 ,1,5
,1,5Changes of LncRNA Expression Profile in Spleen of Diarrhea and Non-diarrhea Individuals in F17 of Hu Sheep Lamb
HUANG SaiNan1, JIN ChengYan1, BAO JianJun2, WANG Yue1, CHEN WeiHao1, WU TianYi1, WANG LiHong1, Lü XiaoYang1, GAO Wen1, WANG BuZhong3, ZHU GuoQiang4, DAI GuoJun1, SUN Wei,1,5通讯作者:
收稿日期:2018-09-18接受日期:2018-12-3网络出版日期:2019-04-01
| 基金资助: |
Received:2018-09-18Accepted:2018-12-3Online:2019-04-01
作者简介 About authors
黄赛男,E-mail:
金澄艳,E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (3268KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
黄赛男, 金澄艳, 鲍建军, 王悦, 陈炜昊, 吴天弋, 王利宏, 吕晓阳, 高雯, 王步忠, 朱国强, 戴国俊, 孙伟. F17大肠杆菌在湖羊羔羊个体脾脏中LncRNA表达谱变化[J]. 中国农业科学, 2019, 52(7): 1282-1294 doi:10.3864/j.issn.0578-1752.2019.07.015
HUANG SaiNan, JIN ChengYan, BAO JianJun, WANG Yue, CHEN WeiHao, WU TianYi, WANG LiHong, Lü XiaoYang, GAO Wen, WANG BuZhong, ZHU GuoQiang, DAI GuoJun, SUN Wei.
0 引言
【研究意义】羊大肠杆菌病是规模化羊场最为常见高发的细菌性疾病之一,传统沿用的抗生素治疗方案存在诸多缺陷。检测影响绵羊大肠杆菌非腹泻型相关基因的表达情况是分析绵羊抗病分子机制的基础,从而可以发现与抗大肠杆菌性状相关的候选基因。【前人研究进展】1966年,ORSKOV等[1]首先报道了猪源大肠杆菌黏附性抗原K88,后证实黏附性抗原K88是一种依赖于质粒表达的蛋白质抗原[2],电子显微镜下观察到其形态为暴露在菌体表面的丝状物。目前已经鉴定出多种动物源产肠毒素大肠杆菌(ETEC)菌毛,包括K88、K99、987P、F17和F41等都是动物源ETEC重要的黏附性毒力因子。lncRNA(Long non-coding RNA)是长度大于200个核苷酸的非编码RNA。大量的研究表明,lncRNA 与人类各种肿瘤、心血管疾病和代谢疾病的发生具有密切的联系和调控作用。值得注意的是,近年来越来越多的研究表明,lncRNA在抗病毒等天然免疫反应中也具有重要的调控作用[3,4,5,6,7],相比于人的lncRNA,羊的lncRNA功能研究较滞后,而且大多集中在分析lncRNA对羊肌肉生长发育、睾丸发育、毛囊发育等性状的调控作用[8,9,10]。【本研究切入点】目前,关于绵羊抗病方面研究还不多,少量的研究主要集中在疾病防治方面[11,12],而绵羊抗病的分子遗传学基础研究鲜有报道。【拟解决的关键问题】利用RNA-seq,对大肠杆菌F17菌毛非腹泻型与腹泻型绵羊个体脾脏中差异表达lncRNA进行筛选,并基于顺式(cis)机制进行靶基因预测以及功能注释分析,筛选出关键lncRNA。在此基础上利用qPCR进行验证。本研究从lncRNA层面上,加深了对绵羊非腹泻组大肠杆菌F17菌毛的认识,同时有望确定绵羊非腹泻型大肠杆菌F17菌毛的功能基因,解决中国地方羊品种对大肠杆菌病的抗病育种关键问题,为今后制订抗大肠杆菌病遗传选育策略奠定基础和提供理论依据。1 材料与方法
1.1 试验设计和样本采集
试验用绵羊于2016年12月购自泰州市西来原生态农业有限公司。随机选择生长发育良好、日龄体重相近的18只3日龄羔羊,并将羔羊全部隔离饲养。先用10%羔羊奶粉(表1)饲喂,以确保在试验前适应饮食需要。5日龄时饲喂12.5%羔羊奶粉和大肠杆菌F17菌液(4.6×108 CFU/mL),同时保证自由饮水。每天记录羔羊粪便形态(表2),在有一些羔羊持续腹泻2 d后,将羔羊分为非腹泻组和腹泻组,并对羊进行安乐死。用4%多聚甲醛收集肠道组织。用RNAlater收集每只羔羊的肝脏、脾脏、十二指肠、空肠和回肠,并立即在液氮中冷冻以进行RNA提取。Table 1
表1
表1奶粉成分(每100克奶粉)
Table 1
| 成分 Ingredient | 水分 Moisture | 粗蛋白 Crude protein | 粗脂肪 Crude fat | 粗纤维 Crude fiber | 粗灰分 Coarse ash | 钙 Calcium | 总磷 Total phosphorus | 赖氨酸 Lysine |
|---|---|---|---|---|---|---|---|---|
| 比例 Proportion | ≤6 | ≥22 | ≥18 | ≤5 | ≤8 | 0.9-3.0 | ≥0.5 | ≥1.0 |
新窗口打开|下载CSV
Table 2
表2
表2粪便形式量[20]
Table 2
| 形态 Form | 描述 Describe |
|---|---|
| 第1种形态 Type 1 | 分散的干球便,如坚果,很难排出 Separate hard lumps, like nuts |
| 第2种形态Type 2 | 香肠状,多块的 Sausage-shaped but lumpy |
| 第3种形态 Type 3 | 腊肠样,但表面有裂缝Like a sausage or snake but with cracks on its surface |
| 第4种形态Type 4 | 腊肠状或蛇状,表面光滑 Like a sausage or snake, smooth and soft |
| 第5种形态Type 5 | 柔软成块,边缘清楚,容易排出Soft blobs with clear-cut edges |
| 第6种形态Type 6 | 软片状,边缘毛糙或是糊状便 Fluffy pieces with ragged edges, a mushy stool |
| 第7种形态 Type 7 | 水样便,无团状成分 Watery, no solid pieces |
新窗口打开|下载CSV
1.2 H.E.染色
用0.9%盐水洗涤空肠组织,并在室温下用4%多聚甲醛固定48 h,然后进行组织学分析。接下来,用苏木精-伊红染色7 μm切片,并在显微镜下观察空肠上皮形态。1.3 文库建设和测序
从每组中的3只绵羊脾脏提取总RNA,并使用NanoDrop 2000超微量分光光度计和Agilent 2100生物分析仪进行质控。用Ribo-Zero TM试剂盒(Epicenter, Madison, WI, USA)除去核糖体RNA。将RNA片段化(平均长度约为200 bp),然后通过逆转录合成和纯化cDNA。使用Qubit?dsDNA HS测定试剂盒进行PCR扩增和纯化后,选择使用NEBNext?Ultra?RNA文库制备试剂盒进行文库构建。在上海欧易生物医学科技有限公司使用Illumina HiSeq 2500平台,对文库进行末端配对测序(测序读长为150 bp)。1.4 鉴定lncRNA和mRNA
过滤原始数据以消除低质量读长,使用映射到参照基因组(Ovis aries v4.0)的干净读长进行组装。用CPC[13]、CNCI[14]、Pfam[15]、PLEK[16]四种编码潜能分析方法,将来自未知转录物中推定的编码RNA和非编码RNA候选物进行分类。将最小长度和外显子数设置为阈值,滤出推定的编码RNA,长度超过200 nt的两个外显子的转录物被选为lncRNA候选物。使用cuffcompare归类不同类型的lncRNA,主要包括intergenic lncRNA(字符为u),intronic lncRNA(字符为i), anti-sense lncRNA(字符为x),sense- overlapping lncRNA(字符为o)。1.5 差异表达分析
由于FPKM法[17]同时考虑了测序深度和转录本长度对fragments计数的影响,因此使用FPKM值(fragments Per kb per Million reads,是每百万fragments中来自某一基因每千碱基长度的fragments数目)估计lncRNA和mRNA转录物的表达水平。使用DESeq[18]软件检测两组之间DE基因的数量和FPKM值。在利用RNA-seq数据比较分析两个样品中同一个转录本是否存在差异表达的时候,选取两个标准:一是FoldChange,就是两样品中同一个转录本表达水平的变化倍数;二是P-value或FDR(padjust),FDR值的计算方法先要对每个转录本计算P-value,再用FDR错误控制法对P-value作多重假设检验校正。默认筛选差异的条件为P<0.05且| log2(倍数变化)|>1。1.6 GO和KEGG通路分析
得到差异表达转录本之后,对差异表达转录本进行GO富集分析,对其功能进行描述(结合GO注释结果)。统计每个GO条目中所包括的差异转录本个数,并用Fisher's exact test计算每个GO条目中差异转录本富集的显著性(默认筛选差异的条件为P<0.05)。KEGG[19]是有关通路的主要公共数据库,利用KEGG数据库对差异转录本进行通路分析(结合KEGG注释结果),并用Fisher's exact test计算每个通路条目中差异转录本富集的显著性(默认筛选差异的条件为P<0.05),通路分析对试验结果有提示作用,通过差异转录本的通路分析,可以找到富集差异转录本的通路条目,寻找不同样品的差异转录本可能和哪些细胞通路的改变有关。1.7 预测DE lncRNA的靶基因
通过计算多个基因之间的Pearson相关系数和P值预测DE lncRNA的靶基因。使用|corelation|≥0.7并且P≤0.05来过滤转录物,选择与黏附功能相关通路的DE lncRNA,并通过顺式和反式作用预测所有DE lncRNA的靶基因。1.8 验证DE lncRNA的表达水平
为验证筛选的DE lncRNA在非腹泻组过程中发挥作用,用q-PCR检测了非腹泻组和腹泻组羔羊脾脏组织中一共12种DE lncRNA和DE mRNA的表达水平,使用2-ΔΔCt方法将每个RNA的相对定量归一化为GAPDH,lncRNA的引物见表3。Table 3
表3
表3GAPDH,DE lncRNA和mRNA的引物
Table 3
| 基因符号Gene symbol | 引物序列Primer sequence | 产物长度Product length (bp) | |
|---|---|---|---|
| 10个DE mRNA的引物Primers of ten DE mRNA | XM_015103189.1 | F:AGCACTTCCTCCTGTCCG | 132 |
| R:CAGCACAGAAGGCAAAGTC | |||
| XM_012106626.2 | F:CCGAGTTTGCAGGTACCCAAC | 121 | |
| R:TTTTGGCGCATGTATACCTG | |||
| XM_012095567.2 | F:TGAGACTCTACTTCGCTGC | 102 | |
| R:TTGCCCATCCTTAATAGCTG | |||
| XM_012181407.2 | F:ACGACGGTGGTTAAATACTC | 125 | |
| R:AGTTGCCCATAGTCACTGGTC | |||
| XM_012181919.2 | F:TCAACCATATGCTGACGGAC | 119 | |
| R:ATGCCGCCTATCAAGGTC | |||
| XM_012152506.2 | F:CCACCTGCGGTTCAAGTTAC | 157 | |
| R:TGCCTGAATCACCTTGTC | |||
| 10个DE lncRNA的引物 Primers of ten DE lncRNA | TCONS_00053949 | F:CCTGGCTATATCCTTACATCAC | 100 |
| R:AAGTTCAAACTCCGCTGCAC | |||
| TCONS_00059692 | F:AATTTCTTCTCGTTCCAAGGC | 131 | |
| R:CCAACAGGGAGCCAACTTC | |||
| TCONS_00089751 | F:AGAAGGCTTTGACCGAAC | 153 | |
| R:TCAATGCCCTCCACGAGAC | |||
| TCONS_00099925 | F:AGTGCCACATGTACCTAGCAG | 146 | |
| R:ACGACAGGCATTTTAACCCATG | |||
| TCONS_00107469 | F:TAGTACAGCCCATATTTATCG | 115 | |
| R:ATTTTCTTTCCACAGGGACG | |||
| TCONS_00099739 | F:CCGACGCTGTCATGATGC | 159 | |
| R:TCCGTCTCCAGAACCAAGGC | |||
| GAPDH的引物 Primers of GAPDH | GAPDH-F | F:GTTCCACGGCACAGTCAAGG | 127 |
| R:ACTCAGCACCAGCATCACCC |
新窗口打开|下载CSV
1.9 统计分析
使用SPSS软件(版本20.0)分析所有数据,使用单因素方差分析(ANOVA)分析差异转录本的相对表达量,并使用Tukey检验进行多重比较。P<0.05被认为具有统计学意义。每组包含3个样品,每个试验重复3次。2 结果
2.1 非腹泻组和腹泻组肠道细菌数量比较及组织学形态观察
根据粪便形态[20],将试验个体分为非腹泻组(12、13、14p)与腹泻组(15、16、17p)。在腹泻组中,细菌计数在4.7×108至1.9×109之间,但在非腹泻组中细菌计数下降到了5.1×106至9.0×107。与非腹泻组相比,腹泻组羔羊肠道细菌数量明显较高(P<0.05,表4,图1),腹泻组羔羊空肠黏膜组织出现不同程度的损伤,色泽暗沉,细胞死亡、裂解,黏膜下层留下较大间隙,肠绒毛部分脱落,肠道黏膜中丰富的毛细血管都被极大的破坏,难以在切片中找到相应结构(图2)。图1
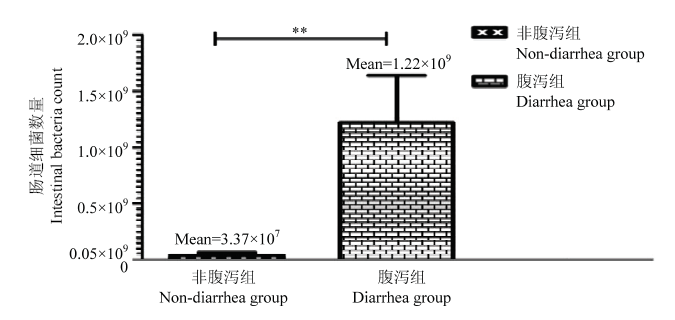 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1非腹泻组与腹泻羔羊肠道细菌数量对比
Fig. 1Comparison of intestinal bacteria count between non-diarrhea group and diarrhea lamb
Table 4
表4
表4非腹泻组与腹泻羔羊肠道细菌数量对比
Table 4
| 组别 Group | 肠道 Intestinal tract | 稀释倍数 Dilution multiple | 细菌数量 Count of bacterial (CFU/mL) | |||
|---|---|---|---|---|---|---|
| 105 | 106 | 107 | 108 | |||
| 非腹泻组 Non-diarrhea group | 十二指肠 Duodenum | 70 | 6 | NG | NG | 6.0×106 |
| 空肠 Jejunum | 51 | NG | NG | NG | 5.1×106 | |
| 回肠 Ileum | >500 | 170 | 9 | NG | 9.0×107 | |
| 腹泻组 Diarrhea group | 十二指肠 Duodenum | >1000 | >500 | 128 | 13 | 1.3×109 |
| 空肠 Jejunum | >1000 | 272 | 47 | NG | 4.7×108 | |
| 回肠 Ileum | >1000 | >500 | 176 | 19 | 1.9×109 | |
新窗口打开|下载CSV
图2
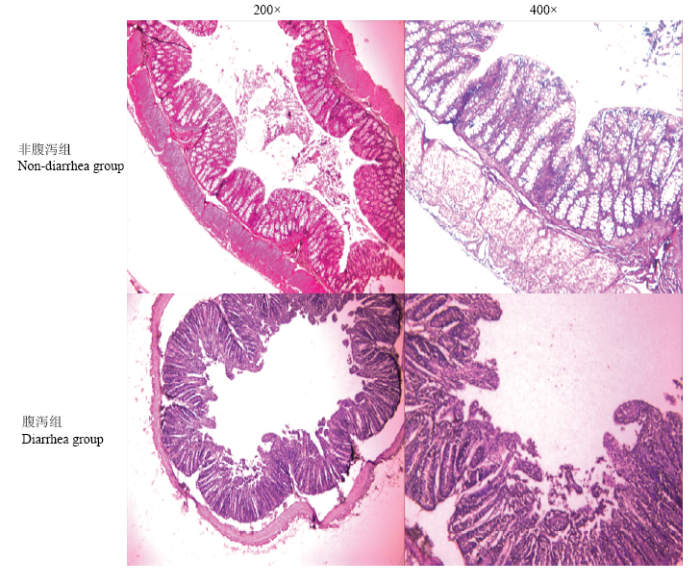 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2非腹泻组和腹泻组羔羊空肠组织学形态观察
Fig. 2Histological observation of jejunum in lambs of non-diarrhea group and diarrhea group
2.2 绵羊脾脏RNA测序概述
构建非腹泻组和腹泻组羔羊脾脏的cDNA文库,使用Illumina HiSeq 2500平台进行测序,分别产生了354 943 820和370 616 990个原始读长。文库的GC含量分别为48.33%和49.67%。将清洁读长中的有效读长映射到ovis aries v4.0参考基因组,超过73.5%的读长被映射到基因组。映射到参考序列多个位置的序列数目低于4.5%,超过70%的读长被唯一映射到参考序列。大约35%的读长映射到基因组的正、负链。此外,通过注释分析映射到外显子区域(约60%)的读长数量高于基因间和内含子区域。这些结果表明,匹配效率很高,大多数读长映射到外显子区域。2.3 绵羊脾脏中转录本的鉴定
在绘制参考序列后,我们从42 460个汇编的转录物中鉴定出已知的1 988个lncRNA和38 843个mRNA。lncRNA长度主要分布于200—5 000 bp,平均长度为2 124 bp。此外,lncRNA主要为intergenic lncRNA(字符为u),intronic lncRNA(字符为i),包含2—3个外显子(图3)。图3
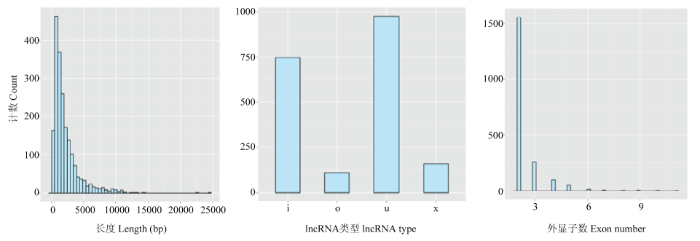 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3lncRNA长度、类型、预测lncRNA的外显子数目统计
Fig. 3lncRNA length, type, predicting the number of exons in lncRNA
2.4 分析和验证差异转录本
利用FPKM值估计lncRNA和mRNA转录物的表达水平,其中lncRNA转录本表达水平相对较低(图4)。图4
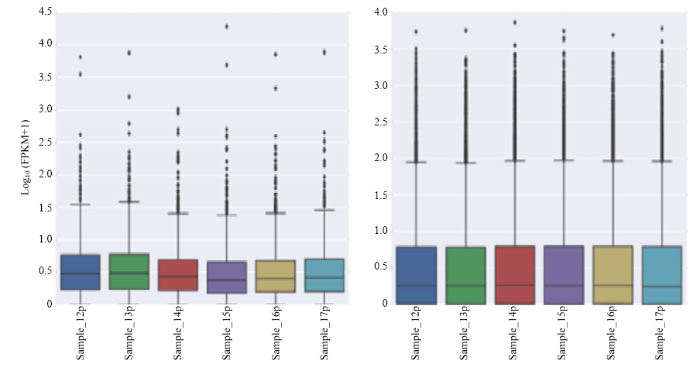 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4lncRNA(左)和mRNA(右)转录本表达水平箱线图
Fig. 4LncRNA (left ) and mRNA (right) transcript expression level box plot
在P<0.05和| log2(倍数变化)|>1的条件下,筛选出14个上调和20个下调的DE lncRNA,370个上调和333个下调的DE mRNA(图5)。
图5
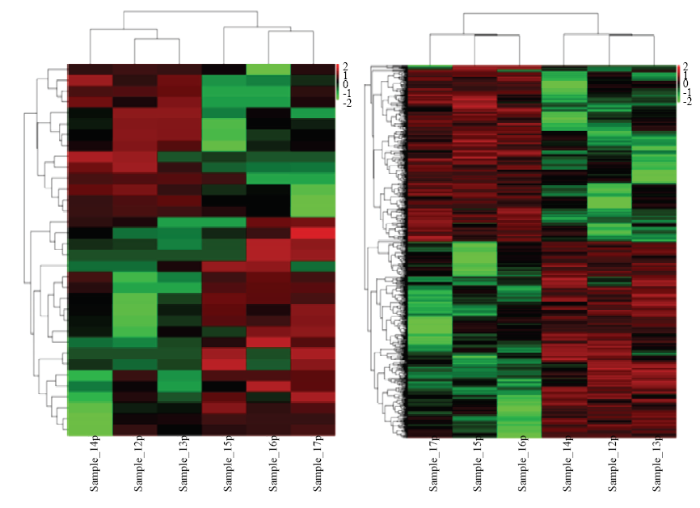 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5非腹泻组和腹泻组型羔羊之间差异表达的lncRNAs(左)和mRNAs(右)
Fig. 5Differentially expressed lncRNAs (left ) and mRNAs (right) between non-diarrhea and diarrhea lambs
为了进一步验证RNA-seq的可靠性,随机选择一共12个DE lncRNA和DE mRNA,用q-PCR验证它们在非腹泻组和腹泻组羔羊体内的相对表达水平,发现与RNA-seq结果一致(图6),表明RNA-seq数据是可靠的。 这些分析还表明,高通量测序具有检测低表达水平(0<FPKM<1)基因的优点。
图6
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图6DE lncRNA和mRNA在非腹泻组和腹泻组羔羊体内的相对表达水平
Fig. 6Relative expression levels of DE lncRNA and mRNA in non-diarrhea and diarrhea lambs
2.5 DE lncRNA的GO功能注释和KEGG 通路富集分析
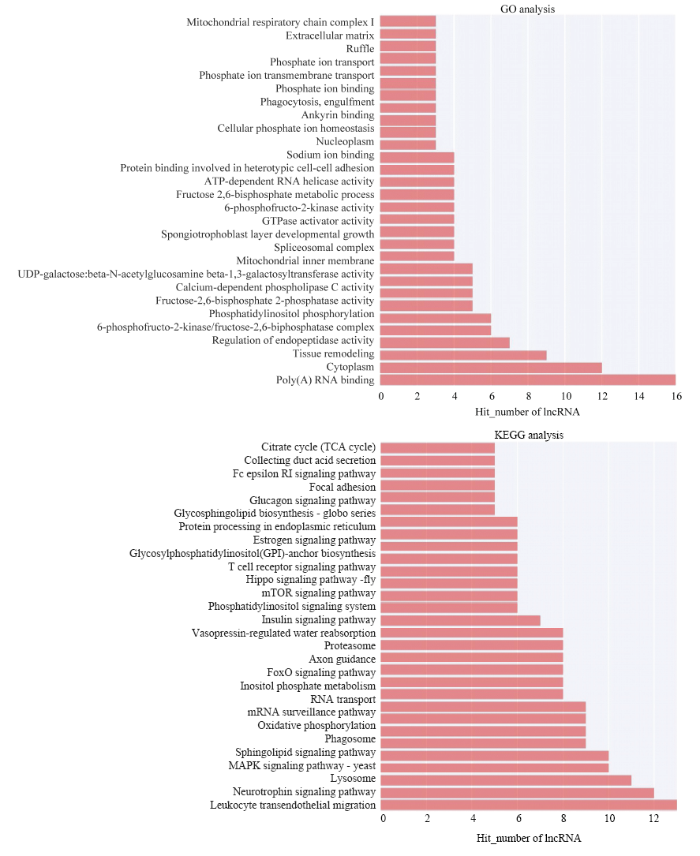
在“lncRNA名称--功能预测Term”之间的对应关系中(附件),分别选取预测可信度(按P-value排序)最高的Top 500个预测关系,对其中各个功能预测Term进行频次计数,统计功能注释较多的GO(或通路)term,反映该试验中得到的差异lncRNAs功能分布的整体情况(图7)。图7
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图7lncRNA的GO功能注释KEGG 通路富集分析
Fig. 7Gene Ontology and KEGG pathway enrichment analysis of DE lncRNAs
DE lncRNA与GO 数据库进行比对的结果表明,一共有34条lncRNA被注释和分类到302个功能亚类中。图7显示了DE lncRNA数量排名前30 位的功能亚类。结果显示,绵羊蛋白质结合(GO:0005515)、细胞核(GO:0005634)、poly(A)RNA结合(GO:0044822)、细胞质(GO:0005737)、组织重塑(GO:0048771)、内肽酶活性的调节(GO:0052548))、6-磷酸果糖-2-激酶/果糖-2,6-双磷酸酶复合物(GO:0043540)、磷脂酰肌醇磷酸化(GO:0046854)、果糖-2,6-二磷酸2-磷酸酶活性(GO:0004331)、钙依赖性磷脂酶C活性(GO:0050429)等10 个功能亚类的lncRNA较多,而其余的功能亚类的lncRNA分布较少。
DE lncRNA与KEGG 通路 数据库进行比对的结果表明,一共有34条lncRNA被注释和归类到149个KEGG 通路中。图7显示了DE lncRNA数量排名前30 位的KEGG 通路。结果显示,绵羊甲状腺激素信号通路(路径:ko04919)、Spliceosome(路径:ko03040)、白细胞跨内皮迁移(路径:ko04670)、神经营养因子信号通路(路径:ko04722)、溶酶体(路径:ko04142)、MAPK信号通路-途径(路径:ko04011)、鞘脂信号通路(路径:ko04071)、吞噬体(路径:ko04145)、氧化磷酸化(路径:ko00190)等9 个KEGG 通路的lncRNA较多,而其余的KEGG 通路的lncRNA分布较少。
2.6 LncRNAs与其临近编码基因的基因组共表达结果
对于感兴趣的DE lncRNAs,笔者搜索其上下游100 kb范围内的所有编码基因,并与该lncRNA有显著共表达(皮尔森相关性计算)的基因取交集。这些在基因组上临近且表达模式上存在共表达的基因很可能被该lncRNA所调控。因此,发现6个基因可能被该相关lncRNA所调控(表5)。Table 5
表5
表5差异lncRNAs的cis调控
Table 5
| lncRNA | 表达趋势 Regulation | mRNA | 基因 Gene | 表达趋势 Regulation | 染色体编号 Chrom | Pearson P值 P value | Pearson 相关系数Correlation | 顺距 cis distance |
|---|---|---|---|---|---|---|---|---|
| TCONS_00053949 | Down | XM_012176963.2 | MYO1G | Up | NC_019461.2 | 0.034 | -0.845 | 1670 |
| TCONS_00059692 | Up | XM_015095635.1 | TIMM29 | Down | NC_019462.2 | 0.01 | -0.916 | 16037 |
| TCONS_00059692 | Up | XM_015095835.1 | CARM1 | Down | NC_019462.2 | 0.033 | -0.848 | 22513 |
| TCONS_00089751 | Up | XM_015097644.1 | ADGRB1 | Up | NC_019466.2 | 0.0027 | 0.957 | 8560 |
| TCONS_00099925 | Down | XM_015098457.1 | SEPT4 | Up | NC_019468.2 | 0.049 | -0.812 | 98788 |
| TCONS_00107469 | Up | XM_012187185.1 | DESI2 | Up | NC_019469.2 | 0.0026 | 0.958 | 94714 |
新窗口打开|下载CSV
3 讨论
由于转录组分析的快速发展,lncRNAs在过去几年作为细胞发育中的新型调节剂受到广泛关注[21]。目前已经鉴定的lncRNAs主要与癌症相关,如前列腺癌[22]、胃癌[23]、肺癌[24]以及乳腺癌[25]等,以及与生殖功能相关的lncRNA[26,27,28,29]。然而,迄今为止,关于羔羊腹泻的lncRNA,特别是绵羊的研究报道很少。湖羊是一种具有高繁殖力和对湿热气候适应性较强的中国特有品种,可以全年在室内饲养。在这项研究中,不仅提供了绵羊腹泻过程中lncRNA的第一个概况,还研究了lncRNA在抗病过程中的可能作用。长期以来,羔羊腹泻对牧场造成了严重的经济损失。在研究中,发现lncRNA的表达水平低于mRNA,与绵羊睾丸组织一致[30],并且lncRNAs和mRNAs的平均长度比猪更长(分别为1 713和1 983 bp[27]。搜索其上下游100 kb范围内的所有编码基因,并与该lncRNAs有显著共表达(皮尔森相关性计算)的基因取交集,发现6个与lncRNA共表达的基因:MYO1G(肌球蛋白IG)、TIMM29(线粒体内膜的转位酶29)、CARM1(共激活因子相关的精氨酸甲基转移酶1)、ADGRB1(黏附G蛋白偶联受体B1)、SEPT4(septin 4)、DESI2(脱嘌呤的肽酶2)。
MYO1G在维持B细胞淋巴细胞的细胞刚度(cell stiffness)方面扮演了很重要的角色,MYO1G的缺失降低细胞刚度,影响细胞黏附,扩散,吞噬作用和B细胞淋巴细胞的內吞过程[31]。关于TIMM29的研究报道比较少,最近研究报道TIMM29被鉴定为哺乳动物TIMM22蛋白复合物第一个特异性组分,同时其在TIMM23蛋白的组装过程中扮演重要角色[32,33]。CARM1,是蛋白质精氨酸甲基转移酶家族(protein arginine methyltransferase,PRMTs)的成员之一,含 有高度保守结构域的具有甲基转移酶活性的酶。CARM1 敲除的小鼠出生时就死亡[34],这表明CARM1为产后生存所必须。后来研究还发现,CARM1抑制能够促进HIV-1活化[35]。ADGRB1属于跨膜蛋白家族——黏附G蛋白偶联受体(aGPCRs)的成员之一,aGPCR家族的定义特征是保守的GAIN结构域,具有自体蛋白水解活性并且可以切割第一跨膜结构域附近的受体,研究发现通过GAIN结构域切割显示的新的N末端茎可以直接激活作为束缚激动剂的ADGRB1[36]。Septins是一个具有GTP 酶活性的高度保守的细胞骨架蛋白家族,肿瘤抑制因子SEPT4是Septins 家族成员之一,其可诱导癌细胞凋亡[37]。研究发现,SEPT4 基因突变的小鼠精子环及其邻近的皮质结构紊乱,引起精子活力低下甚至不运动,最终导致不育症的发生[38,39]。DESI2 基因是一种促凋亡基因,体外试验表明DESI2基因过表达可诱导胰腺癌等肿瘤细胞凋亡,能有效抑制部分癌细胞增殖,其与IP10的组合基因治疗能显著抑制肿瘤生长,并有效延长了患肿瘤小鼠的存活时间[40,41]。
总共703个mRNA和34个已知的lncRNA在非腹泻组和腹泻组之间显着差异表达,其中14个上调和20个下调DE lncRNA。另外,确定了两组中共有1 942个新的lncRNA。搜索lncRNA上下游100 kb范围内的所有编码基因,并与该lncRNA有显著共表达(皮尔森相关性计算)的基因取交集,发现6个基因可能被该相关lncRNA所调控。为了进一步验证RNA-Seq结果,采用q-PCR验证12种已知的lncRNA和mRNA的表达水平,结果一致。
GO是一种广泛用于研究基因功能关系的生物信息学工具。对34个DE lncRNA进行GO分析,在Top 500的预测关系对中,有16个DE lncRNA富集到蛋白结合(GO:0005515)条目。KEGG 通路分析表明,信号通路如Sphingolipid signaling 通路(path:ko04071)、Axon guidance(path:ko04360)、Glycosylphosphatidylinositol (GPI)- anchor biosynthesis(path:ko00563)可能是DE lncRNA共表达基因的重要KEGG途径,相关的lncRNA可能潜在地参与菌毛黏附肠道黏膜的过程。然而,这些通路在抗病过程中的作用很大程度上仍然是未知的。
4 结论
研究了对于腹泻产生非腹泻型和腹泻型羔羊脾脏中lncRNA的表达谱,以进一步了解其在绵羊抗病发生过程中的调控作用。发现了非腹泻组和腹泻组羔羊脾脏组织中差异表达的lncRNA。有助于找出羔羊如何抵抗腹泻的发生机制。 此外,进一步研究这些lncRNA可以为羔羊抵抗腹泻提供科学的依据。(责任编辑 林鉴非)
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1385/0-89603-480-1:425URLPMID:315911 [本文引用: 1]

rskov, Ida (Statens Seruminstitut, Copenhagen, Denmark), and Frits rskov. Episome-carried surface antigen K88 of Escherichia coli. I. Transmission of the determinant of the K88 antigen and influence on the transfer of chromosomal markers. J. Bacteriol. 91:69-75. 1966.-The transmission of the determinant of the Escherichia coli K88 antigen in mixed cultures of E. coli strains is described. The K88 factor could not be transferred by filtrates, nor could responsible phages or colicines be detected. Acriflavine was shown to "cure" the bacteria for the K88 antigen. Generally, the strains having acquired the K88 antigen also acquired the ability to transfer chromosomal markers, but this ability was in some cases retained by segregants which had lost the K88 antigen. Introduction into an F(+) strain caused reduction of the recombination frequency and disappearance of the f(+) antigen. Not all wild-type strains with the K88 antigen are genetic donors of this antigen, at least not to a discernible degree. It was concluded that the K88 antigen determinant is carried by an episome.
[本文引用: 1]
DOI:10.1093/jnci/dju505URLPMID:4402359 [本文引用: 1]
The functions of long noncoding RNAs (lncRNAs) have been identified in several cancers, but the roles of lncRNAs in colorectal cancer (CRC) are less well understood. The transcription factor MYC is known to regulate lncRNAs and has been implicated in cancer cell proliferation and tumorigenesis. CRC cells and tissues were profiled to identify lncRNAs differentially expressed in CRC, from which we further selected MYC-regulated lncRNAs. We used luciferase promoter assay, ChIP, RNA pull-down assay, deletion mapping assay, LC-MS/MS and RNA immunoprecipitation to determine the mechanisms of MYC regulation of lncRNAs. Moreover, soft agar assay and in vivo xenograft experiments (four athymic nude mice per group) provided evidence of MYC-regulated lncRNAs in cancer cell transformation and tumorigenesis. The Kaplan-Meier method was used for survival analyses. All statistical tests were two-sided. We identified lncRNAs differentially expressed in CRC (P < .05, greater than two-fold) and verified four lncRNAs upregulated and two downregulated in CRC cells and tissues. We further identified MYC-regulated lncRNAs, named MYCLos. The MYC-regulated MYCLos may function in cell proliferation and cell cycle by regulating MYC target genes such as CDKN1A (p21) and CDKN2B (p15), suggesting new regulatory mechanisms of MYC-repressed target genes through lncRNAs. RNA binding proteins including HuR and hnRNPK are involved in the function of MYCLos by interacting with MYCLo-1 and MYCLo-2, respectively. Knockdown experiments also showed that MYCLo-2, differentially expressed not only in CRC but also in prostate cancer, has a role in cancer transformation and tumorigenesis. Our results provide novel regulatory mechanisms in MYC function through lncRNAs and new potential lncRNA targets of CRC.
DOI:10.1016/j.chom.2014.10.001URLPMID:25525793 [本文引用: 1]
Ouyang et聽al. identify NRAV, a functional long noncoding RNA that negatively regulates the initial transcription of multiple critical interferon-stimulated antiviral genes, and can promote influenza virus replication and virulence. Host cells dramatically reduce NRAV levels during viral infection. These findings position lncRNAs as modulators of antiviral innate immunity.
DOI:10.4161/rna.29937URLPMID:4615744 [本文引用: 1]
The innate immune system is the first line of defense against microbial pathogens, but tight regulation of gene expression is necessary to prevent the detrimental effects of unrestrained activation. Although the functions of most long noncoding RNAs (lncRNAs; >200 nucleotides) are unknown, many have been shown to regulate diverse cellular activities. Recent reports by us and others have suggested that lncRNAs may also play critical roles in transcriptional regulation of gene expression during innate immune responses. Following engagement of Toll-like receptors, lncRNAs form functional RNA090009protein complexes that recruit activators or remove repressors of transcription, leading to rapid expression of inflammatory mediators. These discoveries suggest that lncRNAs may contribute to the gene regulatory networks that govern host090009pathogen interactions.
DOI:10.1038/ni.2887URLPMID:24840979 [本文引用: 1]
Abstract The rapid changes in gene expression that accompany developmental transitions, stress responses and proliferation are controlled by signal-mediated coordination of transcriptional and post-transcriptional mechanisms. In recent years, understanding of the mechanics of these processes and the contexts in which they are employed during hematopoiesis and immune challenge has increased. An important aspect of this progress is recognition of the importance of RNA-binding proteins and noncoding RNAs. These have roles in the development and function of the immune system and in pathogen life cycles, and they represent an important aspect of intracellular immunity.
DOI:10.1016/j.it.2014.07.005URLPMID:25113636 [本文引用: 1]
It is increasingly clear that long non-coding RNAs (IncRNAs) regulate a variety biological responses, and that they do so by a diverse range of mechanisms. In the field of immunology, recent publications have shown widespread changes in the expression of IncRNAs during the activation of the innate immune response and T cell development, differentiation, and activation. These IncRNAs control important aspects of immunity such as production of inflammatory mediators, differentiation, and cell migration through regulating protein-protein interactions or via their ability to basepair with RNA and DNA. We review the current understanding of the mechanism of action of these immune-related IncRNAs, discuss their impact on physiological and pathological processes, and highlight important areas of inquiry at the intersection between immunology and IncRNA biology.
DOI:10.3390/genes8100283URLPMID:5664133 [本文引用: 1]
Inherited retinal diseases (IRDs) are often associated with variable clinical expressivity (VE) and incomplete penetrance (IP). Underlying mechanisms may include environmental, epigenetic, and genetic factors.Cis-acting expression quantitative trait loci (cis-eQTLs) can be implicated in the regulation of genes by favoring or hampering the expression of one allele over the other. Thus, the presence of such loci elicits allelic expression imbalance (AEI) that can be traced by massive parallel sequencing techniques. In this study, we performed an AEI analysis on RNA-sequencing (RNA-seq) data, from 52 healthy retina donors, that identified 194 imbalanced single nucleotide polymorphisms(SNPs) in 67 IRD genes. Focusing on SNPs displaying AEI at a frequency higher than 10%, we found evidence of AEI in several IRD genes regularly associated with IP and VE (BEST1,RP1,PROM1, andPRPH2). Based on these SNPs commonly undergoing AEI, we performed pyrosequencing in an independent sample set of 17 healthy retina donors in order to confirm our findings. Indeed, we were able to validateCDHR1,BEST1, andPROM1to be subjected tocis-acting regulation. With this work, we aim to shed light on differentially expressed alleles in the human retina transcriptome that, in the context of autosomal dominant IRD cases, could help to explain IP or VE.
DOI:10.1038/s41598-017-05443-5URLPMID:5507887 [本文引用: 1]
Abstract Spermatogenesis can be affected by nutrition, which operates through normal physiological processes by changing the testicular mass and hormone levels profoundly. However, little is known regarding how testis development is regulated by long noncoding RNA (lncRNA). In this study, we investigated the effects of high-grain (HG) feeding on testis development during sexual maturation mediated by lncRNA. The HG diet group showed an increase in growth hormone (GH), insulin-like growth factor-1 (IGF-1) and testosterone (T) levels, and in the number of sperm in the seminiferous tubules compared with the hay-fed group (p < 0.05). Moreover, we found 59 differentially expressed (DE) lncRNAs and 229 DE mRNAs in sheep testis between the two groups. qRT-PCR results of 20 randomly selected DE lncRNAs and mRNAs were also consistent with the RNA-seq data. Through functional enrichment analysis and lncRNA-mRNA interaction network analysis, we screened several lncRNAs that may be enriched for male reproduction such as spermatogenesis, sperm motility, steroid hormones, MAPK and ErbB signaling pathways. This study provides a first insight into the development of the testis with HG feeding in sheep and shows that these changes are associated with alterations in lncRNA expression.
.
DOI:10.1371/journal.pone.0156890URLPMID:27276011 [本文引用: 1]
Initiation of hair follicle (HF) is the first and most important stage of HF morphogenesis. However the precise molecular mechanism of initiation of hair follicle remains elusive. Meanwhile, in previous study, the more attentions had been paid to the function of genes, while the roles of non-coding RNAs (such as long noncoding RNA and microRNA) had not been described. Therefore, the roles of long noncoding RNA(LncRNA) and coding RNA in sheep skin during the initiation of sheep secondary HF were integrated and analyzed, by using strand-specific RNA sequencing (ssRNA-seq).A total of 192 significant differentially expressed genes were detected, including 67 up-regulated genes and 125 down-regulated genes between stage 0 and stage 1 of HF morphogenesis during HF initiation. OnlyWnt2,FGF20were just significant differentially expressed among Wnt, Shh, Notch and BMP signaling pathways. Further expression profile analysis of lncRNAs showed that 884 novel lncRNAs were discovered in sheep skin expression profiles. A total of 15 lncRNAs with significant differential expression were detected, 6 up-regulated and 9 down-regulated. Among of differentially expressed genes and LncRNA, XLOC002437 lncRNA and potential target geneCOL6A6were all significantly down-regulated in stage 1. Furthermore, by using RNAhybrid, XLOC005698 may be as a competing endogenous RNA ponges oar-miR-3955-5p activity. Gene Ontology and KEGG pathway analyses indicated that the significantly enriched pathway was peroxisome proliferator-activated receptors (PPARs) pathway (corrected P-value < 0.05), indicating that PPAR pathway is likely to play significant roles during the initiation of secondary HF.Results suggest that the key differentially expressed genes and LncRNAs may be considered as potential candidate genes for further study on the molecular mechanisms of HF initiation, as well as supplying some potential values for understanding human hair disorders.
DOI:10.3969/j.issn.1673-4556.2017.04.109URL [本文引用: 1]
羊大肠杆菌病是规模养羊场最为常见高发的细菌性疾病之一,传统沿用的抗生素治疗方案存在诸多缺陷.基于对致病原生物学特性及流行病学特点深入研究的基础上,采取早期准确诊断及科学防治措施,在药物防治上达到理想效果,为业内防治羊大肠杆菌病提供借鉴.
DOI:10.3969/j.issn.1673-4556.2017.04.109URL [本文引用: 1]
羊大肠杆菌病是规模养羊场最为常见高发的细菌性疾病之一,传统沿用的抗生素治疗方案存在诸多缺陷.基于对致病原生物学特性及流行病学特点深入研究的基础上,采取早期准确诊断及科学防治措施,在药物防治上达到理想效果,为业内防治羊大肠杆菌病提供借鉴.
DOI:10.3969/J.ISSN.1671-6027.2017.06.071URL [本文引用: 1]
羊大肠杆菌病也称为新生羔羊腹泻病,是一种传染性极强的病症。大肠杆菌是羔羊体内常在的条件致病菌,当外界环境发生突变时极易引发大肠杆菌病,应加以重视。
DOI:10.3969/J.ISSN.1671-6027.2017.06.071URL [本文引用: 1]
羊大肠杆菌病也称为新生羔羊腹泻病,是一种传染性极强的病症。大肠杆菌是羔羊体内常在的条件致病菌,当外界环境发生突变时极易引发大肠杆菌病,应加以重视。
DOI:10.1093/nar/gkm391URLPMID:17631615 [本文引用: 1]
Recent transcriptome studies have revealed that a large number of transcripts in mammals and other organisms do not encode proteins but function as noncoding RNAs (ncRNAs) instead. As millions of transcripts are generated by large-scale cDNA and EST sequencing projects every year, there is a need for automatic methods to distinguish protein-coding RNAs from noncoding RNAs accurately and quickly. We developed a support vector machine-based classifier, named Coding Potential Calculator (CPC), to assess the protein-coding potential of a transcript based on six biologically meaningful sequence features. Tenfold cross-validation on the training dataset and further testing on several large datasets showed that CPC can discriminate coding from noncoding transcripts with high accuracy. Furthermore, CPC also runs an order-of-magnitude faster than a previous state-of-the-art tool and has higher accuracy. We developed a user-friendly web-based interface of CPC at http://cpc.cbi.pku.edu.cn. In addition to predicting the coding potential of the input transcripts, the CPC web server also graphically displays detailed sequence features and additional annotations of the transcript that may facilitate users' further investigation.
DOI:10.1093/nar/gkt646URLPMID:3783192123979317936517431704729 [本文引用: 1]
It is a challenge to classify protein-coding or non-coding transcripts, especially those re-constructed from high-throughput sequencing data of poorly annotated species. This study developed and evaluated a powerful signature tool, Coding-Non-Coding Index (CNCI), by profiling adjoining nucleotide triplets to effectively distinguish protein-coding and non-coding sequences independent of known annotations. CNCI is effective for classifying incomplete transcripts and sense鈥揳ntisense pairs. The implementation of CNCI offered highly accurate classification of transcripts assembled from whole-transcriptome sequencing data in a cross-species manner, that demonstrated gene evolutionary divergence between vertebrates, and invertebrates, or between plants, and provided a long non-coding RNA catalog of orangutan. CNCI software is available athttp://www.bioinfo.org/software/cnci.
DOI:10.1093/nar/gkt1223URLPMID:3965110 [本文引用: 1]
Pfam, available via servers in the UK (http://pfam.sanger.ac.uk/) and the USA (http://pfam.janelia.org/), is a widely used database of protein families, containing 14 831 manually curated entries in the current release, version 27.0. Since the last update article 2 years ago, we have generated 1182 new families and maintained sequence coverage of the UniProt Knowledgebase (UniProtKB) at nearly 80%, despite a 50% increase in the size of the underlying sequence database. Since our 2012 article describing Pfam, we have also undertaken a comprehensive review of the features that are provided by Pfam over and above the basic family data. For each feature, we determined the relevance, computational burden, usage statistics and the functionality of the feature in a website context. As a consequence of this review, we have removed some features, enhanced others and developed new ones to meet the changing demands of computational biology. Here, we describe the changes to Pfam content. Notably, we now provide family alignments based on four different representative proteome sequence data sets and a new interactive DNA search interface. We also discuss the mapping between Pfam and known 3D structures.
DOI:10.1186/1471-2105-15-311URLPMID:4177586 [本文引用: 1]
Background High-throughput transcriptome sequencing (RNA-seq) technology promises to discover novel protein-coding and non-coding transcripts, particularly the identification of long non-coding RNAs (lncRNAs) from de novo sequencing data. This requires tools that are not restricted by prior gene annotations, genomic sequences and high-quality sequencing. Results We present an alignment-free tool called PLEK (predictor of long non-coding RNAs and messenger RNAs based on an improved k-mer scheme), which uses a computational pipeline based on an improved k-mer scheme and a support vector machine (SVM) algorithm to distinguish lncRNAs from messenger RNAs (mRNAs), in the absence of genomic sequences or annotations. The performance of PLEK was evaluated on well-annotated mRNA and lncRNA transcripts. 10-fold cross-validation tests on human RefSeq mRNAs and GENCODE lncRNAs indicated that our tool could achieve accuracy of up to 95.6%. We demonstrated the utility of PLEK on transcripts from other vertebrates using the model built from human datasets. PLEK attained >90% accuracy on most of these datasets. PLEK also performed well using a simulated dataset and two real de novo assembled transcriptome datasets (sequenced by PacBio and 454 platforms) with relatively high indel sequencing errors. In addition, PLEK is approximately eightfold faster than a newly developed alignment-free tool, named Coding-Non-Coding Index (CNCI), and 244 times faster than the most popular alignment-based tool, Coding Potential Calculator (CPC), in a single-threading running manner. Conclusions PLEK is an efficient alignment-free computational tool to distinguish lncRNAs from mRNAs in RNA-seq transcriptomes of species lacking reference genomes. PLEK is especially suitable for PacBio or 454 sequencing data and large-scale transcriptome data. Its open-source software can be freely downloaded from https://sourceforge.net/projects/plek/files/ webcite.
DOI:10.1038/nbt.1621URLPMID:20436464 [本文引用: 1]
High-throughput mRNA sequencing (RNA-Seq) promises simultaneous transcript discovery and abundance estimation. However, this would require algorithms that are not restricted by prior gene annotations and that account for alternative transcription and splicing. Here we introduce such algorithms in an open-source software program called Cufflinks. To test Cufflinks, we sequenced and analyzed >430 million paired 75-bp RNA-Seq reads from a mouse myoblast cell line over a differentiation time series. We detected 13,692 known transcripts and 3,724 previously unannotated ones, 62% of which are supported by independent expression data or by homologous genes in other species. Over the time series, 330 genes showed complete switches in the dominant transcription start site (TSS) or splice isoform, and we observed more subtle shifts in 1,304 other genes. These results suggest that Cufflinks can illuminate the substantial regulatory flexibility and complexity in even this well-studied model of muscle development and that it can improve transcriptome-based genome annotation.
DOI:10.1186/gb-2010-11-10-r106URLPMID:3218662 [本文引用: 1]
High-throughput sequencing assays such as RNA-Seq, ChIP-Seq or barcode counting provide quantitative readouts in the form of count data. To infer differential signal in such data correctly and with good statistical power, estimation of data variability throughout the dynamic range and a suitable error model are required. We propose a method based on the negative binomial distribution, with variance and mean linked by local regression and present an implementation, DESeq, as an R/Bioconductor package.
DOI:10.1093/nar/gkm882URLPMID:18077471 [本文引用: 1]
KEGG (http://www.genome.jp/kegg/) is a database of biological systems that integrates genomic, chemical and systemic functional information. KEGG provides a reference knowledge base for linking genomes to life through the process of PATHWAY mapping, which is to map, for example, a genomic or transcriptomic content of genes to KEGG reference pathways to infer systemic behaviors of the cell or the organism. In addition, KEGG provides a reference knowledge base for linking genomes to the environment, such as for the analysis of drug-target relationships, through the process of BRITE mapping. KEGG BRITE is an ontology database representing functional hierarchies of various biological objects, including molecules, cells, organisms, diseases and drugs, as well as relationships among them. KEGG PATHWAY is now supplemented with a new global map of metabolic pathways, which is essentially a combined map of about 120 existing pathway maps. In addition, smaller pathway modules are defined and stored in KEGG MODULE that also contains other functional units and complexes. The KEGG resource is being expanded to suit the needs for practical applications. KEGG DRUG contains all approved drugs in the US and Japan, and KEGG DISEASE is a new database linking disease genes, pathways, drugs and diagnostic markers.
DOI:10.3109/00365529709011203URL [本文引用: 3]
[本文引用: 1]
DOI:10.1016/j.cca.2017.08.009URL [本文引用: 1]
DOI:10.1177/1010428317699798URLPMID:28618943 [本文引用: 1]
Abstract Gastric cancer remains the third leading cause of cancer-related mortality worldwide, and proliferation of gastric cancer represents the major reason for its poor prognosis. Recent evidence indicates that long non-coding RNAs play crucial roles in development and progression of gastric cancer. Long non-coding RNA differentiation antagonizing non-protein coding RNA is upregulated in hepatic cell carcinoma, but the role of lncRNA differentiation antagonizing non-protein coding RNA in gastric cancer has not been explored. In this article, we found that differentiation antagonizing non-protein coding RNA is also upregulated in gastric cancer. Experiments revealed that silencing differentiation antagonizing non-protein coding RNA significantly inhibited gastric cancer cell proliferation in vitro and in vivo. Overexpression of differentiation antagonizing non-protein coding RNA notably increases gastric cancer cell proliferation. From RNA-seq and gene ontology annotations, we found that differentiation antagonizing non-protein coding RNA influences the gene expression programs in cell metabolic and cycle process. Taken together, our findings suggest that the long non-coding RNA differentiation antagonizing non-protein coding RNA promotes the proliferation of gastric cancer and is a potential prognostic biomarker and therapeutic target in gastric cancer.
DOI:10.1038/s41598-017-09818-6URLPMID:5570922 [本文引用: 1]
react-text: 462 Environmental risks of organic chemicals have been greatly determined by their persistence, bioaccumulation, and toxicity (PBT) and physicochemical properties. Major regulations in different countries and regions identify chemicals according to their bioconcentration factor (BCF) and octanol–water partition coefficient (Kow), which frequently displays a substantial correlation with the... /react-text react-text: 463 /react-text [Show full abstract]
DOI:10.1002/1878-0261.12133URLPMID:28846832 [本文引用: 1]
react-text: 359 Histone deacetylase 6 (HDAC6) is a member of class IIb HDAC family. HDAC6 exists predominantly in the cytoplasm and deacetylates mainly non-histone proteins in the cytoplasm. Via its deacetylase and ubiquitin binding domains, HDAC6 regulates microtubules, cytoskeleton, intracellular trafficking, and cellular responses to stress. HDAC6 plays a central role in physiology and pathobiology in... /react-text react-text: 360 /react-text [Show full abstract]
DOI:10.1002/mrd.22650URLPMID:27111572 [本文引用: 1]
SUMMARY Spermatogenesis is regulated by many meiotic stage-specific genes, but how they coordinate the many individual processes is not fully understood. The Prss/Tessp gene cluster is located on mouse chromosome 9F2-F3, and the three genes at this site ( Prss42/Tessp-2 , Prss43/Tessp-3 , and Prss44/Tessp-4 ) are specifically activated during meiosis in pachytene spermatocytes. We searched for DNase I hypersensitive sites (HSs) and long noncoding RNAs (lncRNAs) at the Prss/Tessp locus to elucidate how they are activated. We found eight DNase I HSs, three of which were testis germ cell-specific at or close to the Prss42/Tessp-2 promoter, and a testis-specific lncRNA, lncRNA-HSVIII , that was transcribed from a region adjacent to the Prss42/Tessp-2 gene. lncRNA-HSVIII transcripts localized to nuclei of most pachytene spermatocytes and the cytosol of stage-X pachytene spermatocytes and spermatids. Chromosome conformation capture revealed that the lncRNA-HSVIII locus specifically interacted with the Prss42/Tessp-2 promoter in primary and secondary spermatocytes. A 5.8-kb genome sequence, encompassing the entire lncRNA-HSVIII sequence and its flanking regions, significantly increased Prss42/Tessp-2 promoter activity using a reporter-gene assay, yet this construct did not change lncRNA-HSVIII expression, indicating that the elevated promoter activity was likely through enhancer activity. Indeed, both upstream and downstream regions of the lncRNA-HSVIII sequence significantly increased Prss42/Tessp-2 promoter activity. Our data therefore identified the direct interaction of a genomic region in the lncRNA-HSVIII locus with the Prss42/Tessp-2 promoter in spermatocytes, and suggested that sequences adjacent to the lncRNA function as enhancers for the Prss42/Tessp-2 gene. Mol. Reprod. Dev. 83: 541 557, 2016. 2016 Wiley Periodicals, Inc .
DOI:10.1095/biolreprod.115.136911URLPMID:26935596 [本文引用: 2]
Abstract Thousands of long non-coding RNAs (lncRNAs) are identified in mouse, rat and human testes, some of which play important roles in testis development and spermatogenesis. However, systematic analysis of lncRNAs expressed in postnatal pig testes has not been reported. Thus, in this study, we presented the expression and characterization of lncRNAs in immature (30-day-old, D30) and mature (180-day-old, D180) pig testes. A total of 90,440,168 (85.75%) and 97,001,700 (95.35%) 150 bp paired-end clean reads were generated in D30 and D180 cDNA libraries using the Illumina HiSeq 4000 platform, respectively. 36,727 transcripts were assembled in those two libraries. 777 lncRNA transcripts from 752 lncRNA gene loci were identified using the highly stringent pipeline, and 101 of those lncRNA transcripts were significantly differentially expressed. Those lncRNAs shared some characteristics with other mammals, which included fewer exons, shorter length and exon length, and lower expression level compared with those of protein-coding genes. 402 protein-coding genes (10 kb) were found as nearest neighbors of 294 out of 752 lncRNA genes, and the Gene ontology (GO) enrichment showed that they were enriched in transcription and development related processes. Fifteen differentially expressed and ten novel lncRNAs were randomly selected and validated by qPCR and RT-PCR. In addition, one of the ten novel lncRNAs was further confirmed using RACE clone technology. This study provided a catalog of porcine testes lncRNAs for further understanding their regulation roles in pig testis development and spermatogenesis. Copyright 2016 by The Society for the Study of Reproduction.
[本文引用: 1]
DOI:10.1101/gr.132159.111URLPMID:22955988 [本文引用: 1]
The human genome contains many thousands of long noncoding RNAs (lncRNAs). While several studies have demonstrated compelling biological and disease roles for individual examples, analytical and experimental approaches to investigate these genes have been hampered by the lack of comprehensive lncRNA annotation. Here, we present and analyze the most complete human lncRNA annotation to date, produced by the GENCODE consortium within the framework of the ENCODE project and comprising 9277 manually annotated genes producing 14,880 transcripts. Our analyses indicate that lncRNAs are generated through pathways similar to that of protein-coding genes, with similar histone-modification profiles, splicing signals, and exon/intron lengths. In contrast to protein-coding genes, however, lncRNAs display a striking bias toward two-exon transcripts, they are predominantly localized in the chromatin and nucleus, and a fraction appear to be preferentially processed into small RNAs. They are under stronger selective pressure than neutrally evolving sequences-particularly in their promoter regions, which display levels of selection comparable to protein-coding genes. Importantly, about one-third seem to have arisen within the primate lineage. Comprehensive analysis of their expression in multiple human organs and brain regions shows that lncRNAs are generally lower expressed than protein-coding genes, and display more tissue-specific expression patterns, with a large fraction of tissue-specific lncRNAs expressed in the brain. Expression correlation analysis indicates that lncRNAs show particularly striking positive correlation with the expression of antisense coding genes. This GENCODE annotation represents a valuable resource for future studies of lncRNAs.
.
[本文引用: 1]
DOI:10.1002/cm.21299URLPMID:27106882 [本文引用: 1]
B-lymphocytes are migrating cells that specialize in antigen presentation, antibody secretion, and endocytosis; these processes implicate the modulation of plasma membrane elasticity. Cell stiffness is a force generated by the interaction between the actin-cytoskeleton and the plasma membrane, which requires the participation of several proteins. These proteins include class I myosins, which are now considered to play a role in controlling membrane–cytoskeleton interactions. In this study, we identified the motor protein Myosin 1g (Myo1g) as a mediator of this phenomenon. The absence of Myo1g decreased the cell stiffness, affecting cell adhesion, cell spreading, phagocytosis, and endocytosis in B-lymphocytes. The results described here reveal a novel molecular mechanism by which Myo1g mediates and regulates cell stiffness in B-lymphocytes. 08 2016 Wiley Periodicals, Inc.
DOI:10.1002/1873-3468.12450URLPMID:5215392 [本文引用: 1]
Hydrophobic inner mitochondrial membrane proteins with internal targeting signals, such as the metabolite carriers, use the carrier translocase (TIM22 complex) for transport into the inner membrane. Defects in this transport pathway have been associated with neurodegenerative disorders. While the TIM22 complex is well studied in baker's yeast, very little is known about the mammalian TIM22 complex. Using immunoprecipitation, we purified the human carrier translocase and identified a mitochondrial inner membrane protein TIM29 as a novel component, specific to metazoa. We show that TIM29 is a constituent of the 440 kDa TIM22 complex and interacts with oxidized TIM22. Our analyses demonstrate that TIM29 is required for the structural integrity of the TIM22 complex and for import of substrate proteins by the carrier translocase.
DOI:10.7554/eLife.17463URLPMID:27554484 [本文引用: 1]
10.7554/eLife.17463.001The TIM22 complex mediates the import of hydrophobic carrier proteins into the mitochondrial inner membrane. While the TIM22 machinery has been well characterised in yeast, the human complex remains poorly characterised. Here, we identify Tim29 (C19orf52) as a novel, metazoan-specific subunit of the human TIM22 complex. The protein is integrated into the mitochondrial inner membrane with it???s C-terminus exposed to the intermembrane space. Tim29 is required for the stability of the TIM22 complex and functions in the assembly of hTim22. Furthermore, Tim29 contacts the Translocase of the Outer Mitochondrial Membrane, TOM complex, enabling a mechanism for transport of hydrophobic carrier substrates across the aqueous intermembrane space. Identification of Tim29 highlights the significance of analysing mitochondrial import systems across phylogenetic boundaries, which can reveal novel components and mechanisms in higher organisms.DOI: http://dx.doi.org/10.7554/eLife.17463.001
DOI:10.1073/pnas.1232272100URL [本文引用: 1]
DOI:10.1093/nar/gkx550URLPMID:28637181 [本文引用: 1]
Abstract In eukaryotic cells, the gene expression status is strictly controlled by epigenetic modifications on chromatin. The repressive status of chromatin largely contributes to HIV latency. Studies have shown that modification of histone H3K27 acts as a key molecular switch for activation or suppression of many cellular genes. In this study, we found that K27-acetylated histone H3 specifically recruited Super Elongation Complex (SEC), the transcriptional elongation complex essential for HIV-1 long terminal repeat (LTR)-mediated and general cellular transcription. Interestingly, H3K27 acetylation further stimulates H3R26 methylation, which subsequently abrogates the recruitment of SEC, forming a negative feedback regulatory loop. Importantly, by inhibiting methyltransferase activity of CARM1, the enzyme responsible for H3R26 methylation, HIV-1 transcription is reactivated in several HIV latency cell models, including a primary resting CD4+ T cell model. When combined with other latency disrupting compounds such as JQ1 or vorinostat/SAHA, the CARM1 inhibitor achieved synergistic effects on HIV-1 activation. This study suggests that coordinated and dynamic modifications at histone H3K27 and H3R26 orchestrate HIV-1 LTR-mediated transcription, and potentially opens a new avenue to disrupt latent HIV-1 infection by targeting specific epigenetic enzymes. The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
DOI:10.1074/jbc.M115.689349URLPMID:26710850 [本文引用: 1]
The adhesion (aGPCRs) are a large yet poorly understood family of seven-transmembrane . A defining characteristic of the aGPCR family is the conserved GAIN domain, which has autoproteolytic activity and can cleave the receptors near the first transmembrane domain. Several aGPCRs, including ADGRB1 (or B1) and ADGRG1 (or G1), have been found to exhibit significantly increased constitutive activity when truncated to mimic GAIN domain cleavage ("NT"). Recent reports have suggested that the new N-terminal stalk, which is revealed by GAIN domain cleavage, can directly activate aGPCRs as a tethered agonist. We tested this hypothesis in studies on two distinct aGPCRs, B1 and G1, by engineering mutant receptors lacking the entire NT including the stalk (B1- and G1-SL, with "SL" indicating "stalkless"). These receptors were evaluated in a battery of assays and compared to full-length WT and cleavage-mimicking (NT) forms of the two receptors. We found that B1-SL, in multiple assays, exhibited robust activity, suggesting that the -proximal stalk region is not necessary for its activation. For G1, however, the results were mixed, with the SL mutant exhibiting robust activity in several assays (including TGF伪 shedding, activation of NFAT and beta-arrestin recruitment) but reduced activity relative to NT in a distinct assay (activation of ). These data support a model in which the activation of certain pathways downstream of aGPCRs is stalk-dependent, whereas to other pathways is stalk-independent.
DOI:10.3109/02656736.2015.1131338URLPMID:26794618 [本文引用: 1]
Introduction to the special issue dedicated to the 30th Annual Meeting of the European Society for Hyperthermic Oncology – ESHO 2015. . ???aop.label???. doi: 10.3109/02656736.2015.1131338
DOI:10.1016/j.devcel.2004.12.005URLPMID:15737930 [本文引用: 1]
Septins are polymerizing GTP binding proteins required for cortical organization during cytokinesis and other cellular processes. A mammalian septin gene 61/61 male mice are sterile due to defective morphology and motility of the sperm flagellum. In Sept4 null spermatozoa, the annulus is replaced by a fragile segment lacking cortical material, beneath which kinesin-mediated intraflagellar transport stalls. The sterility is rescued by injection of sperm into oocytes, demonstrating that each Sept4 null spermatozoon carries an intact haploid genome. The annulus/septin ring is also disorganized in spermatozoa from a subset of human patients with asthenospermia syndrome. Thus, cortical organization based on circular assembly of the septin cytoskeleton is essential for the structural and mechanical integrity of mammalian spermatozoa.
DOI:10.1016/j.devcel.2005.01.021URLPMID:15737931 [本文引用: 1]
The murine septin4 gene ( Sept4) has been implicated in diverse cellular functions, including cytokinesis, apoptosis, and tumor suppression. Here, we investigated the function of Sept4 proteins during mouse development by creating a targeted deletion of the Sept4 genomic locus. Sept4 mutant mice are viable but male sterile due to immotile and structurally defective sperm. During spermatogenesis, Sept4 proteins were essential for proper mitochondrial architecture and establishment of the annulus, a ring-like structure in the tail region of sperm. In addition, Sept4 mutant sperm showed defects in the elimination of residual cytoplasm during sperm maturation and had increased staining for the caspase inhibitor XIAP. This is consistent with a role of the proapoptotic Sept4 protein ARTS in promoting caspase-mediated removal of cytoplasm via inhibition of XIAP. Our results indicate that Sept4 proteins play distinct but evolutionarily conserved functions in different cellular compartments.
DOI:10.1007/s12253-014-9817-3URLPMID:25079376 [本文引用: 1]
Abstract Desumoylating isopeptidase 2 (DESI2) is a recently identified protein with unclear functions. In this study, a total of 132 tissue samples of pancreatic ductal adenocarcinoma and 73 samples of pancreatic normal tissues were explored to assess DESI2 expression and its implications to AKT/mTOR signal. Immunohistochemistry showed DESI2 expression is significantly decreased in cancer tissues versus normal tissues, presenting lowest level in poorly differentiated cancer. Unlike DESI2, the key factors in AKT/mTOR pathway including p-AKT, mTOR, p-mTOR and p-P70S6K present high expression in pancreatic cancer. It is notable that p-mTOR is significantly increased in DESI2-lower cancer compared with DESI2-higher cancer, although mTOR presents no difference in the two groups. The relative p-mTOR/mTOR ratio is also significantly elevated in DESI2-lower cancer. Moreover, the samples whose p-AKT and p-mTOR scores both exceed the median are obviously increased in DESI2-lower cancer compared with DESI2-higher cancer. As a downstream molecule of AKT/mTOR pathway, p-P70S6K was found to display higher level in DESI2-lower pancreatic cancer. High phosphorylation status of those proteins in DESI2-reduced pancreatic cancer indicates that there is high activity of AKT/mTOR signal in condition of DESI2 reduction, which could provide clues to reveal the implications of DESI2 in carcinogenesis.
DOI:10.18632/oncotarget.17623URLPMID:28915590 [本文引用: 1]
Abstract DESI2 (also known as PNAS-4) is a novel pro-apoptotic gene activated during the early response to DNA damage. We previously reported that overexpression of DESI2 induces S phase arrest and apoptosis by activating checkpoint kinases. The present study was designed to test whether combination of DESI2 and IP10 could improve the therapy efficacy in vitro and in vivo . The recombinant plasmid co-expressing DESI2 and IP10 was encapsulated with DOTAP/Cholesterol nanoparticle. Immunocompetent mice bearing CT26 colon carcinoma and LL2 lung cancer were treated with the complex. We found that, in vitro , the combination of DESI2 and IP10 more efficiently inhibited proliferation of CT26, LL2, SKOV3 and A549 cancer cells via apoptosis. In vivo , the combined gene therapy more significantly inhibited tumor growth and efficiently prolonged the survival of tumor bearing mice. Mechanistically, the augmented antitumor activity in vivo was associated with induction of apoptosis and inhibition of angiogenesis. The anti-angiogenesis was further mimicked by inhibiting proliferation of immortalized HUVEC cells in vitro . Meanwhile, the infiltration of lymphocytes also contributed to the enhanced antitumor effects. Depletion of CD8+ T lymphocytes significantly abrogated the antitumor activity, whereas depletion of CD4+ T cells or NK cells showed partial abrogation. Our data suggest that the combined gene therapy of DESI2 and IP10 can significantly enhance the antitumor activity as apoptosis inducer, angiogenesis inhibitor and immune response initiator. The present study may provide a novel and effective method for treating cancer.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}