 , 张雪海
, 张雪海Genome-wide Association Studies of Seed Germination Related Traits in Maize
TIANRun-Miao, ZHANGXue-Hai通讯作者:
收稿日期:2017-11-2
接受日期:2018-03-15
网络出版日期:2018-03-16
版权声明:2018作物学报编辑部作物学报编辑部
基金资助:
作者简介:
-->
展开
摘要
关键词:
Abstract
Keywords:
-->0
PDF (4410KB)元数据多维度评价相关文章收藏文章
本文引用格式导出EndNoteRisBibtex收藏本文-->
随着玉米机械化收获程度的提高, 生产上对玉米种子田间出苗率和出苗整齐度的要求越来越高。种子的高效萌发是种子出苗的前提[1]。大量生产实践证明不同玉米自交系的发芽率和出苗率均存在广泛的遗传变异[2,3], 解析玉米种子出苗能力的遗传基础, 发掘玉米萌发相关性状的优良等位基因, 对玉米产量的提高及农业机械化收获具有重要意义。
全基因组关联分析(genome-wide association studies, GWAS)是研究数量性状的重要方法之一, 最早用于人类疾病研究[4]。近年来, 随着新一代测序技术的发展, GWAS在植物重要性状遗传机制研究中的应用越来越多。Atwell等[5]在拟南芥中首次证明应用GWAS的可行性, 并指出此方法也适用于其他植物。玉米具有丰富的遗传变异和快速的连锁不平衡衰减, 是遗传学研究的重要模式作物。近十年来, 以玉米为材料的GWAS取得了很大进展[7]。以本研究所用关联群体为例, Li等[8]用1.03万个SNP标记对籽粒含油量进行GWAS, 共检测出74个显著位点。Liu等[9]通过整合简化基因组测序、高密度SNP芯片及深度RNA测序数据, 开发一套包含1.25M SNP的标记(MAF≥AFP), 并以此重新检测了关联群体的籽粒含油量, 检测到13个新的显著位点。而利用GWAS对种子萌发相关性状进行遗传分析的报道相对较少。Huang等[10]用5.6万个SNP标记对125个玉米自交系在种子萌发阶段和幼苗期的耐冷性进行GWAS, 共检测到43个显著位点。Shi等[11]利用GWAS对水稻种子在盐胁迫条件下的7个萌发相关性状进行分析, 检测到11个主效位点。Kan等[12]用大豆种子萌发阶段耐盐性的GWAS检测到8个显著SNP。
种子出苗受多种因素影响, 如种子萌发能力、种子活力等。种子萌发包含吸胀和代谢过程的起始、吸水滞后期、进一步吸水后胚根出现3个阶段[13]。目前的研究主要集中在种子萌发的第二和第三阶段。Fu等[14]利用蛋白质组学方法获得了玉米种子萌发第二阶段的杂种优势相关差异表达蛋白。Hu等[15]对玉米种子低温条件下萌发能力的QTL分析, 鉴定出6个种子发芽率的QTL及6个胚根长度的QTL。Han等[16]在人工老化的条件下对玉米种子的发芽率、发芽势、幼苗干重及幼苗根部干重的QTL分析, 共检测到65个QTL, 结合代谢组mQTL分析, 共鉴定出23个候选基因, 这些基因通过调控糖酵解通路和蛋白质代谢进而影响种子的萌发。种子吸胀作为种子休眠结束和种子萌发第一阶段, 对种子的正常萌发具有重要意义, 但相关的遗传研究很少。Farzaneh等[17]利用反向遗传学方法找到一些在拟南芥种子吸胀过程中差异表达的基因, 这些基因的缺失突变体表现出与种子萌发和休眠相关的表型。Li等[18]研究水稻OSCA家族基因时, 发现6个与种子吸胀有关的基因。Noblet等[19]在玉米种子低温吸胀过程中的细胞膜磷脂重组的研究中发现, 玉米种子中饱和脂肪酸与不饱和脂肪酸的差异积累与种子吸胀过程中对低温的敏感性有关。目前有关玉米种子吸胀相关性状的QTL研究尚无报道。为了解玉米种子吸胀相关性状的遗传机制, 本研究对476份玉米自交系种子吸胀前后及吸胀程度的6个相关性状进行测定, 利用1.25M的SNP标记(http://www. maizego.org/Resources.html)对这些性状进行GWAS, 以挖掘影响种子萌发的相关候选基因, 并为相应基因的克隆及功能标记的开发奠定基础。
1 材料与方法
1.1 试验设计与性状调查
476份玉米自交系来自温带、热带及亚热带(http://www.maizego.org/Resources.html), 于2017年春季在室内进行发芽实验, 共3个生物学重复, 每个生物学重复3次抽样测定, 每次抽样均由20粒大小一致的健康种子构成。种子浸泡前, 分别测定每次抽样的20粒干种子的重量(W1)和排水体积(V1); 种子吸胀24 h后(温度26℃, 湿度60%), 用滤纸拭干种子表面的水分, 再次测定每次抽样的重量(W2)和排水体积(V2); 每次抽样的吸胀重量W3和吸胀体积V3分别由W3=W2-W1和V3=V2-V1公式计算得到。以每个生物学重复3次抽样的平均值作为该重复的值, 以每个材料3个生物学重复的平均值作为该性状的值。对每个生物学重复及其平均值分别进行GWAS。测定完种子性状后, 继续培养(室内发芽盒中土培)以确保种子萌发出苗。1.2 统计分析
使用SPSS 23.0软件对各表型数据进行双因素方差分析(two-way ANOVA)、皮尔逊相关性分析(Pearson correlation analysis)及正态性检验(normality test)。1.3 全基因组关联分析
1.25M SNP基因型数据从Maizego网站获得(http://www.maizego.org/Resources.html), 在TASSEL3.0软件中, 分别用只控制群体结构的Q模型、只控制亲缘关系的K模型, 以及同时控制群体结构和亲缘关系的Q+K模型实现关联分析。根据Quantile- Quantile 散点图(QQ plot)对每一个性状在3种模型下的结果进行比较, 并选择最优模型下的GWAS结果作为后续QTL分析的基础。每个性状的曼哈顿图和QQ plot均利用R完成(R Core Team 2012, http:// www.R-project.org/)。以P=2.04×10-6 (1/Ne, Ne为GEC软件计算出的有效标记数490 547)作为显著关联SNP的阈值[20]。1.4 候选基因的筛选
由于该关联群体10条染色体的平均连锁不平衡衰减(LD)距离为30 kb[9], 本研究以显著SNP上下游各延伸30 kb的区间定义为一个QTL, 当相邻QTL物理区间有重叠时(B73 v2版本), 则认为是同一个QTL。以每个QTL内最显著SNP所在基因或邻近基因作为该QTL的首要候选基因, 利用MaizeGDB、NCBI及TAIR网站对QTL内的候选基因进行功能注释和分析。2 结果与分析
2.1 种子萌发相关性状的统计分析
6个性状均表现广泛的遗传变异, 其中, 吸胀前体积(V1)的变异系数最小为17.35%, 吸胀体积的变异系数最大为28.41% (表1)。所有性状的频次分布 (图1)、峰度和偏度(绝对值均小于1)都表现出明显的正态分布特征, 说明这6个性状均是由微效多基因控制的典型数量性状。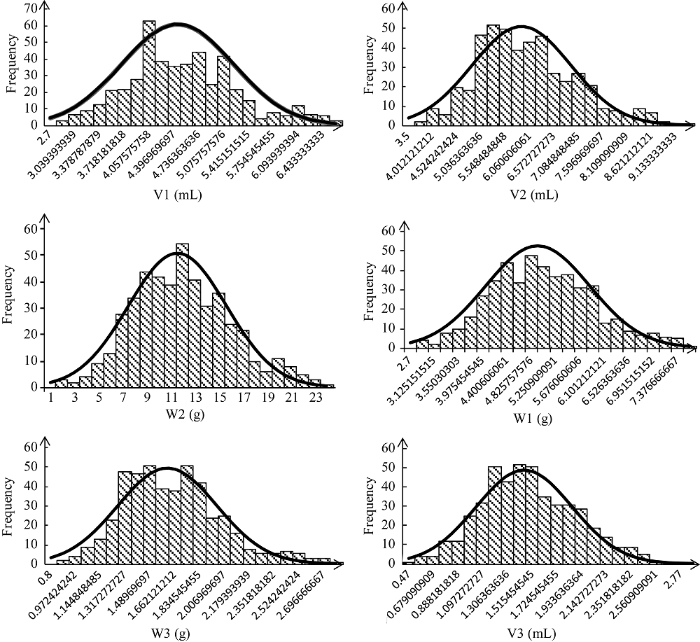 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1种子萌发性状的频次分布图
各性状详细名称见
-->Fig. 1Frequency distribution of seed emergence traits
Name of each trait is given in
-->
Table 1
表1
表1种子萌发性状表型统计分析
Table 1Statistical analysis of seed germination traits
| 性状 Trait | 平均值±标准差 Mean± SD | 变幅 Range | 变异系数 CV (%) | 峰度 Skewness | 偏度 Kurtosis |
|---|---|---|---|---|---|
| W1 (g) | 4.92±0.92 | 1.63-7.40 | 18.77 | 0.22 | 0.07 |
| V1 (mL) | 4.43±0.77 | 2.57-6.47 | 17.35 | 0.42 | -0.03 |
| W2 (g) | 6.52±1.19 | 2.02-9.86 | 18.29 | 0.25 | 0.23 |
| V2 (mL) | 5.87±1.06 | 1.93-9.20 | 18.06 | 0.43 | 0.33 |
| W3 (g) | 1.61±0.35 | 0.38-2.72 | 21.77 | 0.55 | 0.54 |
| V3 (mL) | 1.44±0.41 | 0.35-2.76 | 28.41 | 0.25 | -0.13 |
新窗口打开
方差分析和相关性分析结果表明, 6个性状在不同材料间均存在极显著差异(表2), 且6个性状两两之间均极显著相关(表3), 说明这些性状彼此影响, 且基因型的差异是导致其性状差异的主要原因, 环境因素对性状的表现影响较小。
Table 2
表2
表2种子萌发性状方差分析
Table 2ANOVA of seed germination traits
| 性状 Trait | 变异来源 Variation source | 自由度 df | 均方 MS | F值 F-value |
|---|---|---|---|---|
| W1 | 材料Variety | 475 | 2.56 | 52.99** |
| 重复Repeat | 2 | 0.11 | 2.27 | |
| 误差Error | 940 | 0.05 | ||
| V1 | 材料Variety | 475 | 1.77 | 14.50** |
| 重复Repeat | 2 | 0.96 | 7.85** | |
| 误差Error | 941 | 0.12 | ||
| W2 | 材料Variety | 475 | 4.24 | 32.12** |
| 重复Repeat | 2 | 4.31 | 32.68** | |
| 误差Error | 940 | 0.13 | ||
| V2 | 材料Variety | 475 | 3.35 | 19.35** |
| 重复Repeat | 2 | 0.33 | 1.93 | |
| 误差Error | 93 | 0.17 | ||
| W3 | 材料Variety | 475 | 0.36 | 8.28** |
| 重复Repeat | 2 | 4.02 | 92.23** | |
| 误差Error | 934 | 0.04 | ||
| V3 | 材料Variety | 475 | 0.50 | 2.62** |
| 重复Repeat | 2 | 1.32 | 6.96** | |
| 误差Error | 932 | 0.19 |
新窗口打开
Table 3
表3
表3种子萌发性状皮尔逊关联分析
Table 3Pearson correlation analysis of seed germination traits
| 性状 Trait | W1 | V1 | W2 | V2 | W3 |
|---|---|---|---|---|---|
| V1 | 0.955** | ||||
| W2 | 0.976** | 0.956** | |||
| V2 | 0.935** | 0.942** | 0.966** | ||
| W3 | 0.675** | 0.725** | 0.821** | 0.811** | |
| V3 | 0.606** | 0.543** | 0.682** | 0.793** | 0.724** |
新窗口打开
2.2 关联分析最优模型选择
从3种模型的Q-Q图可以看出(图2), Q模型的假阳性最高(产生更多的I型错误)。对吸胀前重量、吸胀前体积、吸胀后重量、吸胀后体积和吸胀体积性状而言, K模型的拟合效果最好, 即-lg (P)的真实值最接近预测值, 可以很好地控制假阳性, 因此, 选择K模型的GWAS结果对这5个性状进行QTL解析; 对吸胀重量性状而言, Q+K模型拟合效果最好。因此, 选择Q+K模型作为吸胀重量性状GWAS的最终模型。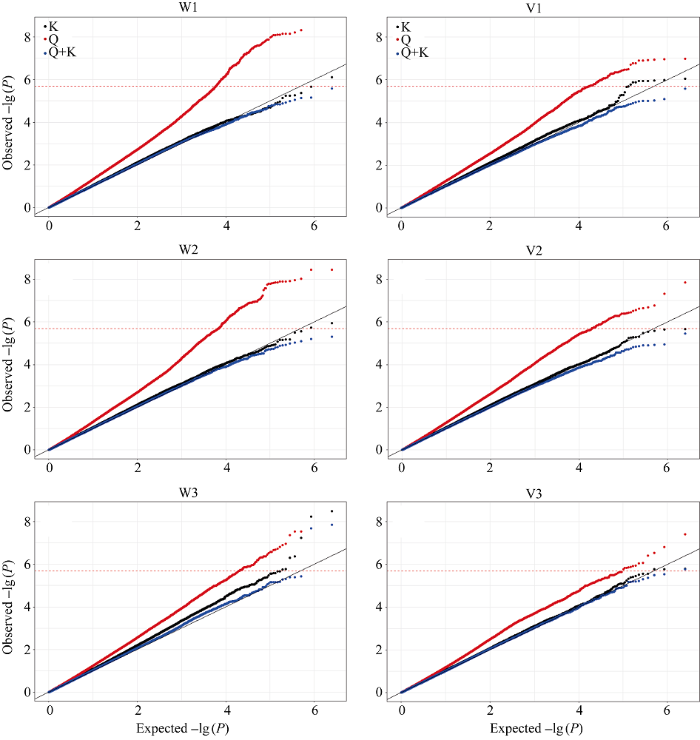 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2种子萌发性状3种模型比较的QQ图
各性状详细名称见
-->Fig. 2Quantile-quantile plots (QQ plots) of estimated -lg(P) from association analysis using three methods for seed emergence traits
Detailed name of each trait was given in
-->
2.3 全基因组关联分析与候选基因筛选
基于最优模型, 在阈值为P≤2.04×10-6 (1/Ne, 1/490 547)时, 共检测到与6个性状显著关联的15个SNP, 分布在玉米第3、第6、第7、第10染色体上(图3和表4)。15个SNP对应6个QTL, 其中, 第7染色体上有2个QTL, 分别影响吸胀前重量(Chr7.S_4828028所在QTL)和吸胀体积(Chr7.S_145970024所在QTL), 可分别解释5.84%和5.13%的表型变异; 第10染色体上的QTL (Chr10.S_6373105所在QTL), 可同时影响吸胀后重量、吸胀重量和吸胀体积, 说明该位点为一因多效位点, 可解释的表型变异为5.17%~6.95%; 第6染色体上检测出2个QTL, 均影响吸胀前体积(Chr6.S_95604386所在QTL和Chr6.S_95604386所在QTL), 可分别解释5.28%和5.93%的表型变异; 第3染色体上检测出1个QTL (Chr3.S_197658474所在QTL), 可解释5.93%的吸涨体积表型变异; 但没有检测到与吸胀后体积显著相关的SNP (图3)。各生物学重复的GWAS结果与各性状均值的GWAS结果高度一致, 除影响吸胀体积的chr3.S_197658474所在QTL外, 其余QTL均在一个或多个生物学重复中被检测到(表5和图4)。 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3种子萌发性状全基因组关联分析曼哈顿图
各性状详细名称见
-->Fig. 3Manhattan of GWAS for seed germination traits
Name of each trait is given in
-->
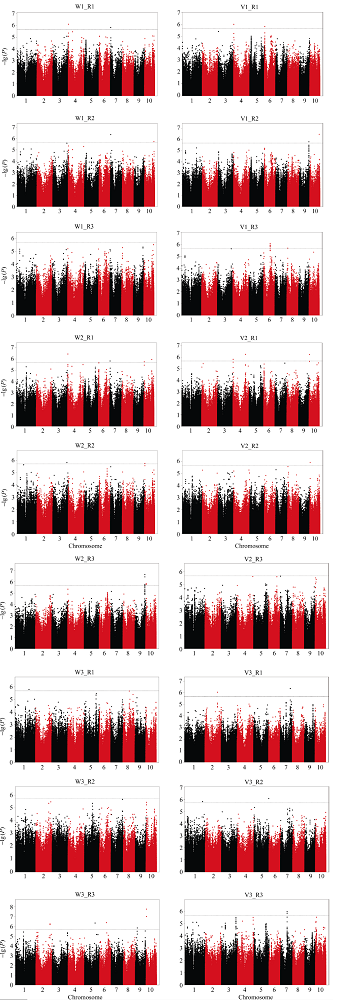 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4种子萌发性状全基因组关联分析曼哈顿图(单个重复)
各性状详细名称见
-->Fig 4Manhattan of GWAS for seed germination traits (single round)
Name of each trait is given in
-->
Table 4
表4
表4种子萌发候选基因及功能注释
Table 4Candidate genes and their anmotations of seed germination traits
 |
新窗口打开
Table 5
表5
表5种子萌发性状显著关联位点
Table 5SNPs associated with seed germination traits
| 性状 Trait | 重复 Repeat | 候选位点 Locus | SNP | 染色体 Chr. | 物理位置 Position | P值 P-value | 贡献率 R2 (%) |
|---|---|---|---|---|---|---|---|
| W1 | 1 | Chr4: 2865435-2865435? | Chr4.S_2925435 | 4 | 2925435 | 7.76×10-7 | 8.33 |
| Chr7: 4798028-4858028 | Chr7.S_4828028 | 7 | 4828028 | 1.48×10-6 | 5.58 | ||
| 2 | Chr10: 139810517-139870517 | Chr10.S_139840517 | 10 | 139840517 | 1.87×10-6 | 9.60 | |
| W1 | Chr7: 4798028-4858028 | Chr7.S_4828028 | 7 | 4828028 | 4.34×10-7 | 6.10 | |
| Chr7.S_4828028 | 7 | 4828028 | 7.66×10-7 | 5.84 | |||
| W2 | 1 | Chr10: 6343038-6403038 | Chr10.S_6373105 | 10 | 6373105 | 1.81×10-6 | 5.16 |
| Chr10: 117510861-117630861 | Chr10.S_117570861 | 10 | 117570861 | 1.16×10-6 | 5.21 | ||
| Chr4: 2865435-2865435 | Chr4.S_2925435 | 4 | 2925435 | 3.83×10-7 | 8.73 | ||
| Chr7: 4798028-4858028 | Chr7.S_4828028 | 7 | 4828028 | 1.57×10-6 | 5.54 | ||
| 2 | Chr10: 6343038-6403038 | Chr10.S_6373105 | 10 | 6373105 | 1.86×10-6 | 5.14 | |
| Chr3: 209027864-209147864 | Chr3.S_209087864 | 3 | 209087864 | 1.58×10-6 | 9.50 | ||
| Chr10: 6343038-6403038 | Chr10.S_6373038 | 10 | 6373038 | 1.75×10-6 | 5.25 | ||
| Chr10.S_6373105 | 10 | 6373105 | 1.50×10-6 | 5.27 | |||
| Chr9: 144257637-144377637 | Chr9.S_144308026 | 9 | 144308026 | 1.77×10-6 | 5.08 | ||
| Chr9.S_144308098 | 9 | 144308098 | 2.01×10-6 | 5.05 | |||
| Chr9.S_144309854 | 9 | 144309854 | 1.77×10-6 | 5.08 | |||
| Chr9.S_144309860 | 9 | 144309860 | 3.69×10-7 | 5.77 | |||
| W2 | Chr9.S_144317637 | 9 | 144317637 | 2.21×10-7 | 6.56 | ||
| Chr10: 6343038-6403038 | Chr10.S_6373038 | 10 | 6373038 | 1.86×10-6 | 5.17 | ||
| Chr10.S_6373105 | 10 | 6373105 | 1.16×10-6 | 5.34 | |||
| W3 | 1 | Chr1: 206063394-206183394 | Chr1.S_206123394 | 1 | 206123394 | 1.55×10-6 | 5.31 |
| 3 | Chr10: 6343038-6403038 | Chr10.S_6373038 | 10 | 6373038 | 1.01×10-7 | 5.92 | |
| Chr10.S_6373105 | 10 | 6373105 | 1.75×10-8 | 6.60 | |||
| Chr2: 152687771-152807771 | Chr2.S_205719834 | 2 | 205719834 | 6.26×10-7 | 7.05 | ||
| Chr2.S_205770041 | 2 | 205770041 | 5.71×10-7 | 5.02 | |||
| Chr2.S_205771072 | 2 | 205771072 | 5.71×10-7 | 5.02 | |||
| Chr2.S_205771170 | 2 | 205771170 | 5.71×10-7 | 5.02 | |||
| Chr2.S_205771560 | 2 | 205771560 | 5.71×10-7 | 5.02 | |||
| Chr2.S_205771697 | 2 | 205771697 | 5.71×10-7 | 5.02 | |||
| Chr2.S_205772111 | 2 | 205772111 | 5.71×10-7 | 5.02 | |||
| Chr2.S_205773201 | 2 | 205773201 | 5.71×10-7 | 5.02 | |||
| Chr2.S_205774142 | 2 | 205774142 | 5.71×10-7 | 5.02 | |||
| Chr5:152687771-152807771 | Chr5.S_152747771 | 5 | 152747771 | 4.70×10-7 | 8.78 | ||
| Chr6: 95574386-95634386 | Chr6.S_98573207 | 6 | 98573207 | 4.22×10-7 | 5.78 | ||
| Chr9: 26957278-27077278 | Chr9.S_27017278 | 9 | 27017278 | 1.47×10-6 | 5.13 | ||
| W3 | Chr10: 6343038-6403038 | Chr10.S_6373038 | 10 | 6373038 | 3.22×10-9 | 8.06 | |
| Chr10.S_6373105 | 10 | 6373105 | 5.75×10-9 | 7.73 | |||
| V1 | 1 | Chr4: 2865435- 2865435? | Chr4.S_2925435 | 4 | 2925435 | 8.89×10-7 | 8.44 |
| Chr6: 6778454-6838454 | Chr6.S_6808454 | 6 | 6808454 | 1.38×10-6 | 8.07 | ||
| 2 | Chr10: 139780517-139900517 | Chr10.S_139840517 | 10 | 139840517 | 3.47×10-7 | 10.56 | |
| Chr9: 144257637- 144377637 | Chr9.S_144309860 | 9 | 144309860 | 1.60×10-6 | 5.12 | ||
| 3 | Chr6: 95574386-95634386 | Chr6.S_95604386 | 6 | 95604386 | 8.22×10-7 | 5.32 | |
| Chr6.S_95604517 | 6 | 95604517 | 1.42×10-6 | 5.08 | |||
| Chr6.S_95604625 | 6 | 95604625 | 1.42×10-6 | 5.08 | |||
| 性状 Trait | 重复 Repeat | 候选位点 Locus | SNP | 染色体 Chr. | 物理位置 Position | P值 P-value | 贡献率 R2 (%) |
| Chr6.S_95604950 | 6 | 95604950 | 1.11×10-6 | 5.20 | |||
| Chr6.S_95605307 | 6 | 95605307 | 1.21×10-6 | 5.14 | |||
| Chr6.S_95605330 | 6 | 95605330 | 1.10×10-6 | 5.22 | |||
| Chr6.S_95605331 | 6 | 95605331 | 1.95×10-6 | 5.04 | |||
| Chr6.S_95605458 | 6 | 95605458 | 1.21×10-6 | 5.14 | |||
| Chr6.S_95605541 | 6 | 95605541 | 1.21×10-6 | 5.14 | |||
| Chr8: 14037401-14157401 | Chr8.S_14097401 | 8 | 14097401 | 1.93×10-6 | 5.71 | ||
| V1 | Chr6: 6778454-6838454 | Chr6.S_6808454 | 6 | 6808454 | 2.01×10-6 | 7.85 | |
| Chr6: 95574386-95634386 | Chr6.S_95604386 | 6 | 95604386 | 8.76×10-7 | 5.28 | ||
| Chr6.S_95604517 | 6 | 95604517 | 1.31×10-6 | 5.10 | |||
| Chr6.S_95604625 | 6 | 95604625 | 1.31×10-6 | 5.10 | |||
| Chr6.S_95604950 | 6 | 95604950 | 1.08×10-6 | 5.19 | |||
| Chr6.S_95605307 | 6 | 95605307 | 1.14×10-6 | 5.16 | |||
| Chr6.S_95605330 | 6 | 95605330 | 1.03×10-6 | 5.23 | |||
| Chr6.S_95605331 | 6 | 95605331 | 1.67×10-6 | 5.09 | |||
| Chr6.S_95605458 | 6 | 95605458 | 1.14×10-6 | 5.16 | |||
| Chr6.S_95605541 | 6 | 95605541 | 1.14×10-6 | 5.16 | |||
| V2 | 1 | Chr10: 6343038-6403038 | Chr10.S_6373038 | 10 | 6373038 | 5.73×10-7 | 5.73 |
| Chr4: 2865435-2865435 | Chr4.S_2925435 | 4 | 2925435 | 1.49×10-6 | 7.54 | ||
| Chr4: 185676597-185796597 | Chr4.S_185736597 | 4 | 185736597 | 5.59×10-7 | 7.89 | ||
| 2 | Chr10: 6147016-6267016 | Chr10.S_6207016 | 10 | 6207016 | 1.14×10-6 | 8.84 | |
| V3 | 1 | Chr2: 182455178-182575178 | Chr2.S_182515178 | 2 | 182515178 | 9.03×10-7 | 5.98 |
| Chr7: 145940024-146000024 | Chr7.S_145970024 | 7 | 145970024 | 4.26×10-7 | 5.75 | ||
| 2 | Chr1: 262661257-262781257 | Chr1.S_262721257 | 1 | 262721257 | 1.59×10-6 | 5.55 | |
| Chr5: 213497149-213617149 | Chr5.S_213557149 | 5 | 213557149 | 8.90×10-7 | 5.68 | ||
| 3 | Chr7: 95428593-95548593 | Chr7.S_95488593 | 7 | 95488593 | 1.05×10-6 | 7.48 | |
| Chr7.S_95488613 | 7 | 95488613 | 1.43×10-6 | 6.80 | |||
| V3 | Chr10: 6343038-6403038 | Chr10.S_6373038 | 10 | 6373038 | 1.60×10-6 | 5.21 | |
| Chr3: 197628474-197688474 | Chr3.S_197658474 | 3 | 197658474 | 1.72×10-6 | 5.93 | ||
| Chr7: 145940024-146000024 | Chr7.S_145970024 | 7 | 145970024 | 1.70×10-6 | 5.13 |
新窗口打开
6个QTL内共涉及到44个候选基因(表4), 其中仅有34% (15/44)有功能注释, 根据候选基因的功能注释, 选择最显著SNP所在基因或邻近基因作为首要候选基因(表4)。其中, 吸胀前重量候选基因GRMZM2G167892编码籽粒成熟蛋白(seed mature protein)。同时影响吸胀后重量、吸胀重量和吸胀体积的候选基因GRMZM2G148411 (第10染色体上), 编码一个包含TLD结构域的钙离子结合蛋白。吸胀前体积的候选基因GRMZM2G318592和吸胀体积的候选基因GRMZM2G146173均编码包含锌指结构域的蛋白。吸胀前体积的候选基因GRMZM2G 449489和吸胀体积的候选基因GRMZM2G544352分别编码硒富集相关蛋白和未知功能蛋白。
3 讨论
不同的分析模型影响关联分析的结果。为确保关联结果的准确性, 最大程度降低假阳性结果, GWAS前要对每个性状进行最优模型的选择。Wang等[21]在玉米丝黑穗病的全基因组关联分析中发现, Q+K模型能更好地降低假阳性的发生。本研究对每个性状进行了3种模型(Q, K, Q+K)的分析, 发现Q+K模型对吸胀前重量、吸胀前体积、吸胀后重量、吸胀后体积和吸胀体积这5个性状的假阳性控制过于严格, 产生了一些假阴性的结果, K模型的拟合效果最好; 而Q+K模型则能更好的地控制吸胀重量性状的假阳性。种子的萌发受赤霉素(GA)和脱落酸(ABA)等植物激素、光照、温度等环境因素以及一氧化氮(NO)和活性氧(ROS)等信号分子[22,23,24,25,26,27]在内的多种因素影响。本研究所筛选的候选基因的功能大多与种子内部激素水平、信号分子调控及活性氧有关。Li等[18]对水稻OSCA家族基因的功能研究证明钙离子可以通过调控吸胀种子的水势影响萌发, 因此与钙离子结合相关的结构或蛋白均可能影响种子萌发。GRMZM2G148411编码具有TLD结构域的核仁蛋白(TLD-domain containing nucleolar protein), 在拟南芥中有2个同源基因。其中, AT4G34070编码一种具有EF-hand基序的钙结合蛋白(Calcium-binding EF-hand family protein), 可能与NADPH氧化酶的合成有关; AT5G06260编码一种与影响钙离子结合的具有TLD结构域的核仁蛋白。GA可以作为主要的钙离子感受器, 增加钙离子和钙调蛋白, 对大麦糊粉层中钙离子信号传导起重要作用; ABA的作用与GA相反[28]。因此, 我们推测GRMZM2G148411可能作为NADPH氧化酶的组分, 调节种子内部ROS的浓度; 或作为钙离子调控的信号分子, 通过调节钙离子与钙调节蛋白的浓度影响种子内部GA和ABA的相对水平, 进而影响种子萌发。吸胀前体积候选基因GRMZM2G318592和吸胀体积候选基因GRMZM2G146173, 均编码一种含有锌指结构的蛋白。Baek等[29]发现, 拟南芥中的ARS1基因对ABA和氧化胁迫下的种子萌发和ROS含量具有重要作用, 该基因编码一种包含锌指结构域的核蛋白。Joseph等[30]研究拟南芥种子中ABA的合成与信号转导时, 发现ZFP3 (锌指蛋白3)是种子萌发过程中抑制ABA的调控因子, 赋予种子对ABA的不敏感性。因此, 推测本研究中的2个编码锌指蛋白的候选基因, 可能是通过调控玉米种子对ABA的敏感程度进一步影响种子的休眠和萌发。上述3个候选基因均作为信号分子直接或间接地参与种子萌发期间内部GA与ABA的相对水平以及ROS含量的调控, 进而影响种子的休眠与萌发。Yazdanpanah等[17]通过反向遗传学方法找到的种子休眠与萌发有关基因中, AT3G22490编码种子成熟蛋白(seed maturation protein), 该基因的敲除突变导致种子提前终止休眠。本研究中的候选基因GRMZM2G167892也编码一种种子成熟蛋白, 该基因可能与种子的萌发及休眠有关, 具体作用机制还有待进一步研究。
4 结论
共检测到15个显著的SNP-性状关联, 涉及6个QTL, 并鉴定出6个最可能的候选基因, 其中, 3个可能通过调节种子萌发过程中激素及ROS水平影响种子萌发, 1个可能通过调控种子成熟影响种子萌发, 另2个影响种子萌发的机制未知。The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
| [1] | . In theory, seed germination, vigour and size (three aspects of seed quality) may influence crop yield through both indirect and direct effects. The indirect effects include those on percentage emergence and time from sowing to emergence. These influence yield by altering plant population density, spatial arrangement, and crop duration. Direct effects on subsequent plant performance are more difficult to discern. A number of different approaches to testing the hypothesis that seed quality affects subsequent plant performance (implicit in some definitions of vigour) are illustrated. The results show that it is possible to detect such effects in some circumstances. |
| [2] | . Abstract Maize (Zea mays) is an excellent model for basic research. Genetic screens have informed our understanding of developmental processes, meiosis, epigenetics and biochemical pathways--not only in maize but also in other cereal crops. We discuss the forward and reverse genetic screens that are possible in this organism, and emphasize the available tools. Screens exploit the well-studied behaviour of transposon systems, and the distinctive chromosomes allow an integration of cytogenetics into mutagenesis screens and analyses. The imminent completion of the maize genome sequence provides the essential resource to move seamlessly from gene to phenotype and back. |
| [3] | . In: Janick J ed. 136 F. BELOW, J. SEEBAUER, M. URIBELARREA, M. SCHNEERMAN, AND S. MOOSE 0 ofILP, and this difference widens during grain development so that by final harvest IHP 150 LSD100 110 120 130 90 120- Stover LSD ILP 20- 40- 60- 80 100- 90- 1 1.2 1.4 1.6 1.8 R e d u |
| [4] | . Age-related macular degeneration (AMD) is a major cause of blindness in the elderly. We report a genome-wide screen of 96 cases and 50 controls for polymorphisms associated with AMD. Among 116,204 single-nucleotide polymorphisms genotyped, an intronic and common variant in the complement factor H gene (CFH) is strongly associated with AMD (nominal P value <10-7). In individuals homozygous for the risk allele, the likelihood of AMD is increased by a factor of 7.4 (95% confidence interval 2.9 to 19). Resequencing revealed a polymorphism in linkage disequilibrium with the risk allele representing a tyrosine-histidine change at amino acid 402. This polymorphism is in a region of CFH that binds heparin and C-reactive protein. The CFH gene is located on chromosome 1 in a region repeatedly linked to AMD in family-based studies. |
| [5] | . |
| [6] | . A high-density haplotype map recently enabled a genome-wide association study (GWAS) in a population of indica subspecies of Chinese rice landraces. Here we extend this methodology to a larger and more diverse sample of 950 worldwide rice varieties, including the Oryza sativa indica and Oryza sativa japonica subspecies, to perform an additional GWAS. We identified a total of 32 new loci associated with flowering time and with ten grain-related traits, indicating that the larger sample increased the power to detect trait-associated variants using GWAS. To characterize various alleles and complex genetic variation, we developed an analytical framework for haplotype-based de novo assembly of the low-coverage sequencing data in rice. We identified candidate genes for 18 associated loci through detailed annotation. This study shows that the integrated approach of sequence-based GWAS and functional genome annotation has the potential to match complex traits to their causal polymorphisms in rice. |
| [7] | . This review summarizes recent progress02in maize GWAS02to establish new insights of functional genomics in the omics era. Particularly, potential contributions from over-genomic variants, innovations for statistical methods, and distinctive02population designs are highlighted to jointly address the missing heritability issue. |
| [8] | . Maize kernel oil is a valuable source of nutrition. Here we extensively examine the genetic architecture of maize oil biosynthesis in a genome-wide association study using 1.03 million SNPs characterized in 368 maize inbred lines, including 'high-oil' lines. We identified 74 loci significantly associated with kernel oil concentration and fatty acid composition (P < 1.8 x 10(-6)), which we subsequently examined using expression quantitative trait loci (QTL) mapping, linkage mapping and coexpression analysis. More than half of the identified loci localized in mapped QTL intervals, and one-third of the candidate genes were annotated as enzymes in the oil metabolic pathway. The 26 loci associated with oil concentration could explain up to 83% of the phenotypic variation using a simple additive model. Our results provide insights into the genetic basis of oil biosynthesis in maize kernels and may facilitate marker-based breeding for oil quantity and quality. |
| [9] | . |
| [10] | . |
| [11] | . Improving the salt tolerance of direct-seeding rice at the seed germination stage is a major breeding goal in many Asian rice-growing countries, where seedlings must often establish in soils with a high salt content. Thus, it is important to understand the genetic mechanisms of salt tolerance in rice and to screen for germplasm with salt tolerance at the seed germination stage. Here, we investigated seven seed germination-related traits under control and salt-stress conditions and conducted a genome-wide association study based on the re-sequencing of 478 diverse rice accessions. The analysis used a mixed linear model and was based on 6,361,920 single nucleotide polymorphisms in 478 rice accessions grouped into whole,indica, and non-indicapanels. Eleven loci containing 22 significant salt tolerance-associated single nucleotide polymorphisms were identified based on the stress-susceptibility indices (SSIs) of vigor index (VI) and mean germination time (MGT). From the SSI of VI, six major loci were identified, explaining 20.2% of the phenotypic variation. From the SSI of MGT, five major loci were detected, explaining 26.4% of the phenotypic variation. Of these, seven loci on chromosomes 1, 5, 6, 11, and 12 were close to six previously identified quantitative gene loci/genes related to tolerance to salinity or other abiotic stresses. The strongest association region for the SSI of MGT was identified in a02~0213.302kb interval (15450039–15,463,330) on chromosome 1, near salt-tolerance quantitative trait loci controlling the Na+: K+ratio, total Na+uptake, and total K+concentration. The strongest association region for the SSI of VI was detected in a02~02164.202kb interval (526662–690,854) on chromosome 2 harboring two nitrate transporter family genes (OsNRT2.1andOsNRT2.2), which affect gene expression under salt stress. The haplotype analysis indicated thatOsNRT2.2was associated with subpopulation differentiation and its minor/rare tolerant haplotype was detected. These results provide valuable information for salt tolerance-related gene cloning and for understanding the genetic mechanisms of salt tolerance at the seed germination stage. This information will be useful to improve the salt tolerance of direct-seeding rice varieties by genomic selection or marker-assisted selection. The online version of this article (doi:10.1186/s12870-017-1044-0) contains supplementary material, which is available to authorized users. |
| [12] | . Soil salinity is a serious threat to agriculture sustainability worldwide. Seed germination is a critical phase that ensures the successful establishment and productivity of soybeans in saline soils.... |
| [13] | . |
| [14] | . Heterosis is observed for most phenotypic traits and developmental stages in many plants. In this study, the embryos, from germinating seeds after 24 of soaking, for five elite maize hybrids and their parents were selected to unravel the genetic basis of heterosis using 2-D proteomic method. In total, 257 (80.06%), 363 (58.74%), 351 (79.95%), 242 (54.50%), and 244 (46.30%) nonadditively expressed proteins were identified in hybrids Zhengdan 958, Nongda 108, Yuyu 22, Xundan 20, and Xundan 18, respectively. The nonadditive proteins were divided into above high-parent (++; 811, 55.66%), high-parent (+; 121, 8.30%), partial dominance (+-; 249, 17.09%), low-parent (-; 30, 2.06%), below low-parent (- -; 62, 4.26%), and D (different; 184, 12.63%) expression patterns. The observed patterns indicate the important roles of dominance, partial dominance, and overdominance in regulating seed germination in maize. Additionally, 54 different proteins were identified by mass spectrometry and classified into nine functional groups: metabolism (9), cell detoxification (8), unknown functional proteins (8), chaperones (7), signal transduction (6), development process (5), other (5), transporter (3), and stress response (3). Of these, the most interesting are those involved with germination-related hormone signal transduction and the abscisic acid and gibberellin regulation networks. |
| [15] | . Abstract Low temperature is the primary factor to affect maize sowing in early spring. It is, therefore, vital for maize breeding programs to improve tolerance to low temperatures at seed germination stage. However, little is known about maize QTL involved in low-temperature germination ability. 243 lines of the intermated B730103Mo17 (IBM) Syn4 recombinant inbred line (RIL) population was used for QTL analysis of low-temperature germination ability. There were significant differences in germination-related traits under both conditions of low temperature (1200°C/16 h, 1800°C/8 h) and optimum temperature (2800°C/24 h) between the parental lines. Only three QTL were identified for controlling optimum-temperature germination rate. Six QTL controlling low-temperature germination rate were detected on chromosome 4, 5, 6, 7 and 9, and contribution rate of single QTL explained between 3.39%~11.29%. In addition, six QTL controlling low-temperature primary root length were detected in chromosome 4, 5, 6, and 9, and the contribution rate of single QTL explained between 3.96%~8.41%. Four pairs of QTL were located at the same chromosome position and together controlled germination rate and primary root length under low temperature condition. The nearest markers apart from the corresponding QTL (only 0.01 cM) were umc1303 (265.1 cM) on chromosome 4, umc1 (246.4 cM) on chromosome 5, umc62 (459.1 cM) on chromosome 6, bnl14.28a (477.4 cM) on chromosome 9, respectively. A total of 3155 candidate genes were extracted from nine separate intervals based on the Maize Genetics and Genomics Database (http://www.maizegdb.org). Five candidate genes were selected for analysis as candidates putatively affecting seed germination and seedling growth at low temperature. The results provided a basis for further fine mapping, molecular marker assisted breeding and functional study of cold-tolerance at the stage of seed germination in maize. |
| [16] | . High seed vigor is important for agricultural production due to the associated potential for increased growth and productivity. However, a better understanding of the underlying molecular mechanisms is required because the genetic basis for seed vigor remains unknown. We used single-nucleotide polymorphism (SNP) markers to map quantitative trait loci (QTLs) for four seed vigor traits in two connected recombinant inbred line (RIL) maize populations under four treatment conditions during seed germination. Sixty-five QTLs distributed between the two populations were identified and a meta-analysis was used to integrate genetic maps. Sixty-one initially identified QTLs were integrated into 18 meta-QTLs (mQTLs). Initial QTLs with contribution to phenotypic variation values of R2>10% were integrated into mQTLs. Twenty-three candidate genes for association with seed vigor traits coincided with 13 mQTLs. The candidate genes had functions in the glycolytic pathway and in protein metabolism. QTLs with major effects (R2>10%) were identified under at least one treatment condition for mQTL2, mQTL3-2, and mQTL3-4. Candidate genes included a calcium-dependent protein kinase gene (302810918) involved in signal transduction that mapped in the mQTL3-2 interval associated with germination energy (GE) and germination percentage (GP), and an hsp20/alpha crystallin family protein gene (At5g51440) that mapped in the mQTL3-4 interval associated with GE and GP. Two initial QTLs with a major effect under at least two treatment conditions were identified for mQTL5-2. A cucumisin-like Ser protease gene (At5g67360) mapped in the mQTL5-2 interval associated with GP. The chromosome regions for mQTL2, mQTL3-2, mQTL3-4, and mQTL5-2 may be hot spots for QTLs related to seed vigor traits. The mQTLs and candidate genes identified in this study provide valuable information for the identification of additional quantitative trait genes. |
| [17] | . Seed dormancy, defined as the incapability of a viable seed to germinate under favourable conditions, is an important trait in nature and agriculture. Despite extensive research on dormancy and germination, many questions about the molecular mechanisms controlling these traits remain unanswered, likely due to its genetic complexity and the large environmental effects which are characteristic of these quantitative traits. To boost research towards revealing mechanisms in the control of seed dormancy and germination we depend on the identification of genes controlling those traits. We used transcriptome analysis combined with a reverse genetics approach to identify genes that are prominent for dormancy maintenance and germination in imbibed seeds ofArabidopsis thaliana. Comparative transcriptomics analysis was employed on freshly harvested (dormant) and after-ripened (AR; non-dormant) 24-h imbibed seeds of four differentDELAY OF GERMINATIONnear isogenic lines (DOGNILs) and the Landsbergerecta(Ler) wild type with varying levels of primary dormancy. T-DNA knock-out lines of the identified genes were phenotypically investigated for their effect on dormancy and AR. We identified conserved sets of 46 and 25 genes which displayed higher expression in seeds of all dormant and all after-ripenedDOGNILs and Ler, respectively. Knock-out mutants in these genes showed dormancy and germination related phenotypes. Most of the identified genes had not been implicated in seed dormancy or germination. This research will be useful to further decipher the molecular mechanisms by which these important ecological and commercial traits are regulated. The online version of this article (10.1186/s12870-017-1098-z) contains supplementary material, which is available to authorized users. |
| [18] | . Background Reception of and response to exogenous and endogenous osmotic changes is important to sustain plant growth and development, as well as reproductive formation. Hyperosmolality-gated... |
| [19] | . Global warming is a major agricultural issue in the Northern hemisphere where higher temperatures are expected to be associated with restricted water availability. In Europe, for maize, earlier and further northward sowings are forecasted in order to avoid water deficit periods in the crop life cycle. However these conditions may compromise seed germination and stand establishment since they will take place at cold temperatures. It is urgent to better understand the molecular bases of response of germinating maize seeds to cold in order to design genotypes adapted to these novel agricultural practices. Here we have performed a global phospholipidomic study to profile changes in membrane reorganisation during seed imbibition at 1065°C of cold-tolerant and -sensitive maize hybrids. Using a Multiple Reaction Monitoring (MRM-MS/MS) method coupled with HPLC we have identified 80 distinct phospholipids. We show that seed sensitivity to cold temperatures during imbibition relies on the accumulation of saturated or poorly unsaturated fatty acids, whatever the phospholipid class. In contrast seeds of cold-tolerant hybrid accumulated polyunsaturated chains which was associated with lower electrolyte leakage during imbibition at 1065°C. The expression of fatty acid desaturase genes provides a molecular model of maize seed sensitivity to imbibitional chilling damage. |
| [20] | . Amino acids are both constituents of proteins, providing the essential nutrition for humans and animals, and signalling molecules regulating the growth and development of plants. Most cultivars of maize are deficient in essential amino acids such as lysine and tryptophan. Here, we measured the levels of 17 different total amino acids, and created 48 derived traits in mature kernels from a maize diversity inbred collection and three recombinant inbred line (RIL) populations. By GWAS, 247 and 281 significant loci were identified in two different environments, 5.1 and 4.4 loci for each trait, explaining 7.44% and 7.90% phenotypic variation for each locus in average, respectively. By linkage mapping, 89, 150 and 165 QTLs were identified in B73/By804, Kui3/B77 and Zong3/Yu87 1 RIL populations, 2.0, 2.7 and 2.8 QTLs for each trait, explaining 13.6%, 16.4% and 21.4% phenotypic variation for each QTL in average, respectively. It implies that the genetic architecture of amino acids is relative simple and controlled by limited loci. About 43.2% of the loci identified by GWAS were verified by expression QTL, and 17 loci overlapped with mapped QTLs in the three RIL populations. GRMZM2G015534, GRMZM2G143008 and one QTL were further validated using molecular approaches. The amino acid biosynthetic and catabolic pathways were reconstructed on the basis of candidate genes proposed in this study. Our results provide insights into the genetic basis of amino acid biosynthesis in maize kernels and may facilitate marker鈥恇ased breeding for quality protein maize. |
| [21] | . Head smut, caused by the fungus Sphacelotheca reiliana (K hn) Clint, is a devastating global disease in maize, leading to severe quality and yield loss each year. The present study is the first to conduct a genome-wide association study (GWAS) of head smut resistance using the Illumina MaizeSNP50 array. Out of 45,868 single nucleotide polymorphisms in a panel of 144 inbred lines, 18 novel candidate genes were associated with head smut resistance in maize. These candidate genes were classified into three groups, namely, resistance genes, disease response genes, and other genes with possible plant disease resistance functions. The data suggested a complicated molecular mechanism of maize resistance against S. reiliana. This study also suggested that GWAS is a useful approach for identifying causal genetic factors for head smut resistance in maize. |
| [22] | . |
| [23] | . |
| [24] | . The seeds of many plant species are dormant at maturity and dormancy loss is a prerequisite for germination. Numerous environmental and chemical treatments are known to lessen or remove seed dormancy, but the biochemical changes that occur during this change of state are poorly understood. Several lines of research have implicated nitric oxide (NO) as a participant in this process. Here, we show that dormant seeds of Arabidopsis thaliana (L.) Heynh. will germinate following treatment with the NO donor sodium nitroprusside (SNP), cyanide (CN), nitrite or nitrate. In all cases, the NO scavenger c-PTIO effectively promotes the maintenance of seed dormancy. c-PTIO does not, however, inhibit germination of fully after-ripened seeds, and c-PTIO does not interact directly with nitrite, nitrate or CN. We also show that volatile CN effectively breaks dormancy of Arabidopsis seeds, and that CN is the volatile compound in SNP that promotes dormancy loss. Our data support the hypothesis that NO is a signaling molecule that plays an important role in the loss of seed dormancy. |
| [25] | . The nitric oxide (NO) donor sodium nitroprusside (SNP) significantly promoted germination of switchgrass (Panicum virgatum L. cv Kanlow) in the light and in the dark at 25 C, across a broad range of concentrations. SNP also promoted seed germination in two other warm-season grasses. A chemical scavenger of NO inhibited germination and blocked SNP stimulation of seed germination. The phenolic (+)-catechin acted synergistically with SNP and nitrite in promoting seed germination. Acidified nitrite, an alternate NO donor also significantly stimulated seed germination. Interestingly, sodium cyanide, potassium ferricyanide and potassium ferrocyanide at 200 M strongly enhanced seed germination as well, whereas potassium chloride was without effect. Ferrocyanide and cyanide stimulation of seed germination was blocked by an NO scavenger. Incubation of seeds with a fluorescent NO-specific probe provided evidence for NO production in germinating switchgrass seeds. Abscisic acid (ABA) at 10 M depressed germination, inhibited root elongation and essentially abolished coleoptile emergence. SNP partially overcame ABA effects on radicle emergence but did not overcome the effects of ABA on coleoptile elongation. Light microscopy indicated extension of the radicle and coleoptiles in seeds maintained on water or on SNP after 2 days. In contrast, there was minimal growth of the radicle and coleoptile in ABA-treated seeds even after 3 4 days. These data indicate that seed germination of warm-season grasses is significantly influenced by NO signaling pathways and document that NO could be an endogenous trigger for release from dormancy in these species. |
| [26] | . A short-term pretreatment of barley seed (Hordeum vulgare cv. Prisma) with hydrogen peroxide leads to dormancy breakage. This powerful oxidant provokes a large increase of glutathione level and of glutathione reductase activity in embryo and storage tissues. Seed scarification results in a similar activation of the glutathione system after 24 h of seed imbibition with distilled water. It is assumed that oxygen diffusion and endogenous hydrogen peroxide formation play a major role in the activation of the glutathione system which might be a key step in dormancy breakage of barley seeds |
| [27] | . Reactive Oxygen Species (ROS) play a key role in various events of seed life. In orthodox seeds, ROS are produced from embryogenesis to germination, i.e., in metabolically active cells, but also in quiescent dry tissues during after ripening and storage, owing various mechanisms depending on the seed moisture content. Although ROS have been up to now widely considered as detrimental to seeds, recent advances in plant physiology signaling pathways has lead to reconsider their role. ROS accumulation can therefore be also beneficial for seed germination and seedling growth by regulating cellular growth, ensuring a protection against pathogens or controlling the cell redox status. ROS probably also act as a positive signal in seed dormancy release. They interact with abscisic acid and gibberellins transduction pathway and are likely to control numerous transcription factors and properties of specific protein through their carbonylation. |
| [28] | . Abstract Reactive oxygen species (ROS) promote the germination of several seeds, and antioxidants suppress it. However, questions remain regarding the role and production mechanism of ROS in seed germination. Here, we focused on NADPH oxidases, which produce ROS. After imbibition, NADPH oxidase mRNAs were expressed in the embryo and in aleurone cells of barley seed; these expression sites were consistent with the sites of ROS production in the seed after imbibition. To clarify the role of NADPH oxidases in barley seed germination, we examined gibberellic acid (GA) / abscisic acid (ABA) metabolism and signaling in barley seeds treated with diphenylene iodonium chloride (DPI), an NADPH oxidase inhibitor. DPI significantly suppressed germination, and suppressed GA biosynthesis and ABA catabolism in embryos. GA, but not ABA, induced NADPH oxidase activity in aleurone cells. Additionally, DPI suppressed the early induction of -amylase by GA in aleurone cells. These results suggest that ROS produced by NADPH oxidases promote GA biosynthesis in embryos, that GA induces and activates NADPH oxidases in aleurone cells, and that ROS produced by NADPH oxidases induce -amylase in aleurone cells. We conclude that the ROS generated by NADPH oxidases regulate barley seed germination through GA / ABA metabolism and signaling in embryo and aleurone cells. |
| [29] | . The phytohormone abscisic acid (ABA) induces accumulation of reactive oxygen species (ROS), which can disrupt seed dormancy and plant development. Here, we report the isolation and characterization of anArabidopsis thalianamutant calledars1(aba androssensitive 1) that showed hypersensitivity to ABA during seed germination and to methyl viologen (MV) at the seedling stage.ARS1encodes a nuclear protein with one zinc finger domain, two nuclear localization signal (NLS) domains, and one nuclear export signal (NES). Thears1mutants showed reduced expression of a gene for superoxide dismutase (CSD3) and enhanced accumulation of ROS after ABA treatment. Transient expression of ARS1 inArabidopsisprotoplasts strongly suppressed ABA-mediated ROS production. Interestingly, nuclear-localized ARS1 translocated to the cytoplasm in response to treatment with ABA, H2O2, or MV. Taken together, these results suggest that ARS1 modulates seed germination and ROS homeostasis in response to ABA and oxidative stress in plants. |
| [30] | . Abstract Seed germination is controlled by environmental signals, including light and endogenous phytohormones. Abscisic acid (ABA) inhibits, whereas gibberellin promotes, germination and early seedling development, respectively. Here, we report that ZFP3, a nuclear C2H2 zinc finger protein, acts as a negative regulator of ABA suppression of seed germination in Arabidopsis (Arabidopsis thaliana). Accordingly, regulated overexpression of ZFP3 and the closely related ZFP1, ZFP4, ZFP6, and ZFP7 zinc finger factors confers ABA insensitivity to seed germination, while the zfp3 zfp4 double mutant displays enhanced ABA susceptibility. Reduced expression of several ABA-induced genes, such as RESPONSIVE TO ABSCISIC ACID18 and transcription factor ABSCISIC ACID-INSENSITIVE4 (ABI4), in ZFP3 overexpression seedlings suggests that ZFP3 negatively regulates ABA signaling. Analysis of ZFP3 overexpression plants revealed multiple phenotypic alterations, such as semidwarf growth habit, defects in fertility, and enhanced sensitivity of hypocotyl elongation to red but not to far-red or blue light. Analysis of genetic interactions with phytochrome and abi mutants indicates that ZFP3 enhances red light signaling by photoreceptors other than phytochrome A and additively increases ABA insensitivity conferred by the abi2, abi4, and abi5 mutations. These data support the conclusion that ZFP3 and the related ZFP subfamily of zinc finger factors regulate light and ABA responses during germination and early seedling development. 2014 American Society of Plant Biologists. All Rights Reserved. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}