 , 王兴春
, 王兴春Genome-wide Identification of Molecular Markers Based on Genomic Re-sequencing of Foxtail Millet Elite Cultivar Jingu 21
ZHAOQing-Ying, WANGXing-Chun通讯作者:
收稿日期:2017-09-11
接受日期:2018-01-8
网络出版日期:2018-01-29
版权声明:2018作物学报编辑部作物学报编辑部
基金资助:
作者简介:
-->
展开
摘要
关键词:
Abstract
Keywords:
-->0
PDF (3503KB)元数据多维度评价相关文章收藏文章
本文引用格式导出EndNoteRisBibtex收藏本文-->
谷子(Setaria italica L.)起源于中国, 是我国过去几千年来的主栽作物和中华民族的哺育作物。脱壳后的谷子称为小米, 富含蛋白质、脂肪、铁、类胡萝卜素、纤维素、维生素等[1,2,3]。近年来, 随着人们生活水平的不断提高及食品的多样化和营养化, 优质小米倍受青睐。然而, 长期以来谷子品质相关的研究进展缓慢, 且主要集中在外观品质和简单的营养成分分析方面。中国作物信息网(http://www. cgris.net/)登记在册的谷子种质资源中呈现出16种不同的米色, 其中黄色米占77%。小米中的黄色物质主要是类胡萝卜素, 其含量与外观品质显著相关, 是小米品质育种的重要指标。黄米中类胡萝卜素的含量与绿米相当, 但显著高于白色品种[4]。王润奇等[5]利用三体分析法将白米和青米基因分别定位于第4和第6染色体, 为米色基因的克隆奠定了基础。最近, 我们发现谷子番茄红素β-环化酶(lycopene beta cyclase, LCYB)可能通过影响β-胡萝卜素和总类胡萝卜素在籽粒中的积累, 进而影响小米的米色[6]。蒸煮品质和食用品质是小米品质构成的最重要方面, 主要由直链淀粉和支链淀粉的含量及比例决定。直链淀粉含量较低的小米米饭柔软且口感好, 相反, 直链淀粉含量较高的小米蒸煮后米饭干燥、蓬松、色暗, 有回生现象[7]。遗憾的是, 目前有关淀粉合成和调控机制的认识主要来自拟南芥和水稻等模式植物, 谷子相关研究较少[8,9]。
随着高通量测序的简单化和低成本化, 利用二代测序技术大规模开发谷子分子标记并解析小米品质等复杂农艺性状成为可能。2012年, 由美国国家能源部所属的联合基因组研究所和中国华大基因分别进行的豫谷1号和张谷的全基因组测序工作相继完成[10,11], 从此谷子功能基因组学研究进入了一个新的时代。此后, 一大批基于全基因组测序的分子标记相继被开发[12,13,14,15,16]。Bai等[16]对十里香谷子品种进行了基因组重测序, 通过比对发现十里香和豫谷1号基因组之间存在762 082个SNP, 26 802个1~5 bp InDels和10 109个结构变异; 而十里香和张谷基因组之间存在915 434个SNP、28 546个InDels和12 968个结构变异。最近, 王军等[17]利用简化基因组测序技术构建了一张1648.8 cM的谷子高密度遗传图谱, 并定位了8个农艺性状的11个主效QTL。我国谷子种质资源非常丰富, 在长期的自然进化和人工选择中积累了大量自然变异。2013年, 我国科学家联合完成了916份谷子种质资源全基因组低倍重测序和单倍体型图谱的构建工作, 为谷子基因资源的发掘和遗传改良提供了海量的基础数据信息[18]。
晋谷21原名晋汾7号, 由晋汾52经钴60 γ射线辐射和连续单株选育而成[19]。其米色金黄发亮, 细柔光滑, 品质和产量均超过历史名米沁州黄[19]。晋谷21于1992年荣获中国首届农业博览会银奖, 1994年获山西省科技进步一等奖, 1995年被国家科委列入国家成果重点推广项目。本实验室曾对晋谷21进行了转录组测序, 鉴定出614个新基因, 并优化了7175个已注释基因的结构, 为晋谷21优异米质基因的发掘奠定了基础[20]。尽管如此, 由于缺少晋谷21的基因组信息, 限制了晋谷21优异米质连锁分子标记的开发和相关基因的克隆。为此, 本研究利用Illumina HiSeq 2000高通量测序技术对晋谷21进行了全基因组重测序, 并将其与豫谷1号参考基因组比较, 开发了大量InDel和SNP分子标记, 并将其应用于晋谷21衍生种的分析。本研究不仅为晋谷21优异米质相关基因的发掘和分子育种奠定了基础, 同时也为其他谷子品种的相关研究提供了借鉴。
1 材料与方法
1.1 植物材料及培养条件
全基因组重测序材料为大田种植的健康无病虫害的晋谷21单株植株, 选取15份谷子种质资源(表1)进行引物通用性分析。编号7~15的种质资源由中国农业科学院作物科学研究所的刁现民研究员馈赠, 其余资源为本实验室保存或创制。所有材料均种植于山西省太谷县山西农业大学试验田内。Table 1
表1
表115份谷子种质资源信息
Table 1Information of 15 foxtail millet germplasm resources
| 编号 Code | 名称 Name | 原产地 Origin | 编号 Code | 名称 Name | 原产地 Origin |
|---|---|---|---|---|---|
| 1 | 晋谷21 Jingu 21 | 山西 Shanxi | 9 | 酒谷 Jiugu | 河北 Hebei |
| 2 | mop1 | 山西 Shanxi | 10 | 碱谷 Jiangu | 内蒙古 Inner Mongolia |
| 3 | mop2 | 山西 Shanxi | 11 | 菠菜腿 Bocaitui | 内蒙古 Inner Mongolia |
| 4 | 青狗尾草 Green foxtail | 山西 Shanxi | 12 | 大青谷 Daqinggu | 内蒙古 Inner Mongolia |
| 5 | 谷莠子 Giant foxtail | 山西 Shanxi | 13 | 龙谷 Longgu | 辽宁 Liaoning |
| 6 | 豫谷1号 Yugu 1 | 河南 Henan | 14 | 谷莠天然杂交种 Guyou natural hybrid | 北京 Beijing |
| 7 | 青谷 Qinggu | 河南 Henan | 15 | 黄粟 Huangsu | 江西 Jiangxi |
| 8 | 黄大粒 Huangdali | 河北 Hebei |
新窗口打开
1.2 基因组DNA的提取、文库构建和高通量测序
采用改良的CTAB法[21]提取用于PCR模板的谷子基因组DNA: 取0.1 g新鲜谷子叶片, 加入0.6 mL CTAB提取液, 利用高通量组织研磨机(宁波新芝SCIENTZ-48)充分破碎; 然后加入500 μL氯仿/异戊醇(24∶1, v/v)进行抽提; 最后取上清液加入等体积的异丙醇以沉淀DNA。重测序用晋谷21基因组的提取采用高效植物基因组DNA提取试剂盒[天根生化科技(北京)有限公司, 货号DP350], 提取操作严格按照试剂盒提供的方法进行。利用配备μCuvette G1.0超微量比色皿的BioSpectrometer分光光度计(PCR用)或者Qubit荧光定量仪(重测序用)测定基因组DNA的浓度和纯度, 并用1% (w/v)的琼脂糖凝胶电泳检测DNA质量。由深圳华大基因科技服务有限公司严格按照Illumina提供的标准方法进行文库的构建和高通量测序: 将5 μg基因组DNA随机打断, 进行片段化; 电泳回收所需长度的DNA片段, 进行末端修复并加上接头制备双端测序文库, 利用Illumina HiSeq 2000测序平台进行高通量测序。
1.3 晋谷21重测序数据分析及InDel和SNP检测
利用Illunima Casava软件对晋谷21经Illumina HiSeq 2000测序平台测序得到的原始图像数据进行碱基识别, 将其转化为原始测序读段(raw reads)。为了保证后续数据分析的质量, 首先将原始测序数据进行过滤, 去除带接头、N所占比例大于5%或质量值≤5的碱基超过50%的低质量测序序列, 从而得到高质量的测序数据。利用BWA软件[22]将晋谷21重测序数据比对到豫谷1号参考基因组[10], 以定位干净读段(clean reads)在参考基因组上的位置, 统计晋谷21基因组测序深度和基因组覆盖度等信息, 并进行InDel和SNP变异的检测。InDel和SNP的检测利用基因组分析工具包(Genome Analysis Toolkit, GATK) GATK[23]进行, 具体流程为: (1)使用Picard软件包(https://sourceforge.net/projects/picard/)的Mark Duplicate工具去除重复序列, 以消除PCR复制的影响; (2)使用基因组分析工具包(Genome Analysis Toolkit, GATK) [23]对SNP和InDel附近位点进行局部重新比对, 校正由于碱基变异产生的比对结果错误; (3)校正碱基质量值; (4)检测InDel和SNP变异位点。
1.4 基于重测序数据的InDel和CAPS分子标记的开发和引物设计
选取晋谷21和豫谷1号差异碱基数在13~50 bp的InDel位点设计InDel引物, 引物设计原则为扩增片段长度为100~250 bp, 且晋谷21和豫谷1号之间的差异在8%以上。基于晋谷21重测序获得的SNP位点信息, 利用在线软件dCAPS Finder 2.0[24]分析最佳酶切位点和选择限制性内切核酸酶, 并设计酶切扩增多态性(cleaved amplified polymorphic sequence, CAPS)标记引物, 引物设计原则为PCR扩增片段长度500~1000 bp。1.5 PCR 扩增及标记验证
PCR体系为20 μL, 包括基因组DNA约50 ng、10× PCR 缓冲液 2 μL, 2 mmol L-1 dNTPs 2 μL, 10 μmol L-1 上下游引物各2 μL和1 U Taq DNA聚合酶。用2.5%琼脂糖凝胶检测PCR或者酶切产物。2 结果与分析
2.1 晋谷21基因组重测序及测序数据统计分析
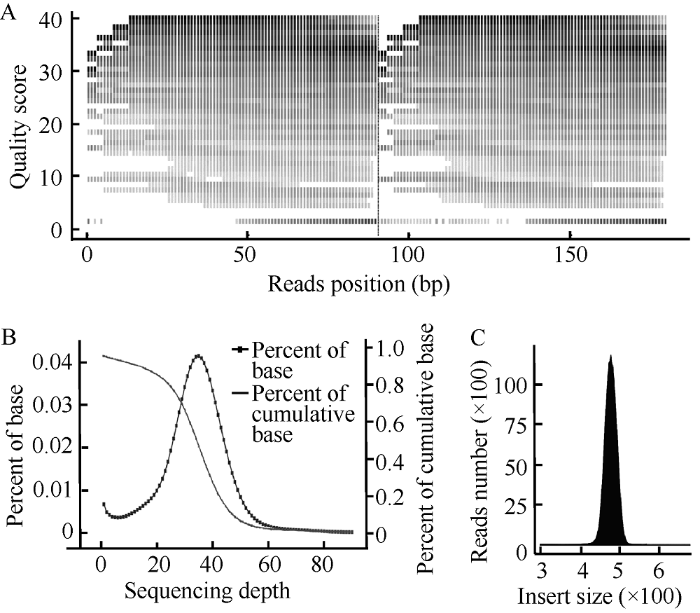
利用Illumina paired-end双端测序, 共获得15.45 Gb的晋谷21基因组原始数据(raw data)。经质量评估和过滤, 最终获得83.05 Mb clean reads, 共计14.95 Gb的高质量测序数据, 测序深度达到36.9×(表2)。其中, 质量值大于等于30的碱基达到92.96%, 表明测序质量较好(表2和图1-A)。为保证结果的可靠性, 所有后续分析都基于上述过滤后的高质量数据。Table 2
表2
表2晋谷21全基因组测序数据信息汇总
Table 2Summary of Jingu 21 whole-genome sequencing information
| 基本信息 Basic information | 测序数据 Sequencing data |
|---|---|
| 清理后读段Clean reads | 83 051 988 bp |
| 清理后碱基Clean bases | 14 949 357 840 bp |
| GC含量GC content | 45.55% |
| Q30值Q30 | 92.96% |
| 匹配读段Mapped reads | 96.16% |
| 1×覆盖度Coverage ratio 1× | 95.34% |
| 5×覆盖度Coverage ratio 5× | 93.39% |
| 10×覆盖度Coverage ratio 10× | 91.54% |
新窗口打开
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图1晋谷21基因组测序质量评估
A: 碱基质量分布图; B: 测序深度分布曲线; C: 测序文库插入片段大小分布图。
-->Fig. 1Evaluation of genome sequencing quality in Jingu 21
A: base quality distribution; B: sequencing-depth distribution curve; C: insert size distribution of the sequencing library.
-->
晋谷21基因组重测序数据与豫谷1号参考基因组平均比对效率为96.16%, 其中双端测序序列均定位到参考基因组上, 且距离符合测序片段长度分布的占93.25%, 基因组覆盖度为95.34% (至少覆盖1×) (表2和图1-B)。为了分析插入片段的实际大小, 检测了双端序列在参考基因组上的起止位置。结果表明插入片段长度分布呈正态分布, 且只有一个单峰, 峰值长度约为480 bp, 这表明测序数据文库构建符合要求(图1-C)。
2.2 基于晋谷21重测序数据的InDel分子标记开发
根据样品晋谷21的测序读段在豫谷1号参考基因组上的定位结果, 利用GATK软件包检测晋谷21与豫谷1号之间是否存在InDel。共发现169 037个InDel位点, 平均密度为417.37个 Mb-1, 这些InDel共导致了1752个基因发生变异。其中, 14 578个InDel位点差异片段长度在13~50 bp之间; 34 733个InDel位点差异片段长度在4~12 bp之间; 117 406个InDel位点差异片段长度在1~3 bp之间(图2)。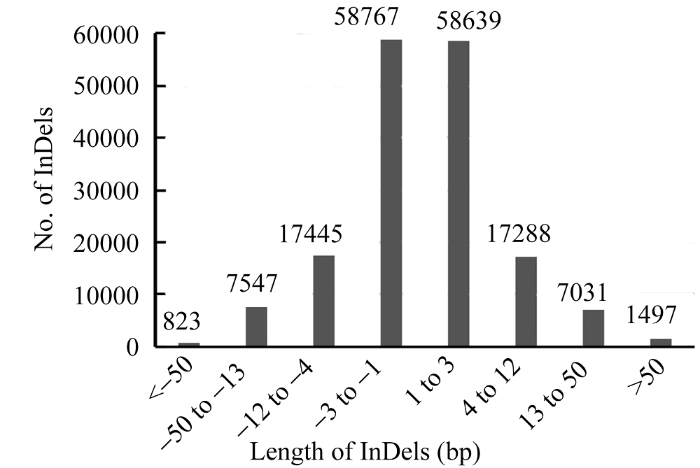 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图2晋谷21和豫谷1号基因组间的InDel统计
正负数分别表示晋谷21基因组中插入(正数)或者缺失(负数)的碱基数。
-->Fig. 2Summary of InDels between Jingu 21 and Yugu 1 genomes
The positive and negative numbers indicate the number of insertion (positive) or deletion (negtive) of base in the Jingu 21 genome.
-->
为了进一步验证候选InDel标记的准确性, 根据其在染色体上的分布情况, 筛选并设计了68对代表性InDel标记引物(附表1)。其中, 8对位于第1染色体, 8对位于第2染色体, 11对位于第3染色体, 8对位于第4染色体, 7对位于第5染色体, 6对位于第6染色体, 6对位于第7染色体, 6对位于第8染色体, 8对位于第9染色体(图3和附表1)。为了检测开发的InDel标记的准确性和上述引物的有效性, 以豫谷1号和晋谷21基因组为模板, 进行了PCR扩增和电泳检测。结果表明, 上述68对引物均能扩增出特异条带。进一步分析表明, 61对引物在豫谷1号和晋谷21之间具有多态性, 且扩增片段大小符合预期结果; 7对引物无多态性或者大小与预期不符(图3和附表1)。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图3InDel标记在晋谷21和豫谷1号的多态性分析
InDel标记详细信息见附
-->Fig. 3Polymorphism analysis of InDel markers between Jingu 21 and Yugu 1
The detailed information of the InDel markers are listed in the supplemental table 1. The first and the second lanes of each marker are Jingu 21 and Yugu 1 respectively. M: D2000 DNA Marker [Tiangen Biotech (Beijing) Co. Ltd., #MD114]. 1~11 represent the order of the markers in supplemental
-->
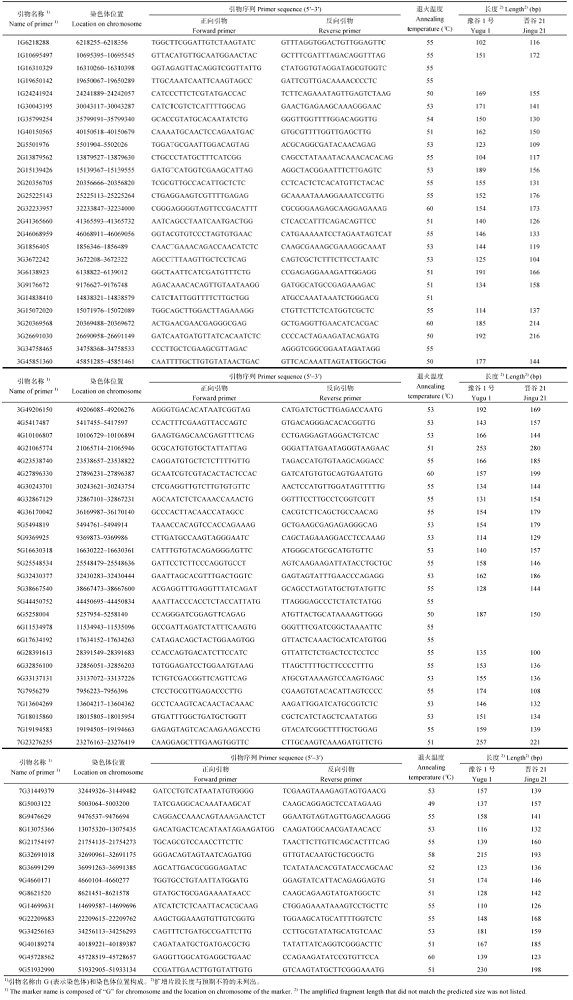
Supplementary table 1
附表1
附表1基于晋谷21基因组重测序的InDel和dCAPS分子标记及其引物
Supplementary table 1Primer sets of InDel markers based on Jingu 21 re-sequencing data
 |
新窗口打开
2.3 基于晋谷21重测序数据的SNP/CAPS分子标记开发与检测
使用GATK软件工具包对晋谷21基因组DNA进行SNP检测, 共获得1 167 555个SNP, 平均密度为2912.33个 Mb-1。其中, 同类型碱基之间突变的转换型SNP位点851 065个, 不同类型碱基之间的颠换型SNP位点 316 490个; 纯合的SNP位点为1 146 969个, 杂合的SNP位点为20 586个, 纯合率为98.24%。利用SnpEff软件[25]对上述SNP进行了注释, 发生在非编码区的突变为1 117 744个, 发生在编码区的突变为49 811个, 这些SNP共导致7098个基因发生非同义突变(表3)。Table 3
表3
表3SNP注释结果
Table 3SNP annotation
| 类型 Type | 数目 Number | 区域 Region |
|---|---|---|
| 基因内Intergenic | 645599 | - |
| 基因间Intragenic | 76 | - |
| 内含子Intron | 83293 | - |
| 基因上游Upstream | 12191 | - |
| 基因下游Downstream | 336911 | - |
| 5′UTR UTR_5′_Prime | 1614 | - |
| 3′UTR UTR_3′_Prime | 3169 | - |
| 剪切受体Splice site acceptor | 111 | - |
| 剪切供体Splice site donor | 88 | - |
| 起始密码子获得Start gained | 831 | - |
| 起始密码子丢失Start lost | 62 | CDS |
| 非同义起始密码子Non synonymous start | 9 | CDS |
| 同义突变Synonymous coding | 21448 | CDS |
| 非同义突变Non synonymous coding | 27694 | CDS |
| 同义终止密码子Synonymous stop | 34 | CDS |
| 终止密码子获得Stop gained | 426 | CDS |
| 终止密码子丢失Stop lost | 138 | CDS |
| 其他Other | 33861 | - |
新窗口打开
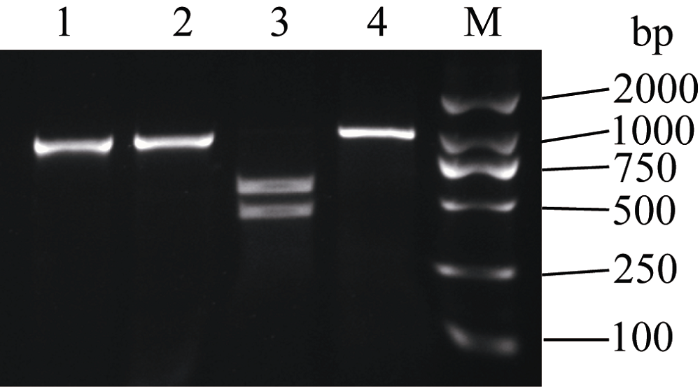
为了验证上述SNP位点的准确性, 我们选取了第3染色体位点14 938 250的SNP进行了验证。在该SNP位点参考基因组序列为C, 而晋谷21为碱基T, 从而导致晋谷21产生了一个新的Nde I酶切位点。据此, 设计CAPS标记引物(5′-TTGAGATTTTGC TGGCGATTGG-3′和5′-CGGCATACTTTTGTCACC CTAC-3′), 并利用晋谷21和豫谷1号基因组DNA进行了PCR验证。如图4所示, 晋谷21和豫谷1号都可以扩增出清晰条带, 且二者条带大小相同。利用Nde I酶切后, 晋谷21片段可以切割成424 bp和538 bp两个片段, 而豫谷1号片段不能被切割(图4)。上述结果与预期相符, 表明基于晋谷21重测序数据开发的该SNP位点是正确的。
 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图4CAPS标记3G14938250对晋谷21和豫谷1号的多态性分析
1: 晋谷21 PCR 扩增产物; 2: 豫谷1号 PCR 扩增产物; 3: 晋谷21 PCR产物的Nde I酶切结果; 4: 豫谷1号PCR 产物Nde I的酶切结果; M: D2000 DNA marker [天根生化科技(北京)有限公司, #MD114]。
-->Fig. 4Polymorphism analysis of CAPS marker 3G14938250 between Jingu 21 and Yugu 1
1: PCR amplicon of Jingu 21; 2: PCR amplicon of Yugu 1; 3: PCR amplicon of Jingu 21 digested with Nde I; 4: PCR amplicon of Yugu 1 digested with Nde I; M: D2000 DNA marker [Tiangen Biotech (Beijing) Co. Ltd., #MD114].
-->
2.4 基于晋谷21重测序数据开发的分子标记的通用性
选取了1份狗尾草、2份谷莠子和12份谷子, 共计15份种质资源验证基于晋谷21重测序数据开发的InDel和SNP/CAPS分子标记的通用性。结果表明, 9对InDel引物都可以在15个谷子种质资源中进行有效的扩增(表1)。其中标记2G5501976仅在晋谷21背景的种质资源和其他13个种质资源之间有多态性, 而在其他13个种质资源之间无多态性; 其余8个InDel标记在所有种质资源中多态性都较好(图5-A)。类似地, 基于SNP重测序数据开发的CAPS标记3G14938250也可以在其他谷子种质资源中有效扩增, 且利用Nde I酶切后表现出较好的多态性(图5-B)。这表明基于晋谷21全基因组重测序数据开发的InDel和SNP/CAPS分子标记在包括狗尾草和谷莠子在内的其他谷子种质资源中具有通用性, 可用于其他谷子种质资源的相关研究。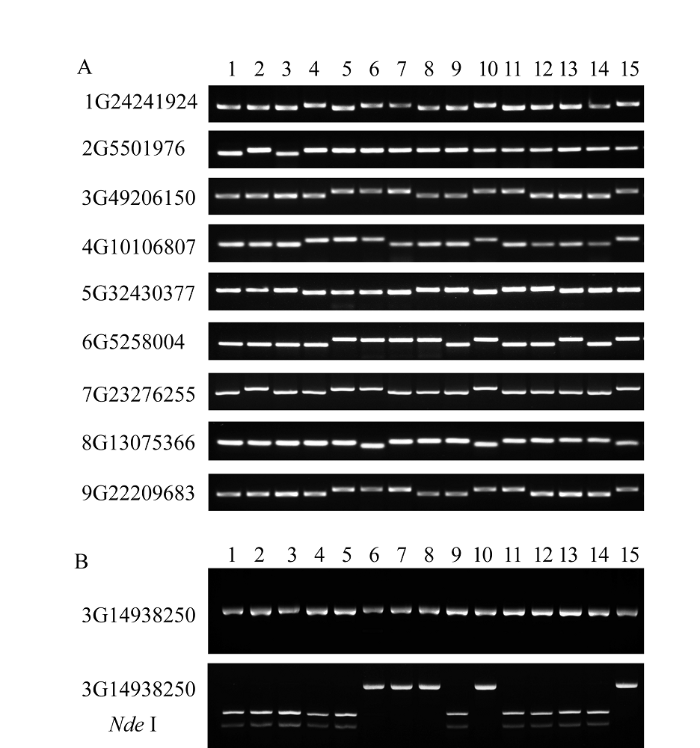 显示原图|下载原图ZIP|生成PPT
显示原图|下载原图ZIP|生成PPT图5InDel和CAPS标记在15个谷子种质资源的多态性分析
A: InDel标记在15个谷子种质资源的多态性分析; B: CAPS标记在15个谷子种质资源的多态性分析。1~15为谷子种质资源, 详细信息见
-->Fig. 5Polymorphic analysis of the InDel and CAPS markers among 15 foxtail millet germplasm resources
A: Polymorphic analysis of InDel markers among 15 foxtail millet germplasm resources; B: Polymorphic analysis of CAPS marker among 15 foxtail millet germplasm resources. The foxtail millet germplasm (1-15) is listed in
-->
3 讨论
3.1 基于二代重测序技术开发晋谷21全基因组分子标记
随着高通量测序技术的发展以及豫谷1号和张谷等谷子基因组测序的完成, 利用二代重测序技术在全基因组水平开发分子标记已经成为可能。Jia等[18]对包括晋谷21在内的916份谷子种质资源进行了重测序, 但测序深度较低, 平均仅为0.7×, 导致晋谷21的SNP数据不全, 且无法开发InDel标记。本研究利用Illumina HiSeq 2000高通量测序平台对名优谷子品种晋谷21进行了36.9×深度测序, 并开发了169 037个InDel标记和1 167 555个SNP标记, 对晋谷21优异米质形成分子机制的解析具有重要价值。与SSR标记一样, InDel标记反映的也是DNA 片段长度的多态性。由于受到测序深度和算法等的限制, 过去往往仅能检测到较小的InDel, 一般小于10 bp[26]。如Bai等[16]利用高通量测序技术开发的谷子InDel标记长度为1~5 bp, 检测这些小的InDel非常困难。为了解决这一问题, 本研究采用了36.9×深度测序, 检测的InDel的长度在1~269 bp之间。其中, 差异片段长度在13~50 bp之间的InDel位点为14 578个, 约占InDel标记总数的8.62% (图2)。这些InDel标记只需要普通琼脂糖凝胶电泳即可检测, 与需要聚丙烯酰胺凝胶电泳检测的SSR标记相比, 可以大大节省人力和物力。
3.2 基于晋谷21重测序数据开发的分子标记的准确性和通用性
设计的68对InDel引物中有61对其扩增片段与预期大小相同, 且呈现出较好的多态性; 而另外7对扩增结果与预期不符, 占所有引物的10.29% (图3)。分别对1G19650142和3G34758465两个标记的豫谷1号和晋谷21扩增片段的测序表明, InDel标记1G19650142 PCR扩增产物为第9染色体23 837 024~ 23 837 239之间216 bp的片段; 而3G34758465 PCR扩增产物为第6染色体10 398 380~10 398 547之间168 bp的片段(王兴春等, 未发表数据)。这表明扩增片段大小与预期不符是非特异扩增而不是高通量测序或者生物信息学分析错误导致的。因此, 建议在设计InDel标记引物时将候选引物序列与豫谷1号参考基因组比对, 选择单一结合位点的引物。分子标记的通用性可以大大节约开发成本, 提高分子标记利用效率。但以往研究多集中在SSR标记的通用性方面, 而有关InDel和SNP等标记通用性的研究较少[27,28]。本研究表明, 基于晋谷21开发的InDel和SNP/CAPS标记也具有较好的通用性, 可用于其他谷子及其近缘野生种狗尾草和谷莠子的相关研究(图5), 但这些标记是否可用于其他禾谷类作物还有待进一步研究。此外, InDel分子标记2G5501976多态性较差, 在除晋谷21背景之外的13个种质资源之间无多态性。因此, 该InDel标记可以作为晋谷21特异标记, 仅用该标记即可高效区分是否为晋谷21及其衍生品种。
3.3 基于晋谷21重测序数据开发的分子标记在小米品质研究和育种中的应用
小米的品质是一个复杂的数量性状, 受众多基因调控。传统方法开发的分子标记数量有限, 难以对品质等复杂农艺性状相关基因进行精细定位。本研究开发了169 037个InDel位点和1 167 555个SNP位点, 平均密度分别为28.58个 Mb-1和2289.32个 Mb-1, 完全可以满足小米品质等复杂农艺性状基因精细定位的需求。InDel标记2G5501976是晋谷21及其衍生品种的特异分子标记, 是这些品种中Seita.2G066500基因启动子缺失14 bp导致的。Seita.2G066500基因编码一个真核生物翻译起始因子, 该基因与晋谷21优异米质的关系还有待进一步研究。此外, 目前水稻中已经克隆了一大批外观品质、营养成分和香味等品质相关的基因[8]。通过序列比对, 筛选谷子中的同源基因, 结合本研究鉴定的InDel和SNP位点, 进一步研究上述品质相关基因在晋谷21中的变异情况, 从而有望揭示晋谷21优异米质形成的分子机制。4 结论
检测到169 037个InDel位点和1 167 555个SNP位点, 初步揭示了晋谷21的基因组信息。开发了InDel和CAPS分子标记, 为深入解析晋谷21优异米质形成的分子机制和分子标记辅助育种奠定了基础。基于晋谷21重测序数据开发的InDel和SNP标记在狗尾草、谷莠子和谷子等谷子种质资源具有通用性, 可用于谷子种质资源鉴定、遗传分析、基因图位克隆和杂交种鉴定。致谢: 感谢中国农业科学院作物科学研究所的刁现民研究员提供了谷子种质资源(表1编号7~15)。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。
参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
| [1] | . 小米在我国起源于黄河流域,栽 培历史悠久,具有生育期短、适应性广、耐干旱、耐贫瘠、耐储存等优点,是北方地区广泛种植的一种小杂粮,它不仅营养丰富、容易消化,还可以进一步加工生产 黄色素、膳食纤维等高附加值产品。对小米在食品、医药和化工行业的应用研究进行总结,并提出下一步的研究方向。 . 小米在我国起源于黄河流域,栽 培历史悠久,具有生育期短、适应性广、耐干旱、耐贫瘠、耐储存等优点,是北方地区广泛种植的一种小杂粮,它不仅营养丰富、容易消化,还可以进一步加工生产 黄色素、膳食纤维等高附加值产品。对小米在食品、医药和化工行业的应用研究进行总结,并提出下一步的研究方向。 |
| [2] | . 采用正相高效液相色谱法测定200份不同省份来源的谷子育成品种籽粒中的4种维生素E(α-VE、β-VE、γ-VE和δ-vE)及叶黄素和玉米黄素2种类胡萝卜素组分的含量,并分析了谷子维生素E含量与类胡萝卜素含量和主要农艺性状的相关性。结果显示,我国谷子育成品种籽粒中VE总量、α-VE含量及(β+γ)-VE含量均呈正态分布,δ-VE含量呈偏态分布,且(β+γ)-VE是维生素E的主要组分;陕西和吉林谷子的VE总量和(β+γ)-VE含量显著高于其他省份品种,东北乏省谷子的α—VE含量显著高于其他省份,而不同省份谷子品种中δ-VE含量差异不显著;不同省份谷子品种间叶黄素含量差异达极显著(P〈0.01),玉米黄素含量差异不显著,类胡萝卜素总含量差异显著(P〈0.05)。相关分析表明,参试品种籽粒中叶黄素含量与VE总量呈显著正相关,与α-VE含量呈极显著正相关,与δ-VE含量呈极显著负相关,与(β+γ)-VE含量不相关;玉米黄素与VE总量和α-VE含量呈显著正相关,与δ-VE含量呈极显著负相关,与(β+γ)-VE含量不著相关;VE总量和(β+γ)-VE含量与千粒重呈显著负相关,δ-VE含量与粒重和穗重呈极显著正相关,类胡萝卜素各组分与草重呈极显著正相关。 . 采用正相高效液相色谱法测定200份不同省份来源的谷子育成品种籽粒中的4种维生素E(α-VE、β-VE、γ-VE和δ-vE)及叶黄素和玉米黄素2种类胡萝卜素组分的含量,并分析了谷子维生素E含量与类胡萝卜素含量和主要农艺性状的相关性。结果显示,我国谷子育成品种籽粒中VE总量、α-VE含量及(β+γ)-VE含量均呈正态分布,δ-VE含量呈偏态分布,且(β+γ)-VE是维生素E的主要组分;陕西和吉林谷子的VE总量和(β+γ)-VE含量显著高于其他省份品种,东北乏省谷子的α—VE含量显著高于其他省份,而不同省份谷子品种中δ-VE含量差异不显著;不同省份谷子品种间叶黄素含量差异达极显著(P〈0.01),玉米黄素含量差异不显著,类胡萝卜素总含量差异显著(P〈0.05)。相关分析表明,参试品种籽粒中叶黄素含量与VE总量呈显著正相关,与α-VE含量呈极显著正相关,与δ-VE含量呈极显著负相关,与(β+γ)-VE含量不相关;玉米黄素与VE总量和α-VE含量呈显著正相关,与δ-VE含量呈极显著负相关,与(β+γ)-VE含量不著相关;VE总量和(β+γ)-VE含量与千粒重呈显著负相关,δ-VE含量与粒重和穗重呈极显著正相关,类胡萝卜素各组分与草重呈极显著正相关。 |
| [3] | . (2017). Foxtail millet: Properties, processing, health benefits, and uses. Food Reviews International. Ahead of Print. doi: 10.1080/87559129.2017.1290103 |
| [4] | . Abstract he millet, containing abundant vitamin, mineral and meal fabric, is a kind of important nutrition and health food. The major constituents of millet yellow pigment are kinds of natural carotenes. The 169 foxtail millet cultivars, including 154 yellow millets, 12 white millets and 3 green millets from different regions, were planted on Nanbin farm in Sanya city, Hainan province in 2009. Yellow pigment content of millet was measured and the appearance quality was appraised to find out the difference of yellow pigment content of millet from different regions and the relation of yellow pigment and appearance quality. The results indicated that yellow pigment content had large range, and that of yellow millets was from 5.40 to 19.55 mg?kg-1 and that of green millets was from 10.14 to 16.44 mg?kg-1 and that of white millets was lower from 1.10 to 2.49 mg?kg-1. The average contents of yellow pigment from different regions had clearly differences, and that of millet from Chifeng, Datong, Lanzhou and Huhehaote was less than 10 mg?kg-1, and that of millet from Fenyang, Baoding, Anyang, Shijiazhuang was higher than 13 mg?kg-1. The millet appearance quality from different regions had a clearly difference, and that of millet from Taiyuan, Changzhi, Datong and Yan n was poorer, and the proportion of quality millets was lower, while that of millet from Hengshui, Cangzhou, Baoding, Anyang and Shijiazhuang was better, and the proportion of quality millets was higher. The relation of millet appearance quality and yellow pigment content is significant. The yellow pigment content is an important factor of millet appearance quality, and may be used as an important indicator of quality breeding. . Abstract he millet, containing abundant vitamin, mineral and meal fabric, is a kind of important nutrition and health food. The major constituents of millet yellow pigment are kinds of natural carotenes. The 169 foxtail millet cultivars, including 154 yellow millets, 12 white millets and 3 green millets from different regions, were planted on Nanbin farm in Sanya city, Hainan province in 2009. Yellow pigment content of millet was measured and the appearance quality was appraised to find out the difference of yellow pigment content of millet from different regions and the relation of yellow pigment and appearance quality. The results indicated that yellow pigment content had large range, and that of yellow millets was from 5.40 to 19.55 mg?kg-1 and that of green millets was from 10.14 to 16.44 mg?kg-1 and that of white millets was lower from 1.10 to 2.49 mg?kg-1. The average contents of yellow pigment from different regions had clearly differences, and that of millet from Chifeng, Datong, Lanzhou and Huhehaote was less than 10 mg?kg-1, and that of millet from Fenyang, Baoding, Anyang, Shijiazhuang was higher than 13 mg?kg-1. The millet appearance quality from different regions had a clearly difference, and that of millet from Taiyuan, Changzhi, Datong and Yan n was poorer, and the proportion of quality millets was lower, while that of millet from Hengshui, Cangzhou, Baoding, Anyang and Shijiazhuang was better, and the proportion of quality millets was higher. The relation of millet appearance quality and yellow pigment content is significant. The yellow pigment content is an important factor of millet appearance quality, and may be used as an important indicator of quality breeding. |
| [5] | . 利用三体分析法进行谷子染色体基因定位。以豫谷1号三体1~7、四体8和四体9为母本,显性矮秆、法谷56-81、马青苗为父本,配置组合分别进行显性矮秆基因、白米基因和青米基因染色体定位。根据三体1~7植株形态特征和父本标志性状鉴定各自的杂种F1,采用细胞学方法,通过检查植株根尖染色体数来鉴定杂种F1的三体8和三体9。经调查和分析各种三体杂种F2性状分离情况,把显性矮秆基因定位在3号染色体,白米基因定位在4号染色体,青米基因定位在6号染色体。对不同区域的9个青米品种等位检测表明,这些青米基因都是等位基因。以两点测验法,配置组合1066A×法谷56-81和1066A×马青苗进行基因连锁分析。结果表明,4号染色体上糯性胚乳基因与白米基因间的交换值为(28.9±4.4)cM;6号染色体上1066A不育基因与青米基因间的交换值为(23.2±1.8)cM。 . 利用三体分析法进行谷子染色体基因定位。以豫谷1号三体1~7、四体8和四体9为母本,显性矮秆、法谷56-81、马青苗为父本,配置组合分别进行显性矮秆基因、白米基因和青米基因染色体定位。根据三体1~7植株形态特征和父本标志性状鉴定各自的杂种F1,采用细胞学方法,通过检查植株根尖染色体数来鉴定杂种F1的三体8和三体9。经调查和分析各种三体杂种F2性状分离情况,把显性矮秆基因定位在3号染色体,白米基因定位在4号染色体,青米基因定位在6号染色体。对不同区域的9个青米品种等位检测表明,这些青米基因都是等位基因。以两点测验法,配置组合1066A×法谷56-81和1066A×马青苗进行基因连锁分析。结果表明,4号染色体上糯性胚乳基因与白米基因间的交换值为(28.9±4.4)cM;6号染色体上1066A不育基因与青米基因间的交换值为(23.2±1.8)cM。 |
| [6] | . 谷子是中国北方重要杂粮作物,米色是评价谷子品质的重要指标,目前关于谷子米色的形成机制仍不明确。本研究选用米色分别为深黄、浅黄、白色和绿色的谷子品种各2个,对这8个谷子品种进行了总类胡萝卜素含量、β-胡萝卜素含量与米色差异间关系的分析,以及分子水平4个β-胡萝卜素代谢相关基因在籽粒不同灌浆阶段的表达模式分析,结果表明:CCI指数作为米色测定的综合指标,可对不同品种米色差异进行鉴定,且分别与总类胡萝卜素、β-胡萝卜素含量呈显著与极显著正相关。通过对8个品种SiLCYB基因组DNA克隆发现,只有深黄品种JG21中出现序列变异,有两个SNP位点,且第二处单核苷酸变异使相应氨基酸由谷氨酰胺变为精氨酸。SiLCYB表达不具有组织特异性,在叶中表达最高,茎中最低。通过对籽粒不同灌浆阶段β-胡萝卜素合成基因SiLCYB与另外3个β-胡萝卜素代谢相关基因(SiLCYBLCYE,SiHYD,SiCCD1)的表达分析发现:SiLCYB在大多数品种中表达基本恒定,且与β-胡萝卜素积累没有表现出显著相关性;而SiLCYE在不同品种中普遍呈现出与SiLCYB同增同减的表达模式,但表达水平低于SiLCYB;同时发现2个降解相关基因(SiHYD,SiCCD1)的表达与β-胡萝卜素积累在灌浆特定阶段表现出了显著或极显著负相关。因此,推测SiLCYB与降解基因SiHYD和SiCCD1共同作用,通过影响β-胡萝卜素和总类胡萝卜素在籽粒中的积累,进而影响米色的形成。 . 谷子是中国北方重要杂粮作物,米色是评价谷子品质的重要指标,目前关于谷子米色的形成机制仍不明确。本研究选用米色分别为深黄、浅黄、白色和绿色的谷子品种各2个,对这8个谷子品种进行了总类胡萝卜素含量、β-胡萝卜素含量与米色差异间关系的分析,以及分子水平4个β-胡萝卜素代谢相关基因在籽粒不同灌浆阶段的表达模式分析,结果表明:CCI指数作为米色测定的综合指标,可对不同品种米色差异进行鉴定,且分别与总类胡萝卜素、β-胡萝卜素含量呈显著与极显著正相关。通过对8个品种SiLCYB基因组DNA克隆发现,只有深黄品种JG21中出现序列变异,有两个SNP位点,且第二处单核苷酸变异使相应氨基酸由谷氨酰胺变为精氨酸。SiLCYB表达不具有组织特异性,在叶中表达最高,茎中最低。通过对籽粒不同灌浆阶段β-胡萝卜素合成基因SiLCYB与另外3个β-胡萝卜素代谢相关基因(SiLCYBLCYE,SiHYD,SiCCD1)的表达分析发现:SiLCYB在大多数品种中表达基本恒定,且与β-胡萝卜素积累没有表现出显著相关性;而SiLCYE在不同品种中普遍呈现出与SiLCYB同增同减的表达模式,但表达水平低于SiLCYB;同时发现2个降解相关基因(SiHYD,SiCCD1)的表达与β-胡萝卜素积累在灌浆特定阶段表现出了显著或极显著负相关。因此,推测SiLCYB与降解基因SiHYD和SiCCD1共同作用,通过影响β-胡萝卜素和总类胡萝卜素在籽粒中的积累,进而影响米色的形成。 |
| [7] | . 采用快速黏度分析仪(RVA)分析全国27个小米品种的RVA谱线和各特征值间的相关性。结果表明:小米品种间的RVA谱特征值差异显著,小米衰减值的测定对于小米的品种鉴别具有较强的区分能力;直链淀粉含量与衰减值呈极显著负相关,与回生值呈极显著的正相关。可以利用直链淀粉含量并辅助RVA谱特征值进行小米品质的鉴定。 . 采用快速黏度分析仪(RVA)分析全国27个小米品种的RVA谱线和各特征值间的相关性。结果表明:小米品种间的RVA谱特征值差异显著,小米衰减值的测定对于小米的品种鉴别具有较强的区分能力;直链淀粉含量与衰减值呈极显著负相关,与回生值呈极显著的正相关。可以利用直链淀粉含量并辅助RVA谱特征值进行小米品质的鉴定。 |
| [8] | . 水稻是中国重要的粮食作物之一,高产与优质一直是品种改良的主要目标。目前,中国稻米品质表现总体偏低,在一定程度上影响了其市场竞争力。稻米品质属综合性状,是指稻米或稻米相关产品满足消费者或生产加工需求的各种特性,主要涉及稻米的物理和化学特性,包括精米率、米粒形状、透明度、蒸煮时间、米饭质地与香味、冷饭质地以及营养成分等指标。通常用碾磨品质、外观品质、蒸煮与食味品质和营养品质4个方面来评价稻米品质。近10年来,在上述稻米品质性状相关基因的克隆与功能研究领域已取得了长足的进展。水稻粒形不仅是重要的产量性状也是碾磨和外观品质的重要决定因素,目前已克隆了多个粒形相关的QTL和基因。根据粒形相关基因的表型效应可将其分为3类,即伴随植株矮化的小粒控制基因(第一类,包括D1、D2、D11、D61和SMG1等)、粒形特异基因(第二类,如GS3、GL3.1、GW7、GW2、GW5、GS5、GS6、TGW6、GW8、BG2、GW6a和GS2等)和小圆粒基因(第三类,即SRS),其中只有第二类基因具有较好的育种利用价值。垩白是决定稻米外观品质的首要性状,同时也会影响碾磨品质。目前尽管已经鉴定了大量QTL,但只有少数QTL被精细定位和克隆,如Chalk5、cyPPDK、G1F1、OsRab5a、FLOURYENDOSPERM2、PDIL1-1和SSG4等主要通过调控胚乳灌浆和储藏物积累而影响稻米外观表现。淀粉占精米胚乳干重的90%以上,其组成与结构是决定稻米外观和蒸煮与食味品质的最重要因素。淀粉的合成是由多基因参与的复杂调控网络,直接参与淀粉合成的淀粉合成酶类基因的功能已经比较清楚;此外,参与胚乳淀粉代谢的一些转录因子如Dull、OsEBP89、OsEBP5、OsRSR1和OsbZIP58等也已被陆续鉴定和克隆。蛋白质是稻米的第二大成分,目前已克隆了众多的贮藏蛋白编码基因,并且已鉴定克隆了多个与蛋白质 . 水稻是中国重要的粮食作物之一,高产与优质一直是品种改良的主要目标。目前,中国稻米品质表现总体偏低,在一定程度上影响了其市场竞争力。稻米品质属综合性状,是指稻米或稻米相关产品满足消费者或生产加工需求的各种特性,主要涉及稻米的物理和化学特性,包括精米率、米粒形状、透明度、蒸煮时间、米饭质地与香味、冷饭质地以及营养成分等指标。通常用碾磨品质、外观品质、蒸煮与食味品质和营养品质4个方面来评价稻米品质。近10年来,在上述稻米品质性状相关基因的克隆与功能研究领域已取得了长足的进展。水稻粒形不仅是重要的产量性状也是碾磨和外观品质的重要决定因素,目前已克隆了多个粒形相关的QTL和基因。根据粒形相关基因的表型效应可将其分为3类,即伴随植株矮化的小粒控制基因(第一类,包括D1、D2、D11、D61和SMG1等)、粒形特异基因(第二类,如GS3、GL3.1、GW7、GW2、GW5、GS5、GS6、TGW6、GW8、BG2、GW6a和GS2等)和小圆粒基因(第三类,即SRS),其中只有第二类基因具有较好的育种利用价值。垩白是决定稻米外观品质的首要性状,同时也会影响碾磨品质。目前尽管已经鉴定了大量QTL,但只有少数QTL被精细定位和克隆,如Chalk5、cyPPDK、G1F1、OsRab5a、FLOURYENDOSPERM2、PDIL1-1和SSG4等主要通过调控胚乳灌浆和储藏物积累而影响稻米外观表现。淀粉占精米胚乳干重的90%以上,其组成与结构是决定稻米外观和蒸煮与食味品质的最重要因素。淀粉的合成是由多基因参与的复杂调控网络,直接参与淀粉合成的淀粉合成酶类基因的功能已经比较清楚;此外,参与胚乳淀粉代谢的一些转录因子如Dull、OsEBP89、OsEBP5、OsRSR1和OsbZIP58等也已被陆续鉴定和克隆。蛋白质是稻米的第二大成分,目前已克隆了众多的贮藏蛋白编码基因,并且已鉴定克隆了多个与蛋白质 |
| [9] | . Foxtail millet is a minor yet important crop in some areas of the world, particularly northern China. It has strong adaptability to abiotic stresses, especially drought, and poor soil. It also has high nutritional value. Foxtail millet is rich in essential amino acids, fatty acids and minerals, and is considered to be one of the most digestible and non-allergenic grains available and has significant importance for human health. Given foxtail millet ability to adapt to abiotic stresses associated with climate change, it is more important than ever to develop breeding strategies that facilitate the increasing demand for high quality grain that better satisfies consumers. Here we review research on foxtail millet quality evaluation, appearance, cooking and eating quality at the phenotypic level. We review analysis of the main nutrients in foxtail millet, their relationships and the biochemical and genetic factors affecting their accumulation. In addition, we review past progress in breeding this regionally important crop, outline current status of breeding of foxtail millet, and make suggestions to improve grain quality. |
| [10] | . We generated a high-quality reference genome sequence for foxtail millet (Setaria italica). The 65400-Mb assembly covers 6580% of the genome and >95% of the gene space. The assembly was anchored to a 992-locus genetic map and was annotated by comparison with >1.3 million expressed sequence tag reads. We produced more than 580 million RNA-Seq reads to facilitate expression analyses. We also sequenced Setaria viridis, the ancestral wild relative of S. italica, and identified regions of differential single-nucleotide polymorphism density, distribution of transposable elements, small RNA content, chromosomal rearrangement and segregation distortion. The genus Setaria includes natural and cultivated species that demonstrate a wide capacity for adaptation. The genetic basis of this adaptation was investigated by comparing five sequenced grass genomes. We also used the diploid Setaria genome to evaluate the ongoing genome assembly of a related polyploid, switchgrass (Panicum virgatum). |
| [11] | . |
| [12] | . Background Foxtail millet ( Setaria italica(L.) Beauv.) is an important gramineous grain-food and forage crop. It is grown worldwide for human and livestock consumption. Its small genome and diploid... |
| [13] | . Background Foxtail millet [ Setaria italica(L.) P. Beauv.], a crop of historical importance in China, has been adopted as a model crop for studying C-4 photosynthesis, stress biology and biofuel... |
| [14] | . Abstract The prominent attributes of foxtail millet (Setaria italica L.) including its small genome size, short life cycle, inbreeding nature, and phylogenetic proximity to various biofuel crops have made this crop an excellent model system to investigate various aspects of architectural, evolutionary and physiological significances in Panicoid bioenergy grasses. After release of its whole genome sequence, large-scale genomic resources in terms of molecular markers were generated for the improvement of both foxtail millet and its related species. Hence it is now essential to congregate, curate and make available these genomic resources for the benefit of researchers and breeders working towards crop improvement. In view of this, we have constructed the Foxtail millet Marker Database (FmMDb; http://www.nipgr.res.in/foxtail.html), a comprehensive online database for information retrieval, visualization and management of large-scale marker datasets with unrestricted public access. FmMDb is the first database which provides complete marker information to the plant science community attempting to produce elite cultivars of millet and bioenergy grass species, thus addressing global food insecurity. |
| [15] | . Transposable elements (TEs) are major components of plant genome and are reported to play significant roles in functional genome diversity and phenotypic variations. Several TEs are highly polymorphic for insert location in the genome and this facilitates development of TE-based markers for various genotyping purposes. Considering this, a genome-wide analysis was performed in the model plant foxtail millet. A total of 30,706 TEs were identified and classified as DNA transposons (24,386), full-length Copia type (1,038), partial or solo Copia type (10,118), full-length Gypsy type (1,570), partial or solo Gypsy type (23,293) and Long- and Short-Interspersed Nuclear Elements (3,659 and 53, respectively). Further, 20,278 TE-based markers were developed, namely Retrotransposon-Based Insertion Polymorphisms (4,801, ???24%), Inter-Retrotransposon Amplified Polymorphisms (3,239, ???16%), Repeat Junction Markers (4,451, ???22%), Repeat Junction-Junction Markers (329, ???2%), Insertion-Site-Based Polymorphisms (7,401, ???36%) and Retrotransposon-Microsatellite Amplified Polymorphisms (57, 0.2%). A total of 134 Repeat Junction Markers were screened in 96 accessions of Setaria italica and 3 wild Setaria accessions of which 30 showed polymorphism. Moreover, an open access database for these developed resources was constructed (Foxtail millet Transposable Elements-based Marker Database; http://59.163.192.83/ltrdb/index.html). Taken together, this study would serve as a valuable resource for large-scale genotyping applications in foxtail millet and related grass species.<br> |
| [16] | . Foxtail millet (Setariaitalica) is a drought-resistant, barren-tolerant grain crop and forage. Currently, it has become a new model plant for cereal crops and biofuel grasses. Although two reference genome sequences were released recently, comparative genomics research on foxtail millet is still in its infancy. Using the Solexa sequencing technology, we performed genome re-sequencing on one important foxtail millet Landrace, Shi-Li-Xiang (SLX). Compared with the two reference genome sequences, the following genetic variation patterns were identified: 762,082 SNPs, 26,802 insertion/deletion polymorphisms of 1 to 5 bp in length (indels), and 10,109 structural variations (SVs) between SLX and Yugu1 genomes; 915,434 SNPs, 28,546 indels and 12,968 SVs between SLX and Zhang gu genomes. Furthermore, based on the Yugu1 genome annotation, we found out that ~ 40% SNPs resided in genes containing NB-ARC domain, protein kinase or leucine-rich repeats, which had higher non-synonymous to synonymous SNPs ratios than average, suggesting that the diversification of plant disease resistance proteins might be caused by pathogen pressure. In addition, out of the polymorphisms identified between SLX and Yugu1, 465 SNPs and 146 SVs were validated with more than 90% accuracy, which could be used as DNA markers for whole-genome genotyping and marker-assisted breeding. Here, we also represented an example of fine mapping and identifying awaxylocus in SLX using these newly developed DNA markers. This work provided important information that will allow a deeper understanding of the foxtail millet genome and will be helpful for dissecting the genetic basis of important traits in foxtail millet. |
| [17] | . Abstract Foxtail millet (Setaria italica), a very important grain crop in China, has become a new model plant for cereal crops and biofuel grasses. Although its reference genome sequence was released recently, quantitative trait loci (QTLs) controlling complex agronomic traits remains limited. The development of massively parallel genotyping methods and next-generation sequencing technologies provides an excellent opportunity for developing single-nucleotide polymorphisms (SNPs) for linkage map construction and QTL analysis of complex quantitative traits. In this study, a high-throughput and cost-effective RAD-seq approach was employed to generate a high-density genetic map for foxtail millet. A total of 2,668,587 SNP loci were detected according to the reference genome sequence; meanwhile, 9,968 SNP markers were used to genotype 124 F2 progenies derived from the cross between Hongmiaozhangu and Changnong35; a high-density genetic map spanning 1648.8 cM, with an average distance of 0.17 cM between adjacent markers was constructed; 11 major QTLs for eight agronomic traits were identified; five co-dominant DNA markers were developed. These findings will be of value for the identification of candidate genes and marker-assisted selection in foxtail millet. |
| [18] | . Foxtail millet (Setaria italica) is an important grain crop that is grown in arid regions. Here we sequenced 916 diverse foxtail millet varieties, identified 2.58 million SNPs and used 0.8 million common SNPs to construct a haplotype map of the foxtail millet genome. We classified the foxtail millet varieties into two divergent groups that are strongly correlated with early and late flowering times. We phenotyped the 916 varieties under five different environments and identified 512 loci associated with 47 agronomic traits by genome-wide association studies. We performed a de novo assembly of deeply sequenced genomes of a Setaria viridis accession (the wild progenitor of S. italica) and an S. italica variety and identified complex interspecies and intraspecies variants. We also identified 36 selective sweeps that seem to have occurred during modern breeding. This study provides fundamental resources for genetics research and genetic improvement in foxtail millet. |
| [19] | . 正 1 选育经过晋谷21号原名晋汾7号,经过14年的选育历程。1972年由中国科学院原子能研究所利用钴60咖玛(Y)射线处理晋汾52干种子,M-1代三码 取样混脱留种,F-3经南繁北育单株选择,1975年升鉴定圃,1976年参加品种比较试验,1979年开始在交口回龙乡大面积种植。群众认为,咖玛谷 (指晋汾7号)不 . 正 1 选育经过晋谷21号原名晋汾7号,经过14年的选育历程。1972年由中国科学院原子能研究所利用钴60咖玛(Y)射线处理晋汾52干种子,M-1代三码 取样混脱留种,F-3经南繁北育单株选择,1975年升鉴定圃,1976年参加品种比较试验,1979年开始在交口回龙乡大面积种植。群众认为,咖玛谷 (指晋汾7号)不 |
| [20] | . 尽管谷子(Setaria italica)全基因组序列图谱已经公布,但其基因注释很不完善。为此,本文应用RNA-Seq技术开展了谷子新基因发掘和已注释基因结构优化工作。以‘晋谷21’谷子叶片为材料提取总RNA,构建测序文库并利用Illumina HiSeq2500测序平台进行双端测序,最终获得37 072 949条高质量的干净读段(clean reads)。将其进一步与‘豫谷1号’谷子参考基因组进行序列比对,鉴定出614个新基因。在此基础上,利用COG、GO、KEGG、Swiss-Prot和NR等数据库对其进行了功能注释,获得了438个新基因的注释信息。此外,还优化了7 175个已注释基因的结构,延伸了4 330个基因的5′端和5 362个基因的3′端。本研究旨在为后续谷子功能基因组学研究和其他生物基因组注释信息的完善提供有益的借鉴。 . 尽管谷子(Setaria italica)全基因组序列图谱已经公布,但其基因注释很不完善。为此,本文应用RNA-Seq技术开展了谷子新基因发掘和已注释基因结构优化工作。以‘晋谷21’谷子叶片为材料提取总RNA,构建测序文库并利用Illumina HiSeq2500测序平台进行双端测序,最终获得37 072 949条高质量的干净读段(clean reads)。将其进一步与‘豫谷1号’谷子参考基因组进行序列比对,鉴定出614个新基因。在此基础上,利用COG、GO、KEGG、Swiss-Prot和NR等数据库对其进行了功能注释,获得了438个新基因的注释信息。此外,还优化了7 175个已注释基因的结构,延伸了4 330个基因的5′端和5 362个基因的3′端。本研究旨在为后续谷子功能基因组学研究和其他生物基因组注释信息的完善提供有益的借鉴。 |
| [21] | . |
| [22] | . |
| [23] | . |
| [24] | . |
| [25] | . |
| [26] | . AbstractGenome-wide detection of short insertion/deletion length polymorphisms (InDels, |
| [27] | . 藜麦因营养均衡受到越来越多的关注,但尚未深入开展其基础研究。开发微卫星序列重复SSR分子标记将为藜麦的遗传分析提供重要资源。本研究利用NCBI数据库中藜麦RNA测序RNA-Seq及表达序列标签EST数据挖掘、验证及评价藜麦EST-SSR,共发现1862个藜麦非单核苷酸EST-SSR。其中,二核苷酸重复最多(38.3%),六核苷酸重复最少(11.7%)。不同重复类型SSR的数量随着核苷酸数目的增加呈下降趋势。在随机选取验证的119个EST-SSR标记中,66(55.9%)个能够扩增出清晰条带,39个在4份藜麦资源中具有多态性,且其多态性与重复序列长度不具有显著相关性。t测验显示,多态性EST-SSR在藜麦与其他藜科种质间不存在显著差异,说明其具有良好的通用性,可用于藜科物种的遗传关系分析。 . 藜麦因营养均衡受到越来越多的关注,但尚未深入开展其基础研究。开发微卫星序列重复SSR分子标记将为藜麦的遗传分析提供重要资源。本研究利用NCBI数据库中藜麦RNA测序RNA-Seq及表达序列标签EST数据挖掘、验证及评价藜麦EST-SSR,共发现1862个藜麦非单核苷酸EST-SSR。其中,二核苷酸重复最多(38.3%),六核苷酸重复最少(11.7%)。不同重复类型SSR的数量随着核苷酸数目的增加呈下降趋势。在随机选取验证的119个EST-SSR标记中,66(55.9%)个能够扩增出清晰条带,39个在4份藜麦资源中具有多态性,且其多态性与重复序列长度不具有显著相关性。t测验显示,多态性EST-SSR在藜麦与其他藜科种质间不存在显著差异,说明其具有良好的通用性,可用于藜科物种的遗传关系分析。 |
| [28] | . Expressed sequence tags (ESTs) are important resource for gene discovery, gene expression and its regulation, molecular marker development, and comparative genomics. We procured 10000 ESTs and analyzed 267 EST-SSRs markers through computational approach. The average density was one SSR/10.45???kb or 6.4% frequency, wherein trinucleotide repeats (66.74%) were the most abundant followed by di- (26.10%), tetra- (4.67%), penta- (1.5%), and hexanucleotide (1.2%) repeats. Functional annotations were done and after-effect newly developed 63 EST-SSRs were used for cross transferability, genetic diversity, and bulk segregation analysis (BSA). Out of 63 EST-SSRs, 42 markers were identified owing to their expansion genetics across 20 different plants which amplified 519 alleles at 180 loci with an average of 2.88 alleles/locus and the polymorphic information content (PIC) ranged from 0.51 to 0.93 with an average of 0.83. The cross transferability ranged from 25% for wheat to 97.22% for Schlerostachya, with an average of 55.86%, and genetic relationships were established based on diversification among them. Moreover, 10 EST-SSRs were recognized as important markers between bulks of pooled DNA of sugarcane cultivars through BSA. This study highlights the employability of the markers in transferability, genetic diversity in grass species, and distinguished sugarcane bulks. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}