病的持久性进行了鉴定与分析。结果表明,在陇南小麦生产品种太多仅经3~8年即丧失抗务锈性的情况下,里勃留拉和N.斯特拉姆潘列在近30年间表现了突出的持久抗性。里勃留拉一年表现免疫,其余年份均发病,但病情指数仅为0.01%~5%。N.斯特拉姆潘列则以表现免疫为主,少数年份发病,病情指数0.1%~2.5%。成农4号也已大面积种植20余年,表现感病的反应型,少数年份病情指数较高.但多年来均能保持正常落黄和较为稳定的产量。此外,清农1号抗性保持16年,但以后却成为高感品种。本文在上述试验的基础上对小麦蒂锈病抗病性的持久性等问题进行了讨论。
DOI:10.3852/08-098URLPMID:20120228 [本文引用: 2]
Puccinia striiformis f.sp. tritici (PST), a basidiomycota responsible for wheat yellow rust, has a strict clonal behavior and a low genetic diversity in European and Australian populations. On the other hand high diversity has been reported in Chinese populations. Moreover it is thought that in China yellow rust epidemics start recurrently from the western highlands where over-summering occurs. To compare PST genetic diversity in this area to the one described in France seven AFLP primer combinations were used to analyze a sample of 160 isolates collected in 2001 in five counties of Gansu Province. The AFLP data revealed 40 polymorphic bands, discriminating 139 AFLP genotypes. Linkage disequilibrium and phylogeographic analyses support the hypothesis of a reproductive mode that is not strictly clonal. In this regard Chinese isolates from Gansu strongly contrast with the European studies using the same markers. Genetic diversity of this 1 y sampling in Gansu is found to be seven times higher than the one observed in France over 20 y and exhibits lower linkage disequilibrium. The effective population size of the French sample was estimated to be 1000 times smaller than the Gansu population. These results support the hypothesis of large population size as well as the occurrence of genetic recombination, while the importance of Gansu as a main over-summering area requires assessment through larger scale studies.
DOI:10.1016/j.fgb.2008.12.007URLPMID:19570502 [本文引用: 2]
Wheat yellow rust (Puccinia striiformis f.sp. tritici) (PST) has been described as a strongly clonal species in both European and Australian populations, with very limited molecular diversity but rapidly evolving virulences. Contrastingly, marked genetic diversity has been reported in Chinese PST populations. To test whether such variability could originate from oversummering areas, we assessed the diversity of virulence and molecular markers (AFLP and SSR) using 412 PST isolates from the highlands of Tianshui county in Gansu province. Very marked phenotypic and genotypic diversity (38% and 89%, respectively) was found. No genetic structure dependent on the sites sampled (Fst=0.004) or altitude distribution (Fst=0.0098) was detected, indicating important gene flow at the county scale. This study also revealed genetic recombination between molecular markers and thus strongly suggests the existence of a sexual or parasexual cycle in PST in Tianshui county. The observations of higher rates of sexual spore production in genotypes originating from Tianshui are the very first elements suggestive of the existence of a sexual cycle in this species.
DOI:10.1094/PDIS-09-15-1112-REURLPMID:30682289 [本文引用: 1]
Stripe rust, caused by Puccinia striiformis f. tritici, is an important wheat disease in China. P. striiformis f. sp. tritici overwintering and nonoverwintering regions based on the temperature were described elsewhere ( Shi et al. 2005 ). The temperature limit for P. striiformis f. sp. tritici overwintering is derived from field observations. However, P. striiformis f. sp. tritici has recently been observed to overwinter at sites where overwintering is predicted to be unlikely. We studied P. striiformis f. sp. tritici overwintering across several sites in regions close to or further away from the current P. striiformis f. sp. tritici
[本文引用: 1]
DOI:10.1023/A:1015678432355URL [本文引用: 1]
The durability of disease resistance is affected by the evolutionary potential of the pathogen population. Pathogens with a high evolutionary potential are more likely to overcome genetic resistance than pathogens with a low evolutionary potential. We will propose a set of guidelines to predict the evolutionary potential of pathogen populations based on analysis of their genetic structure. Under our model of pathogen evolution, the two most important parameters to consider are reproduction/mating system and gene/genotype flow. Pathogens that pose the greatest risk of breaking down resistance genes are those that possess a mixed reproduction system, with at least one sexual cycle per growing season and asexual reproduction during the epidemic phase, and a high potential for gene flow. The lowest risk pathogens are those with strict asexual reproduction and low potential for gene flow. We will present examples of high- and low-risk pathogens. Knowledge of the population genetic structure of the pathogen may offer insight into the best breeding strategy for durable resistance. We will present broad guidelines suggesting a rational method for breeding durable resistance according to the population genetics of the pathogen.
DOI:10.1007/s11274-012-1170-7URLPMID:23054697 [本文引用: 1]
Population genetic diversity in Tianshui city was analyzed with SSR markers in 605 single-pustule isolates of the stripe rust pathogen, Puccinia striiformis f. sp. tritici (Pst), obtained from 19 varieties of wheat. Significant differences in genetic diversity among populations were defected. Genetic diversity was highest in population on Tian 863-13, a highly resistant variety, whereas genetic diversity was lowest in population on Huixianhong, a highly susceptible variety. Seven populations from seven varieties that carried the common Yr18 resistance gene were clustered as one sub-group at 0.88 similarity coefficient, which showed that resistance gene selection had close relation with pathogen's component. The results of present study can provide a theoretical basis for integrated management of wheat stripe rust and effective deployment of resistance genes in Pst over-summering zones in China.
DOI:10.1094/PHYTO-09-12-0249-RURLPMID:23514262 [本文引用: 2]
The wheat stripe rust pathogen (Puccinia striiformis f. sp. tritici) population in China has been reported to be a distinct genetic group with higher diversity than those in many other countries. Genetic recombination in the P. striiformis f. sp. tritici population has been identified with molecular markers but whether sexual reproduction occurs in China is unknown. In this study, we surveyed barberry plants for infection by rust fungi in the stripe rust
DOI:10.1094/PHYTO-100-5-0432URLPMID:20373963 [本文引用: 1]
The life history of Puccinia striiformis remains a mystery because the alternate host has never been identified. Inoculation of grasses using aeciospores from naturally infected Berberis chinensis and B. koreana resulted in infection on Poa pratensis, producing uredinia typical of stripe rust caused by P. striiformis. Analyses using real-time polymerase chain reaction and DNA sequence confirmed the rust fungus as P. striiformis. Pycnia and aecia were produced on B. chinensis, B. holstii, B. koreana, and B. vulgaris after inoculation using germinating telia of P. striiformis f. sp. tritici. Wheat inoculated with aeciospores from B. chinensis resulted in uredinia, which demonstrated that Berberis spp. also serve as alternate hosts for the wheat stripe rust pathogen. The elucidation of the complete life history for P. striiformis f. sp. tritici will provide a powerful tool to rapidly advance our knowledge of the genetics of this rust fungus, and will lead to the development of improved strategies for a better control of stripe rust.
[D].
[本文引用: 1]
[D].
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}