 ,1,2, 邹枚伶2, 张辰笈2,4, 江思容2,5, Eder Jorge de Oliveira6, 张圣奎7, 夏志强,2,3,4,*, 王文泉,2,3,4,5,*, 李有志,1,*
,1,2, 邹枚伶2, 张辰笈2,4, 江思容2,5, Eder Jorge de Oliveira6, 张圣奎7, 夏志强,2,3,4,*, 王文泉,2,3,4,5,*, 李有志,1,*Genetic diversity and population structure analysis by SNP and InDel markers of cassava in Brazil
SUN Qian,1,2, ZOU Mei-Ling2, ZHANG Chen-Ji2,4, JIANG Si-Rong2,5, Eder Jorge de Oliveira6, ZHANG Sheng-Kui7, XIA Zhi-Qiang,2,3,4,*, WANG Wen-Quan,2,3,4,5,*, LI You-Zhi,1,*通讯作者:
收稿日期:2020-03-13接受日期:2020-08-19网络出版日期:2021-01-12
| 基金资助: |
Received:2020-03-13Accepted:2020-08-19Online:2021-01-12
| Fund supported: |
作者简介 About authors
E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (2162KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
孙倩, 邹枚伶, 张辰笈, 江思容, Eder Jorge de Oliveira, 张圣奎, 夏志强, 王文泉, 李有志. 基于SNP和InDel标记的巴西木薯遗传多样性与群体遗传结构分析[J]. 作物学报, 2021, 47(1): 42-49. doi:10.3724/SP.J.1006.2021.04067
SUN Qian, ZOU Mei-Ling, ZHANG Chen-Ji, JIANG Si-Rong, Eder Jorge de Oliveira, ZHANG Sheng-Kui, XIA Zhi-Qiang, WANG Wen-Quan, LI You-Zhi.
木薯(Manihot esculenta Crantz)是大戟科(Euphorbiaceae)木薯属(Manihot P. Mill.)的多年生灌木植物, 具有高生物量、抗贫瘠、抗病虫能力强等特点, 被广泛种植于亚、非、美三洲等多个国家或地区[1]。木薯起源于亚马逊河流域, 于19世纪20年代传入中国, 最初种植于广东省, 之后逐渐在海南、广西、贵州、云南等地大量种植。它是世界三大薯类作物之一, 同时也是世界上第六大粮食作物, 仅次于小麦、水稻、玉米、马铃薯和大麦[2]。其用途广泛, 除可食用、饲用外, 还可用于生产加工, 如造纸、纺织、生物燃料等[3]。木薯块根还可用于提取淀粉, 加工成薯条、面包, 以及生产燃料乙醇等; 茎秆可用来进行木薯繁殖、粉碎还田、做燃料等; 叶片可作蔬菜食用或喂鱼、养蚕等[4]。
木薯基因组具有高度杂合的特性, 原因是其异花授粉, 且长期进行无性繁殖。由于基因组的高度杂合, 从而增加了木薯遗传变异的多样性, 这些多样性可为木薯育种人员提供更多可选择的良好亲本, 但同时由于木薯的基因组高度杂合、亲缘关系不清晰、遗传改良周期长等特点也增加了育种的工作难度[5]。目前已有一些利用相关序列扩增多态性(sequence-related amplified polymorphism, SRAP)、简单重复序列(simple sequence repeat, SSR)、扩增片段长度多态性 (amplified fragment length polymorphism, AFLP)、单核苷酸多态性(single nucleotide polymorphisms, SNP)等分子标记进行木薯遗传多样性的研究。Fregene等[6]利用SSR标记对来源于哥伦比亚、巴西和秘鲁等地的木薯地方品种的种质资源多样性评价发现, 不同国家来源的木薯种质的遗传多样性水平都很高, 其中来自巴西和哥伦比亚材料的基因多样性水平最高。Alex等[7]利用13对SSR标记对巴西多地的传统甜木薯品种的群体结构和遗传多样性评估结果显示, 该群体的遗传多样性平均值为0.5407, 范围为0.3138 (GA21)~0.6502 (GA140), 表明该群体的遗传变异性宽泛。Carvalho等[8]采用SSR标记和RAPD标记的研究中发现, 巴西的木薯种质资源的地理来源和遗传聚类具有显著正相关关系。
丰富木薯种质资源的遗传多样性, 并对其遗传背景和性状进行综合评价, 发掘控制优良性状的优异等位基因, 对今后木薯育种具有重大意义。全基因组关联分析(genome-wide association study, GWAS)能够鉴定目的表型性状与遗传标记或基因间的关系, 并检测出控制相关性状的优良等位基因位点[9]。而进行关联分析需要先评估实验群体的遗传多样性、遗传结构及亲缘关系[10]。但目前利用SNP和InDel标记对木薯进行遗传多样性、亲缘关系及群体结构分析等的相关研究还鲜为报道。
本研究拟利用SNP和InDel分子标记, 对由巴西Embrapa机构提供的来源于巴西多地的192份木薯种质资源进行遗传多样性和群体结构分析。本研究将为以后木薯育种亲本选配提供材料和理论指导, 也可为下一步通过关联分析发掘控制木薯种质中优良性状的优异等位基因提供理论依据, 从而促进利用分子标记辅助选择技术培育木薯新品种。
1 材料与方法
1.1 试验材料
供试木薯材料共192份, 均为巴西栽培种木薯(表1)。Table 1
表1
表1192份木薯栽培种
Table 1
| 编号 No. | 名称 Name | 编号 No. | 名称 Name | 编号 No. | 名称 Name | 编号 No. | 名称 Name | 编号 No. | 名称 Name | 编号 No. | 名称 Name |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 001 | 9624-09 | 033 | BGM0400 | 065 | BGM0783 | 097 | BGM1291 | 129 | BGM1608 | 161 | BGM1942 |
| 002 | 98150-06 | 034 | BGM0405 | 066 | BGM0788 | 098 | BGM1313 | 130 | BGM1615 | 162 | BGM1957 |
| 003 | BGM0020 | 035 | BGM0408 | 067 | BGM0807 | 099 | BGM1324 | 131 | BGM1622 | 163 | BGM2022 |
| 004 | BGM0042 | 036 | BGM0425 | 068 | BGM0886 | 100 | BGM1327 | 132 | BGM1626 | 164 | BGM2028 |
| 005 | BGM0045 | 037 | BGM0428 | 069 | BGM0889 | 101 | BGM1328 | 133 | BGM1640 | 165 | BGM2041 |
| 006 | BGM0073 | 038 | BGM0436 | 070 | BGM0896 | 102 | BGM1364 | 134 | BGM1662 | 166 | BGM2047 |
| 007 | BGM0087 | 039 | BGM0443 | 071 | BGM0905 | 103 | BGM1370 | 135 | BGM1667 | 167 | BGM2052 |
| 008 | BGM0103 | 040 | BGM0451 | 072 | BGM0917 | 104 | BGM1376 | 136 | BGM1668 | 168 | BGM2061 |
| 009 | BGM0135 | 041 | BGM0452 | 073 | BGM0930 | 105 | BGM1387 | 137 | BGM1672 | 169 | BGM2062 |
| 010 | BGM0145 | 042 | BGM0467 | 074 | BGM0972 | 106 | BGM1396 | 138 | BGM1679 | 170 | BGM2063 |
| 011 | BGM0152 | 043 | BGM0495 | 075 | BGM0976 | 107 | BGM1409 | 139 | BGM1682 | 171 | BGM2071 |
| 012 | BGM0163 | 044 | BGM0501 | 076 | BGM0982 | 108 | BGM1412 | 140 | BGM1684 | 172 | BGM2078 |
| 013 | BGM0179 | 045 | BGM0507 | 077 | BGM0989 | 109 | BGM1429 | 141 | BGM1690 | 173 | BGM2081 |
| 014 | BGM0188 | 046 | BGM0523 | 078 | BGM0993 | 110 | BGM1437 | 142 | BGM1697 | 174 | BGM2082 |
| 015 | BGM0206 | 047 | BGM0540 | 079 | BGM1042 | 111 | BGM1440 | 143 | BGM1698 | 175 | BGM2083 |
| 016 | BGM0209 | 048 | BGM0542 | 080 | BGM1050 | 112 | BGM1458 | 144 | BGM1704 | 176 | BRS Amansa Burro |
| 017 | BGM0211 | 049 | BGM0543 | 081 | BGM1081 | 113 | BGM1485 | 145 | BGM1706 | 177 | BRS Caipira |
| 018 | BGM0213 | 050 | BGM0544 | 082 | BGM1106 | 114 | BGM1487 | 146 | BGM1715 | 178 | BRS Dourada |
| 019 | BGM0226 | 051 | BGM0550 | 083 | BGM1110 | 115 | BGM1490 | 147 | BGM1722 | 179 | BRS Formosa |
| 020 | BGM0250 | 052 | BGM0551 | 084 | BGM1127 | 116 | BGM1495 | 148 | BGM1728 | 180 | BRS Gema Ovo |
| 021 | BGM0261 | 053 | BGM0562 | 085 | BGM1153 | 117 | BGM1510 | 149 | BGM1732 | 181 | BRS Jari |
| 022 | BGM0264 | 054 | BGM0564 | 086 | BGM1164 | 118 | BGM1524 | 150 | BGM1750 | 182 | BRS Kiriris |
| 023 | BGM0269 | 055 | BGM0574 | 087 | BGM1165 | 119 | BGM1535 | 151 | BGM1757 | 183 | BRS Mulatinha |
| 024 | BGM0283 | 056 | BGM0600 | 088 | BGM1171 | 120 | BGM1537 | 152 | BGM1760 | 184 | BRS Tapioqueira |
| 025 | BGM0288 | 057 | BGM0620 | 089 | BGM1174 | 121 | BGM1539 | 153 | BGM1761 | 185 | BRS Verdinha |
| 026 | BGM0331 | 058 | BGM0624 | 090 | BGM1178 | 122 | BGM1549 | 154 | BGM1763 | 186 | Cascuda |
| 027 | BGM0336 | 059 | BGM0640 | 091 | BGM1180 | 123 | BGM1552 | 155 | BGM1794 | 187 | Corrente |
| 028 | BGM0338 | 060 | BGM0654 | 092 | BGM1193 | 124 | BGM1576 | 156 | BGM1814 | 188 | Eucalipto |
| 029 | BGM0352 | 061 | BGM0670 | 093 | BGM1202 | 125 | BGM1581 | 157 | BGM1835 | 189 | Fécula Branca |
| 030 | BGM0361 | 062 | BGM0678 | 094 | BGM1226 | 126 | BGM1590 | 158 | BGM1850 | 190 | IAC90 |
| 031 | BGM0390 | 063 | BGM0726 | 095 | BGM1252 | 127 | BGM1593 | 159 | BGM1883 | 191 | Olho Junto |
| 032 | BGM0394 | 064 | BGM0752 | 096 | BGM1281 | 128 | BGM1596 | 160 | BGM1884 | 192 | Valencia |
新窗口打开|下载CSV
1.2 木薯基因组DNA提取及建库测序
采用改良的CTAB法[11]提取木薯叶片的基因组DNA, 经1%的琼脂糖凝胶电泳检测及浓度测定后, 将工作液浓度稀释到100 ng μL-1, -20℃保存。然后利用AFSM[12]技术对192份木薯DNA样品使用96孔PCR板分别构建EcoR I-Msp I和EcoR I-Hpa II文库, 单克隆检测达到要求后, 将相应的EcoR I- Msp I和EcoR I-Hpa II文库按1∶1比例混合成1个文库, 总共192个样本, 构建了2个AFSM文库, 并利用Hiseq 2500对构建好的测序文库进行双端150 bp测序。1.3 SNP和InDels分子标记检测
利用Perl脚本(1.4 巴西木薯群体结构、遗传多样性及群体分化分析
先利用PHYLIP (此外, 使用ADMIXTURE软件[18]进行群体结构分析, 估算出最佳群体亚群数。先用PLINK软件[19]调整ADMIXTURE软件的输入文件格式, 并输入文件, 然后将亚群数K值范围设置为1~12, 根据得到的cross-validation error值选择合适的亚群数K值, 利用各个材料占各亚群的遗传成分系数(Q)构成群体遗传结构矩阵。
利用VCFtools软件(
2 结果与分析
2.1 巴西木薯群体基因型分析
通过对192份巴西栽培种木薯基因组DNA进行AFSM建库及测序, 总共得到了155 G数据, 过滤后得到134 G数据, 893,020,018条reads。再利用木薯参考基因组AM560 V6.1, 通过SAMtools和VCFtools软件对192份木薯样品基因组进行扫描, 得到796,006个SNPs和116,821个InDels。通过哈迪温伯格检测(HWE)>0.001、次等位基因频率(MAF)≥0.05过滤, 并舍去低质量的变异位点后, 仅保留了9443个高质量的变异位点(7946个SNPs和1997个InDels)用于后续分析。其中, 3287个SNPs和InDels位于基因间隔区, 4005个SNPs和InDels位于基因上游区, 471个SNPs和InDels位于基因下游区, 2个SNPs和InDels位于5°端UTR。845个SNPs和InDels属于错义突变, 745个SNPs和InDels属于同义突变, 417个SNPs和InDels属于移码突变, 另有171个SNPs和InDels属于其他类型突变(表2)。
Table 2
表2
表2SNPs和InDels的统计
Table 2
| 总数 Total | 间隔区 Intergenic region | 基因上游区 Upstream gene region | 基因下游区 Downstream gene region | 5'端UTR 5' prime UTR variant | 错义突变 Missense variant | 同义突变 Synonymous variant | 移码突变 Frame shift variant | 其他类型 Other types |
|---|---|---|---|---|---|---|---|---|
| 9443 | 3287 | 4005 | 471 | 2 | 845 | 745 | 417 | 171 |
新窗口打开|下载CSV
2.2 巴西木薯群体结构分析
通过ADMIXTURE软件利用9943个高质量的SNPs和InDels分子标记对192份巴西栽培种木薯进行群体遗传结构分析。将亚群数K值范围设置为1~12, 计算不同K值下的交叉验证错误率(cross-validation error, CV error)。当K从1到2时, CV error值迅速减小; K从2到4时, CV error值又逐渐增加; 当K从4到9时, CV error值逐渐减小并趋于平缓; 当K大于9时, CV error值又出现一定的增幅(图1-a)。说明在K等于9时, CV error值最小, 因此巴西栽培种木薯群体可分为9个亚群(Subgroup 1~Subgroup 9)。图1
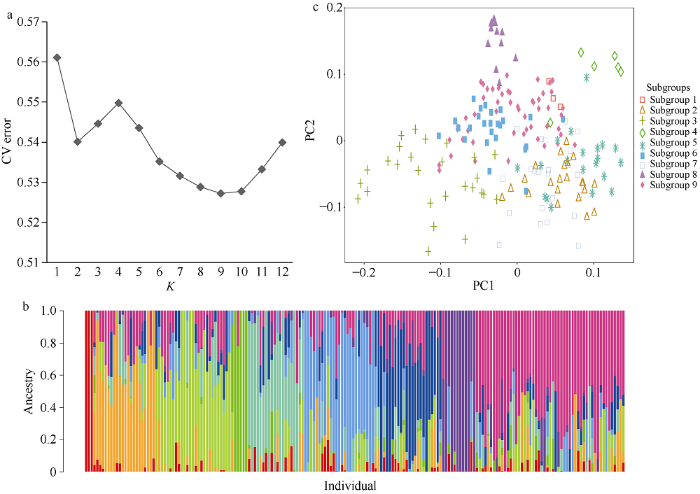 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1192份木薯品种的群体遗传结构
a: 利用ADMIXTURE软件对192份木薯材料进行群体结构分析, 计算K为1~12时的CV error。b: K = 9时的群体结构, 在该群体结构中, 每个个体用一根带有9种不同颜色的线表示, 根据颜色的占比推断该品种属于哪个亚群, 红、橙、黄、绿、蓝绿、湖蓝、深蓝、紫、紫红色分别代表亚群1~9。c: 利用高质量变异位点对192份巴西木薯进行主成分分析, 每一个点代表一份样品, 橙色、桔色、黄色、草绿、深绿、天蓝、深蓝、紫和紫红色分别代表亚群1~9 (根据ADMIXTURE软件推断的结果)。
Fig. 1Population genetic structure of 192 cassava cultivars
a: ADMIXTURE was used to analyze the population structure of 192 cassava materials, and the cross-validation error was calculated when K was 1-12. b: The population structure at K = 9, in this population structure, each individual is represented by a line with multiple different colors, and the subgroup to which the individual belongs is inferred based on the proportion of the color, the nine color regions arranged from left to right in order represent subgroups 1 to 9, respectively. c: Principal component analysis (PCA) of the group, one point represents an individual, nine colors and their corresponding shapes represent different subgroups.
192份巴西木薯可以被分为9个亚群, 再根据每个个体在这9个亚群的Q值, 将每个个体归类到Q值最大所在的亚群(图1-b)。9个亚群中分别含有3份、22份、27份、6份、20份、25份、24份、12份和53份材料。
主成分分析以所有的高质量SNPs和InDels为基础, 通过R软件分析绘图, 得到如下结果: 该木薯群体的9个亚群在PC1轴上可以看出一定的分布差距, 大部分亚群可以聚类在一起, 该结果说明聚类结果与群体结构的划分具有一致性(图1-c)。
由图2可知, 聚类结果与群体结构的划分相一致, 亚群1、亚群2、亚群4、亚群6和亚群8能较好地分别聚在一起, 而其他亚群样品大致能聚在一起, 且样品间有一定的交叉。巴西木薯各栽培种之间并未聚类到一起, 可能是由于木薯栽培历史比较短, 来源于巴西多地的木薯栽培种还未产生明显的分化。
图2
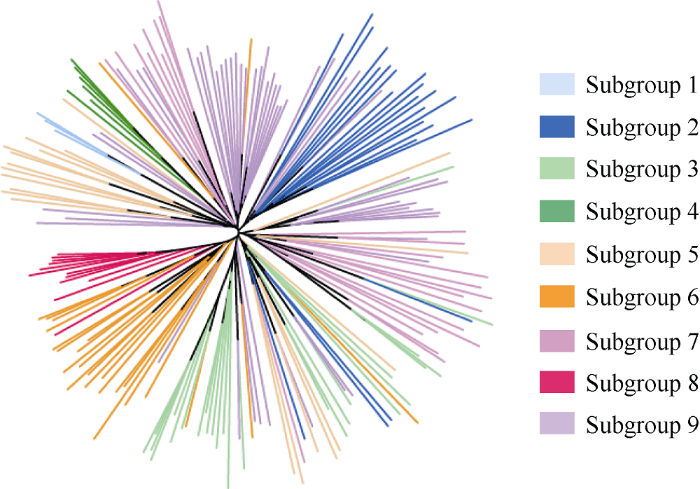 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2192份巴西栽培种木薯系统进化树图
淡蓝色、深蓝色、草绿色、深绿色、肉色、橙色、紫红色、红色和紫色分别代表亚群1~9。
Fig. 2Phylogenetic tree of 192 cassava cultivars from Brazil
Light blue, dark blue, grass green, dark green, flesh, orange, fuchsia, red, and purple represent subgroup 1 to 9, respectively.
2.3 巴西栽培种木薯遗传多样性分析
利用9943个高质量的SNPs和InDels, 通过计算遗传多样性指数(π), 评估巴西栽培种木薯群体和各个亚群的遗传多样性。通过vcftools计算发现, 巴西栽培种木薯群体的遗传多样性指数为0.274, 亚群1~9的遗传多样性指数在0.192~0.289之间, 其中亚群1具有最低的遗传多样性指数(0.192), 亚群7、亚群2、亚群6和亚群3具有相对较高的遗传多样性指数, 分别达到0.284、0.281、0.264和0.261, 而亚群5具有9个亚群中最高的遗传多样性指数(0.289) (表3)。说明巴西栽培种木薯群体具有相对较高的遗传多样性水平。Table 3
表3
表3遗传多样性指数(π)的统计
Table 3
| 亚群 Subgroup | π |
|---|---|
| 1 | 0.192 |
| 2 | 0.281 |
| 3 | 0.261 |
| 4 | 0.221 |
| 5 | 0.289 |
| 6 | 0.264 |
| 7 | 0.284 |
| 8 | 0.209 |
| 9 | 0.234 |
| 整体Entirety | 0.274 |
| 平均Average | 0.248 |
新窗口打开|下载CSV
利用群体分化指数(Fst)评估巴西栽培木薯亚群间的差异程度(表4)发现, 除亚群1和亚群4之间有较强遗传分化外, 其他亚群之间均为中等或较弱遗传分化, 亚群间的遗传分化指数在0.031~0.152之间。其中, 亚群8与其他各亚群之间均为中等分化; 而除亚群4与亚群1外, 亚群4和亚群1分别与其他各亚群之间也均为中等分化。表明, 除亚群1与亚群4间遗传分化较强、亲缘关系较远外, 其余各亚群间的为中等或较弱遗传分化程度, 即亚群间的亲缘关系相对均较近。
另外, 本研究对试验所用的巴西栽培木薯的遗传距离分析发现, 这些木薯种质间的遗传距离为0.084~0.297, 平均遗传距离为0.228。其中, BGM 1883与Valencia遗传距离最近(0.084); BGM0640与BRSJari遗传距离最远(0.297)。
Table 4
表4
表4群体分化指数(Fst)的统计
Table 4
| 亚群Subgroup | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | |||||||||
| 2 | 0.065 | ||||||||
| 3 | 0.081 | 0.061 | |||||||
| 4 | 0.152 | 0.073 | 0.112 | ||||||
| 5 | 0.053 | 0.032 | 0.069 | 0.062 | |||||
| 6 | 0.058 | 0.045 | 0.033 | 0.086 | 0.041 | ||||
| 7 | 0.059 | 0.038 | 0.054 | 0.078 | 0.031 | 0.045 | |||
| 8 | 0.139 | 0.085 | 0.076 | 0.119 | 0.084 | 0.056 | 0.080 | ||
| 9 | 0.058 | 0.043 | 0.046 | 0.083 | 0.048 | 0.034 | 0.038 | 0.061 |
新窗口打开|下载CSV
3 讨论
3.1 木薯群体结构分析
研究表明, 基因型与性状之间会产生假关联, 其原因可能是群体结构分层、等位基因分布不均等[22]。为了消除造成关联分析不准确的因素, 我们需要先对试验群体进行群体结构分析。本研究利用ADMIXTURE软件对巴西木薯自然群体的群体结构分析表明, 当K=9时, CV error值最小, 由此将192份木薯种质划分为9个亚群, 该结果与聚类分析、主成分分析的结果大概相符, 它们之间相互补充及印证, 说明该木薯群体的遗传结构较为可靠。在这9个亚群中, 群体分化指数在0.03~0.15之间, 且大部分亚群间的群体分化指数均小于0.09, 表明该木薯群体存在一个中等偏弱的遗传分化。前人的研究结果显示, 中国热带农业科学院热带品种资源种质圃收集的158份木薯种质的群体分化指数在0.03~0.07之间[23]; 在其他地区的栽培木薯中, 群体分化指数在0.01~0.05之间[14], 表明国内收集的木薯遗传分化程度较低。比较看来, 本研究中的巴西木薯种质的群体分化指数高于国内收集的木薯种质, 可挑选优质巴西木薯品种并引进中国, 从而丰富已有的木薯种质资源。3.2 木薯遗传多样性分析
本研究对遗传多样性指数(π)进行了计算, 从而评估该木薯群体的遗传多样性。Ramu等[14]的研究表明, 来源于不同地区(尼日利亚、哥伦比亚、巴西等地)的国外栽培木薯的遗传多样性指数为0.0036, 低于其祖先(M. esculenta ssp. flabellifolia, π = 0.0051); Fregene等[6]对来源于哥伦比亚、巴西和秘鲁等地的木薯地方品种的种质资源多样性评价发现, 巴西和哥伦比亚的木薯种质具有最高的遗传多样性水平; 在张圣奎对中国热带农业科学院热带品种资源种质圃收集的158份木薯种质的遗传多样性研究中发现, 该群体的遗传多样性指数为1.21×10-4, 表明该群体的遗传多样性较低[23], 同时也表明目前国内的木薯种质资源丰富度较为缺乏。本研究发现, 该巴西木薯群体中各亚群的遗传多样性指数在0.19~0.29之间, 平均遗传多样性指数为0.248, 说明巴西木薯群体的遗传多样性较为丰富, 可引进部分优良巴西种质以丰富国内的木薯种质资源。除此之外, 样本之间的亲缘关系也会对关联分析的结果造成一定的影响。本研究对192份木薯种质间的遗传距离进行分析, 从而评估不同材料之间的亲缘关系, 结果发现这些木薯种质的平均遗传距离为0.228。4 结论
本研究利用9943个高质量的SNPs和InDels对192份巴西Embrapa机构提供的木薯种质进行了和群体遗传结构分析。遗传多样性分析结果显示, 巴西木薯群体的遗传多样性水平较为丰富, 高于中国和哥伦比亚等地区; 群体遗传结构分析结果显示, 该群体被划分为9个亚群, 此结果与主成分分析及聚类分析结果基本一致。另外, 该木薯群体的分化程度较低, 但高于国内的木薯种质资源。遗传距离分析显示, BGM1883与Valencia遗传距离最近, BGM0640与BRSJari遗传距离最远。该研究将为之后关联分析挖掘优良基因及引进优良巴西木薯种质提供依据。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1007/s11103-005-2270-7URL [本文引用: 1]

Cassava or manioc (Manihot esculenta Crantz), a perennial shrub of the New World, currently is the sixth world food crop for more than 500 million people in tropical and sub-tropical Africa, Asia and Latin America. It is cultivated mainly by resource-limited small farmers for its starchy roots, which are used as human food either fresh when low in cyanogens or in many processed forms and products, mostly starch, flour, and for animal feed. Because of its inherent tolerance to stressful environments, where other food crops would fail, it is often considered a food-security source against famine, requiring minimal care. Under optimal environmental conditions, it compares favorably in production of energy with most other major staple food crops due to its high yield potential. Recent research at the Centro Internacional de Agricultura Tropical (CIAT) in Colombia has demonstrated the ability of cassava to assimilate carbon at very high rates under high levels of humidity, temperature and solar radiation, which correlates with productivity across all environments whether dry or humid. When grown on very poor soils under prolonged drought for more than 6 months, the crop reduce both its leaf canopy and transpiration water loss, but its attached leaves remain photosynthetically active, though at greatly reduced rates. The main physiological mechanism underlying such a remarkable tolerance to drought was rapid stomatal closure under both atmospheric and edaphic water stress, protecting the leaf against dehydration while the plant depletes available soil water slowly during long dry periods. This drought tolerance mechanism leads to high crop water use efficiency values. Although the cassava fine root system is sparse, compared to other crops, it can penetrate below 2 m soil, thus enabling the crop to exploit deep water if available. Leaves of cassava and wild Manihot possess elevated activities of the C4 enzyme PEP carboxylase but lack the leaf Kranz anatomy typical of C4 species, pointing to the need for further research on cultivated and wild Manihot to further improve its photosynthetic potential and yield, particularly under stressful environments. Moreover, a wide range in values of Km (CO2) for the C3 photosynthetic enzyme Rubisco was found among cassava cultivars indicating the possibility of selection for higher affinity to CO2, and consequently higher leaf photosynthesis. Several plant traits that may be of value in crop breeding and improvement have been identified, such as an extensive fine root system, long leaf life, strong root sink and high leaf photosynthesis. Selection of parental materials for tolerance to drought and infertile soils under representative field conditions have resulted in developing improved cultivars that have high yields in favorable environments while producing reasonable and stable yields under stress.]]>
URLPMID:22711743 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/s00122-003-1348-3URLPMID:12856084 [本文引用: 2]
Cassava (Manihot esculenta) is an allogamous, vegetatively propagated, Neotropical crop that is also widely grown in tropical Africa and Southeast Asia. To elucidate genetic diversity and differentiation in the crop's primary and secondary centers of diversity, and the forces shaping them, SSR marker variation was assessed at 67 loci in 283 accessions of cassava landraces from Africa (Tanzania and Nigeria) and the Neotropics (Brazil, Colombia, Peru, Venezuela, Guatemala, Mexico and Argentina). Average gene diversity (i.e., genetic diversity) was high in all countries, with an average heterozygosity of 0.5358 +/- 0.1184. Although the highest was found in Brazilian and Colombian accessions, genetic diversity in Neotropical and African materials is comparable. Despite the low level of differentiation [F(st)(theta) = 0.091 +/- 0.005] found among country samples, sufficient genetic distance (1-proportion of shared alleles) existed between individual genotypes to separate African from Neotropical accessions and to reveal a more pronounced substructure in the African landraces. Forces shaping differences in allele frequency at SSR loci and possibly counterbalancing successive founder effects involve probably spontaneous recombination, as assessed by parent-offspring relationships, and farmer-selection for adaptation.
[本文引用: 1]
DOI:10.1023/A:1017548930235URL [本文引用: 1]
URLPMID:27494320 [本文引用: 1]
[本文引用: 1]
DOI:10.1093/nar/8.19.4321URLPMID:7433111 [本文引用: 1]
A method is presented for the rapid isolation of high molecular weight plant DNA (50,000 base pairs or more in length) which is free of contaminants which interfere with complete digestion by restriction endonucleases. The procedure yields total cellular DNA (i.e. nuclear, chloroplast, and mitochondrial DNA). The technique is ideal for the rapid isolation of small amounts of DNA from many different species and is also useful for large scale isolations.
DOI:10.1038/srep07300URLPMID:25466435 [本文引用: 2]
We describe methods for the assessment of amplified-fragment single nucleotide polymorphism and methylation (AFSM) sites using a quick and simple molecular marker-assisted breeding strategy based on the use of two restriction enzyme pairs (EcoRI-MspI and EcoRI-HpaII) and a next-generation sequencing platform. Two sets of 85 adapter pairs were developed to concurrently identify SNPs, indels and methylation sites for 85 lines of cassava population in this study. In addition to SNPs and indels, the simplicity of the AFSM protocol makes it particularly suitable for high-throughput full methylation and hemi-methylation analyses. To further demonstrate the ease of this approach, a cassava genetic linkage map was constructed. This approach should be widely applicable for genetic mapping in a variety of organisms and will improve the application of crop genomics in assisted breeding.
URLPMID:22388286 [本文引用: 1]
URLPMID:33269350 [本文引用: 3]
In order to produce proteins essential for their propagation, many pathogenic human viruses, including SARS-CoV-2 the causative agent of COVID-19 respiratory disease, commandeer host biosynthetic machineries and mechanisms. Three major structural proteins, the spike, envelope and membrane proteins, are amongst several SARS-CoV-2 components synthesised at the endoplasmic reticulum (ER) of infected human cells prior to the assembly of new viral particles. Hence, the inhibition of membrane protein synthesis at the ER is an attractive strategy for reducing the pathogenicity of SARS-CoV-2 and other obligate viral pathogens. Using an in vitro system, we demonstrate that the small molecule inhibitor ipomoeassin F (Ipom-F) potently blocks the Sec61-mediated ER membrane translocation/insertion of three therapeutic protein targets for SARS-CoV-2 infection; the viral spike and ORF8 proteins together with angiotensin-converting enzyme 2, the host cell plasma membrane receptor. Our findings highlight the potential for using ER protein translocation inhibitors such as Ipom-F as host-targeting, broad-spectrum, antiviral agents.
DOI:10.1093/bioinformatics/btp352URLPMID:19505943 [本文引用: 1]
SUMMARY: The Sequence Alignment/Map (SAM) format is a generic alignment format for storing read alignments against reference sequences, supporting short and long reads (up to 128 Mbp) produced by different sequencing platforms. It is flexible in style, compact in size, efficient in random access and is the format in which alignments from the 1000 Genomes Project are released. SAMtools implements various utilities for post-processing alignments in the SAM format, such as indexing, variant caller and alignment viewer, and thus provides universal tools for processing read alignments. AVAILABILITY: http://samtools.sourceforge.net.
URLPMID:22728672 [本文引用: 1]
DOI:10.1016/j.ajhg.2010.11.011URLPMID:21167468 [本文引用: 1]
For most human complex diseases and traits, SNPs identified by genome-wide association studies (GWAS) explain only a small fraction of the heritability. Here we report a user-friendly software tool called genome-wide complex trait analysis (GCTA), which was developed based on a method we recently developed to address the
DOI:10.1101/gr.094052.109URLPMID:19648217 [本文引用: 1]
Population stratification has long been recognized as a confounding factor in genetic association studies. Estimated ancestries, derived from multi-locus genotype data, can be used to perform a statistical correction for population stratification. One popular technique for estimation of ancestry is the model-based approach embodied by the widely applied program structure. Another approach, implemented in the program EIGENSTRAT, relies on Principal Component Analysis rather than model-based estimation and does not directly deliver admixture fractions. EIGENSTRAT has gained in popularity in part owing to its remarkable speed in comparison to structure. We present a new algorithm and a program, ADMIXTURE, for model-based estimation of ancestry in unrelated individuals. ADMIXTURE adopts the likelihood model embedded in structure. However, ADMIXTURE runs considerably faster, solving problems in minutes that take structure hours. In many of our experiments, we have found that ADMIXTURE is almost as fast as EIGENSTRAT. The runtime improvements of ADMIXTURE rely on a fast block relaxation scheme using sequential quadratic programming for block updates, coupled with a novel quasi-Newton acceleration of convergence. Our algorithm also runs faster and with greater accuracy than the implementation of an Expectation-Maximization (EM) algorithm incorporated in the program FRAPPE. Our simulations show that ADMIXTURE's maximum likelihood estimates of the underlying admixture coefficients and ancestral allele frequencies are as accurate as structure's Bayesian estimates. On real-world data sets, ADMIXTURE's estimates are directly comparable to those from structure and EIGENSTRAT. Taken together, our results show that ADMIXTURE's computational speed opens up the possibility of using a much larger set of markers in model-based ancestry estimation and that its estimates are suitable for use in correcting for population stratification in association studies.
URLPMID:17701901 [本文引用: 1]
URLPMID:21653522 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]

{kind=link}
{kind=link}
{kind=link}
{kind=link}