 ,1,2, 王俊1, 刘章雄,2,*, 邱丽娟,1,2,*
,1,2, 王俊1, 刘章雄,2,*, 邱丽娟,1,2,*Mapping of an incomplete dominant gene controlling multifoliolate leaf by BSA-Seq in soybean (Glycine max L.)
ZHANG Zhi-Hao,1,2, WANG Jun1, LIU Zhang-Xiong,2,*, QIU Li-Juan,1,2,*通讯作者:
收稿日期:2020-03-24接受日期:2020-08-19网络出版日期:2020-09-02
| 基金资助: |
Received:2020-03-24Accepted:2020-08-19Online:2020-09-02
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (3196KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
张之昊, 王俊, 刘章雄, 邱丽娟. 基于BSA-Seq技术挖掘大豆中黄622的多小叶基因[J]. 作物学报, 2020, 46(12): 1839-1849. doi:10.3724/SP.J.1006.2020.04075
ZHANG Zhi-Hao, WANG Jun, LIU Zhang-Xiong, QIU Li-Juan.
叶片是植物进行光合作用和呼吸作用的重要器官, 对植物的生长发育起着重要作用。在植物界, 叶片的形态在不同物种间存在广泛的多样性, 单叶植物有一个与叶柄相连的叶片, 如水稻、玉米等, 而复叶植物的叶片由多个着生在叶轴上的小叶组成, 如番茄等。与单叶相比, 复叶虽然在叶片总面积上有所减少, 但其遭受外界的机械压力(如风、雨等)比单叶要小的多, 因此复叶的形成使植物更能适应恶劣的环境[1]。相关研究表明, 大豆多小叶单株全生育期不摘叶处理的部分农艺性状和产量表现均明显优于全生育期摘除多片叶的对照处理[2]。因此, 大豆多小叶相关基因的挖掘和研究有助于改善大豆农艺性状和产量表现。
大豆多小叶的遗传研究可追溯到1972年, Fehr[3]利用七小叶和五小叶大豆材料, 与三出复叶大豆进行两两杂交构建F2群体, 遗传分析表明, 五小叶和七小叶分别受显性基因Lf1和隐性基因lf2控制。Jeong等[4]通过构建遗传图谱精细定位了控制五小叶的基因Lf1, 该基因编码一个假定AP2功能域, 但并没有对该基因进行进一步的功能验证。Devine[5]发现了控制七小叶的隐性基因lf2与控制茸毛密度的基因Pd2存在连锁关系, Seversike等[6]鉴定出与lf2基因连锁的分子标记, 从而将lf2定位在B1连锁群(11号染色体)。傅来卿[7]通过对栽培大豆品种Wilkine进行物理诱变得到多小叶突变体, 该多小叶突变体每个复叶有4到7片小叶, 对该突变体进行遗传分析, 命名了一个控制多小叶的基因lf3; 王克晶等[8]对五小叶的野生大豆进行遗传研究, 命名了基因Lf4和Lf5。总之, 大豆多小叶的相关研究多处于遗传分析和初定位阶段, 通过正向遗传学挖掘多小叶基因的相关研究鲜见报道。
突变体是进行正向遗传学研究的优异材料, 对随机诱变产生的突变体进行研究, 不仅能够挖掘新的基因, 也可以揭示已知基因的新功能[9,10,11,12]。随着新一代测序技术的发展, 基于BSA-Seq的方法挖掘新基因在作物农艺性状相关基因的定位研究中的应用日益广泛[12,13,14,15,16], 该方法在不构建遗传图谱的情况下高效快速地挖掘目的基因。本研究在栽培大豆中品661的突变体库中鉴定出一个多小叶突变体——中黄622, 该突变体在田间达到5个到9个小叶, 是研究大豆小叶数量的优异资源。利用BSA-Seq方法进行定位, 确定了6个候选基因, 为大豆多小叶基因图位克隆奠定了基础。
1 材料与方法
1.1 群体构建与表型鉴定
本试验通过EMS诱变, 并在田间筛选, 在栽培大豆中品661突变体库中鉴定出一个多小叶的不完全显性突变体中黄622, 2016年将中品661与中黄622进行杂交, 得到F0, 2017年6月将F1种植于北京顺义, 并于同年10月收获。2018年6月在顺义种植F2群体, 并于盛花期观察239个F2植株表型, 记录单株每个复叶的小叶数量并做记录。根据多小叶复叶的数目, 将单株分为3个等级: I级为多小叶复叶极多, 整个单株上仅有极少数(0~4个)叶片为正常三出复叶, 其余均为多小叶, 与突变体中黄622的表型相同; II级为多小叶复叶数目介于突变体中黄622和野生型之间, 至少有1个多小叶复叶; III级为整个单株所有叶片均为正常叶片, 与野生型相同。2018年10月收获F2单株的种子, 每个F2单株选取10粒以上的种子于2018年11月在海南种植F2:3群体, 于盛花期观察单株中每个复叶的小叶数量并做记录。通过观察F2在F2:3的分离情况, 来确定每个F2单株表型的准确性和稳定性。1.2 混池测序
根据2个亲本及F2叶片表型, 构建2个亲本池和2个极端表型混池, 亲本池分别为10株中品661与10株突变体中黄622; 在F2植株中, 选择单株所有叶片均为三出复叶的30株个体构建正常叶片混池, 单株复叶均为多小叶30株个体构建多小叶混池。具体操作步骤如下: 首先, 在植株盛花期取植株顶端幼叶, 利用CTAB法分别提取单株叶片 DNA; 然后检测DNA浓度, 将不同池的植株DNA等量混合构建出4个混池。送北京百迈克生物科技有限公司测序。利用Illunima Casava 1.8进行碱基识别分析, 采用双端150 bp测序策略进行基因组测序。亲本池测序深度为10×, 后代混池测序深度为30×。参考基因组为Wm82.a2.v1版本的大豆基因组[17]。使用GATK [18]软件工具来实现SNP的检测, 利用SnpEff [19]软件进行变异注释和预测变异影响。1.3 数据分析
1.3.1 关联分析 在关联分析前, 首先对SNP进行过滤, 过滤标准如下: 首先过滤掉有多个基因型的SNP位点, 其次过滤掉read支持度小于4的SNP位点, 最终得到高质量的可信SNP位点。欧式距离(euclidean distance, ED)算法, 是利用测序数据寻找混池间存在显著差异标记, 并以此评估与性状关联区域的方法[20]。理论上, 多小叶混池和正常叶混池之间除了小叶数目性状相关位点存在差异, 其他位点均趋向于一致, 因此非目标位点的ED值应趋向于0。ED方法的计算公式如下所示, ED值越大, 表明该标记在多小叶混池和正常叶混池之间的差异越大。
$\text{ED=}\sqrt{{{\left( \text{Amut}-\text{Awt} \right)}^{2}}+{{\left( \text{Cmut}-\text{Cwt} \right)}^{2}}+{{\left( \text{Gmut}-\text{Gwt} \right)}^{2}}+{{\left( \text{Tmut}-\text{Twt} \right)}^{2}}}$
式中, Amut表示A碱基在突变混池中的频率, Awt表示A碱基在野生型混池中的频率; Cmut表示C碱基在突变混池中的频率, Cwt表示C碱基在野生型混池中的频率; Gmut表示G碱基在突变混池中的频率, Gwt表示G碱基在野生型混池中的频率; Tmut表示T碱基在突变混池中的频率, Twt表示T碱基在野生型混池中的频率。
在进行分析时, 利用多小叶混池和正常叶混池间基因型存在差异的SNP位点, 统计各个碱基在2个混池中的深度, 并计算每个位点ED值, 为消除背景噪音, 本试验采用原始ED的5次方作为关联值以达到消除背景噪音的功能[20]。
SNP-index是一种通过混池间的基因型频率差异进行标记关联分析的方法[20,21], 主要是寻找混池之间基因型频率的显著差异, 用Δ(SNP-index)统计。标记SNP与性状关联度越强, Δ(SNP-index)越接近于1。Δ(SNP-index)计算公式如下:
SNP-index (Mut) = ρx/(ρX + ρx)
SNP-index (WT) = ρx/(ρX + ρx)
ΔSNP-index=SNP-index(Mut) - SNP-index(WT)
式中, Mut为子代的突变池, 即多小叶混池, WT代表子代的野生池即正常叶混池, ρX和ρx分别为野生型亲本中品661的等位基因和突变型亲本中黄622的等位基因在各自池中出现的read数目。通过ΔSNP-index 可以观察每个位点在多小叶混池和正常叶混池之间的差异。
为了消除假阳性的位点, 利用标记在基因组上的位置, 可对同一条染色体上标记的ΔSNP-index值进行拟合[21], 本研究采用DISTANCE方法对ΔSNP-index进行拟合, 然后根据关联阈值, 选择阈值以上的区域作为与性状相关的区域。
1.3.2 变异位点的鉴定 为了检测过滤SNP方法的可靠性, 选取了1个高质量的SNP位点和其附近的8个低质量的SNP位点进行验证。首先根据比对参考基因组, 在SoyBase (
1.3.3 候选区域基因的功能注释 应用BLAST [22]软件对候选区间内的编码基因进行多个数据库(NR [23]、Swiss-Prot、GO [24]、KEGG [25]、COG [26])的深度注释。本研究的BSA-Seq数据分析流程图如图1所示。
图1
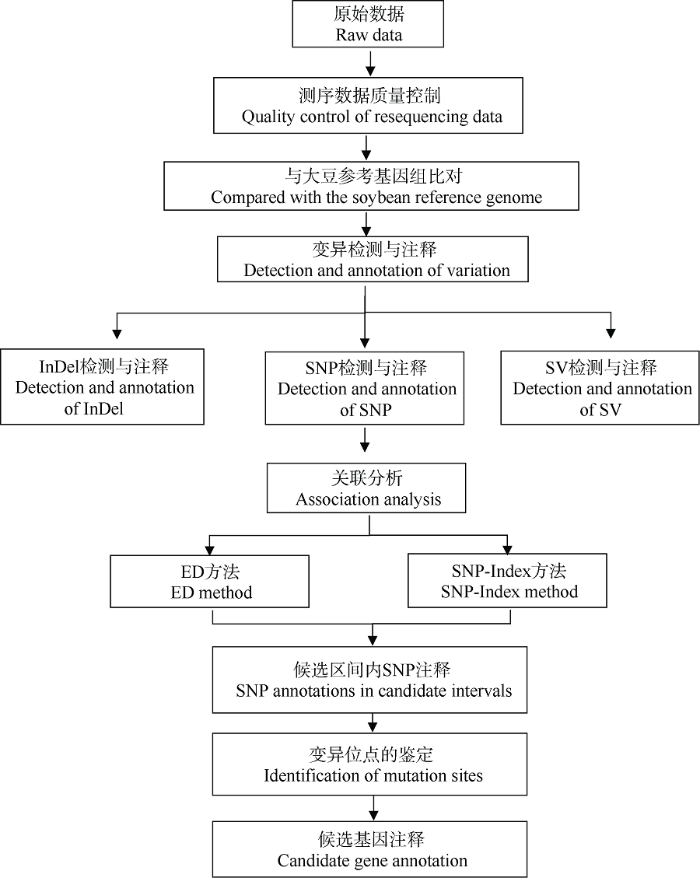 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1BSA-Seq数据分析流程
Fig. 1BSA-seq data analysis process
1.3.4 数据可视化 本研究利用circos软件(
1.4 候选基因表达分析
为了研究关联区域内候选基因在不同组织中的表达情况, 利用Phytozome (2 结果与分析
2.1 多小叶遗传分析
突变体中黄622与野生型中品661相比, 叶片多为5到9片的多小叶复叶, 但也有少量单株出现0~4片正常的三出复叶。通过观察F2:3的表型及分离情况, 确定对应F2单株的基因型。观察发现, 多小叶等位基因纯合基因型(lf a lf a)单株的表型均为I型, 正常叶等位基因纯合基因型(lf b lf b)单株的表型均为III型, 而杂合单株(lf a lf b)的表型绝大多数为II型, 仅有极少量的I型(2个)和III型(4个)(表1), 这说明杂合单株的表型不稳定。表明, 该多小叶性状受1对不完全显性基因控制, 且杂合基因型单株的表型可能受到环境和遗传背景的影响, 表现出不同程度的多小叶性状。卡方检测结果表明, 3种基因型的F2单株数目不符合1:2:1 (χ20.05 = 5.99 < χ2 = 9.18 < χ20.01 = 9.27) (表1)。卡方值偏高可能是群体过小或各种基因型的存活比例不同造成的。Table 1
表1
表1F2群体中不同基因型和表型的单株数目
Table 1
| 表型 Phenotype | 基因型Genotype | 期望比 Expectation ratio | χ2 | P0.05, 0.01 | ||
|---|---|---|---|---|---|---|
| lf alf a | lf alf b | lf blf b | ||||
| I | 54 | 2 | 0 | — | — | — |
| II | 0 | 99 | 0 | — | — | — |
| III | 0 | 4 | 80 | — | — | — |
| 总数Total | 54 | 105 | 80 | 1︰2︰1 | 9.18 | 5.99, 9.27 |
新窗口打开|下载CSV
2.2 测序数据分析
2.2.1 数据质控 对从F2选择30个多小叶单株和30个正常叶单株分别构建混合池, 与2个亲本通过Illumina HiSeq重测序, 过滤后得到的碱基数目为144.84 Gbp, Q30>91.61%, GC含量在35.81%~ 36.94%, 插入片段大小的分布呈单峰的正态分布, 样品与参考基因组平均比对效率为98.83%, 平均覆盖深度为 32.75×, 基因组覆盖度为99.22% (至少1个碱基覆盖)。这些参数说明, 测序数据质量合格, 与大豆参考基因组比对效率较高, 可用于后续变异检测及性状的基因定位。2.2.2 关联分析 进一步过滤SNP, 得到高质量可信SNP位点12,160个。采用ED算法和SNP-index 2种算法对这些高质量SNP进行关联。
利用ED方法计算关联值, 并取原始ED的5次方作为关联值以达到消除背景噪音的功能, 然后采用DISTANCE方法对ED值进行拟合。取所有位点拟合值的median+3SD作为分析的关联阈值[20], 计算得0.49。根据关联阈值判定, ED关联结果如图2-A所示, 在11号染色体共得到2个区域, 总长度为5.29 Mb, 共包含1103个基因(表2)。
图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2利用2种关联方法鉴定多小叶基因候选区间
A: ED关联分析结果, 横坐标为染色体位置, 纵坐标代表拟合后的欧式距离(ED)值的5次方, 黑色的线为拟合后的ED的5次方作关联值, 红色的虚线代表显著性关联阈值。B: SNP-index关联分析结果, 横坐标为染色体位置, 黑色的线为拟合后的ΔSNP-index值, 红色的线代表置信度为0.99的阈值线, 蓝色的线代表置信度为0.95的阈值线, 绿色的线代表置信度为0.90的阈值线。2种关联分析结果均表明, 与多小叶相关的关联区域位于11号染色体末端。
Fig. 2Identification of multiple leaflet gene candidate intervals using two association methods
A: ED correlation analysis results, the abscissa is the chromosome position, the ordinate represents the fifth power of the euclidean distance (ED) value after fitting, the black line is the fifth power of ED after fitting, the dashed line represents the significance association threshold. B: SNP-index correlation analysis results, the abscissa for chromosomal location, the black line for fitting after ΔSNP-index value, the red line represents the confidence level of 0.99 the threshold line, blue line represents the confidence level of 0.95 the threshold line, green line represents the confidence level of the threshold line of 0.90. The results of the two association analysis methods show that the correlation regions associated with the multifoliolate leaf trait is located at the end of chromosome 11.
Table 2
表2
表2利用不同方法在11号染色体获得关联区域
Table 2
| 关联分析方法 Association analysis method | 染色体 Chromosome | 关联区域 起始位置 Start of associated regions | 关联区域 终止位置 End of associated regions | 关联区域 大小 Associated region size (Mb) | 关联区域内的 基因数量 Gene number in the associated regions |
|---|---|---|---|---|---|
| 欧氏距离 Euclidean distance (ED) | Chr. 11 | 0 | 4,150,000 | 4.15 | 896 |
| Chr. 11 | 5,570,000 | 6,710,000 | 1.14 | 207 | |
| 总计Total | — | — | 5.29 | 1103 | |
| SNP-index | Chr. 11 | 0 | 250,000 | 0.25 | 44 |
| Chr. 11 | 1,510,000 | 3,480,000 | 1.97 | 439 | |
| Chr. 11 | 5,570,000 | 6,770,000 | 1.20 | 218 | |
| 总计Total | — | — | 3.42 | 701 | |
| 2种方法交集 Intersection of two methods | Chr. 11 | 0 | 250,000 | 0.25 | 44 |
| Chr. 11 | 1,510,000 | 3,480,000 | 1.97 | 439 | |
| Chr. 11 | 5,570,000 | 6,710,000 | 1.14 | 207 | |
| 总计Total | — | — | 3.36 | 690 |
新窗口打开|下载CSV
2个混池的ΔSNP-index的分布如图2-B所示。当置信度为0.99时, 利用SNP-index方法共关联到3个区域, 总长度为3.42 Mb, 共包含701个基因(表2)。2种关联分析方法得到的结果取交集, 共得到3个区域(表2), 总长度3.36 Mb, 共包含690个基因。结合上述内容, 将样品的变异结果及BSA关联分析结果使用circos软件(http://circos.ca/)作图(图3), 将测序分析结果和关联分析结果可视化。
图3
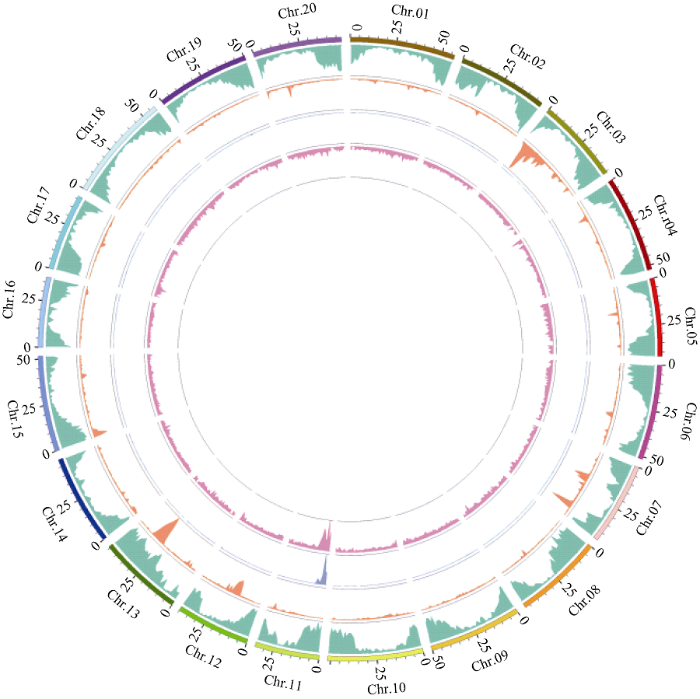 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3样品间SNP及关联信号在染色体上的分布
从外到里依次为: 第1圈: 染色体坐标; 第2圈: 基因分布; 第3圈: SNP密度分布; 第4圈: ED值分布; 第5圈: ΔSNP-index值分布。
Fig. 3Distribution of SNPs and associated signals on chromosomes between samples
From outside to inside in order: the first circle represents chromosome coordinates, the second circle represents gene distribution, the third circle represents SNP density distribution, the fourth circle represents ED value distribution, and the fifth circle represents ΔSNP-index value distribution.
2.2.3 候选基因的筛选 根据原始测序数据, 区间内的SNP有43个, 其中高质量的SNP有6个, 低质量的SNP有37个。为了验证过滤SNP方法的可靠性, 进一步筛选候选基因, 本试验设计了3对引物(表3), 分别用以鉴定1个高质量的SNP及其2侧的低质量SNP位点。其中引物SNP2的扩增产物片段全长7016 bp, 包含1个高质量的SNP位点, 而引物SNP1扩增产物片段全长5633 bp, 包含该高质量SNP上游的5个低质量位点, 引物SNP3扩增产物片段全长1247 bp, 包含该高质量SNP下游的3个低质量位点。对两亲本PCR产物进行测序鉴定。两亲本序列比对结果表明, 中品661和中黄622在8个低质量的SNP位点处均无变异, 而2个亲本在高质量SNP处的测序结果与重测序结果一致(表4)。这些结果为候选基因的筛选提供了参考依据。
Table 3
表3
表3引物序列和信息
Table 3
| 引物名称 Primer ID | 上游引物序列 Forward primer (5°-3°) | 下游引物序列 Reverse primer (5°-3°) | 产物大小 Product size (bp) | 检测位点数目 Number of detection sites |
|---|---|---|---|---|
| SNP 1 | GCCGTAGCACTTCACTCATTCA | ATGTTCTATAAGCCGTTTTTTGGAAG | 5633 | 5 |
| SNP 2 | TCTACTCGCAAAAGGCACAG | CATTTATATGTTCGTCAATATTTAG | 7016 | 1 |
| SNP 3 | GGAATGCCAATGGGCTTTAATT | GATCTTGCTTACCAATTTCTCG | 1247 | 3 |
新窗口打开|下载CSV
Table 4
表4
表4对区间内部分SNP位点进行鉴定
Table 4
| 染色体 Chr. | SNP位点 SNP loci | 参考碱基 Reference base | 突变碱基 Altered base | 混池read值Bulk read value in mixed pool | SNP质量评价 SNP quality evaluation | 鉴定结果 Appraisal results | |||
|---|---|---|---|---|---|---|---|---|---|
| 中品661 Zhongpin 661 | 中黄622 Zhonghuang 622 | 正常叶混池 Normal leaf mixing pool | 多小叶混池 Multi-leaflet mixing pool | ||||||
| Chr.11 | 1738094 | C | T | 14,7 | 8,0 | 32,0 | 42,0 | 低 Low | 假False |
| Chr.11 | 1738120 | C | T | 14,5 | 8,0 | 33,0 | 39,0 | 低Low | 假False |
| Chr.11 | 1738157 | G | A | 10,0 | 9,1 | 32,5 | 38,4 | 低Low | 假False |
| Chr.11 | 1738175 | A | T | 9,0 | 9,1 | 34,9 | 39,6 | 低Low | 假False |
| Chr.11 | 1738511 | A | T | 20,0 | 9,1 | 37,4 | 39,10 | 低Low | 假False |
| Chr.11 | 1947868 | A | T | 12,0 | 0,7 | 27,0 | 0,48 | 高High | 真True |
| Chr.11 | 1964348 | C | T | 9,0 | 5,1 | 42,5 | 41,7 | 低Low | 假False |
| Chr.11 | 1964460 | T | C | 8,0 | 9,5 | 37,28 | 44,28 | 低Low | 假False |
| Chr.11 | 1964710 | T | A | 9,0 | 6,2 | 36,8 | 38,8 | 低Low | 假False |
新窗口打开|下载CSV
根据不同质量SNP的鉴定结果, 本研究对区间内的候选基因进行了进一步的筛选。筛选到定位区间内亲本间存在高质量的SNP的6个基因上(表5)。在这6个基因中, 有3个基因发生了非同义突变, 导致氨基酸序列发生变异, 1个基因发生了同义突变, 其余2个基因的突变分别发生在基因上游和内含子上, 该结果为多小叶候选基因的发掘提供了信息。
Table 5
表5
表52种方法共定位区间内亲本之间SNP类型
Table 5
| SNP位点 SNP loci | 碱基类型 Base type | 突变类型 Mutation type | 基因ID Gene ID | 基因功能注释 Annotated function | |
|---|---|---|---|---|---|
| 中品661 Zhongpin 661 | 中黄622 Zhonghuang 622 | ||||
| 1947868 | A | T | 非同义SNV Nonsynonymous SNV | Glyma.11G027100 | 同源盒蛋白knotted-1-like-7 Homeobox protein knotted-1-like-7 |
| 2489820 | G | A | 同义SNV Synonymous SNV | Glyma.11G034100 | 亮氨酸-tRNA连接酶/亮氨酰-tRNA合成酶 Leucine-tRNA ligase/Leucyl-tRNA synthetase |
| 2954231 | G | A | 非同义SNV Nonsynonymous SNV | Glyma.11G040200 | 无意义转录物1的调节因子(UPF1, RENT1) Regulator of nonsense transcripts 1 (UPF1, RENT1) |
| 3156378 | G | A | 非同义SNV Nonsynonymous SNV | Glyma.11G043100 | At-hook motif核定位蛋白21相关 At-hook motif nuclear localized protein 21- related |
| 3334413 | C | A | 基因上游 Upstream | Glyma.11G045200 | 核基质构成蛋白1蛋白相关 Nuclear matrix constituent protein 1-like protein-related |
| 6288434 | C | T | 内含子 Intron | Glyma.11G083800 | 酰基激活酶1, 过氧化物酶体相关 Acyl-activating enzyme 1, peroxisomal-related |
新窗口打开|下载CSV
2.2.4 候选基因表达的生物信息学分析 为了进一步探究共定位区间内候选基因在各个组织中的表达情况, 利用phytozome (phytozome.jgi.doe. gov/)数据库查询了6个基因在Williams 82中的表达数据, 从构建的表达图谱(图4)可以看出, Glyma. 11G027100的表达谱较广, 在茎端分生组织、叶、花和荚中均有较高表达; Glyma.11G083800和Glyma.11G034100在叶片中的表达都比较高, 但在分生组织中表达量较低, 其中Glyma.11G083800在节和根中也有较高的表达量, 而Glyma.11G034100则在根毛中高表达; Glyma.11G040200在叶和茎中的表达量居中, 在花和荚中有着较高的表达, 在茎端分生组织和其他部位表达较低; Glyma. 11G043100在节、根、根毛表达较高, 在其他组织中表达较低; Glyma.11G045200在荚、节和根毛中表达较高, 在其他部位表达较低。考虑到相关基因在分生组织和叶片中的表达以及表达量的变化对复叶发育的重要性, 推测在分生组织和叶片中表达较高的Glyma.11G027100可能与复叶发育相关的基因, 在分生组织中表达较低, 但在叶片中表达较高的Glyma.11G083800和Glyma.11G034100也有可能是复叶发育相关的基因。但候选基因在中品661与中黄622中是否与Williams 82的表达模式一致, 仍需要后期验证。
图4
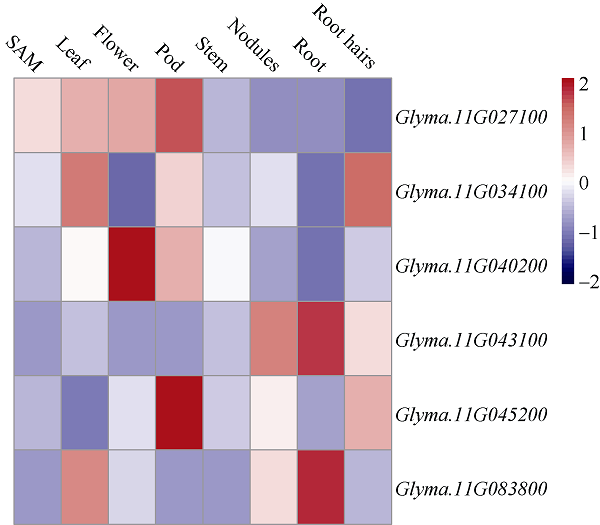 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图46个候选基因的表达图谱
方块内颜色显示候选基因表达水平: 蓝色最低, 白色居中, 红色最高。
Fig. 4Expression profile of six candidate genes
Colors in the square represent the expression level of candidate genes: blue is the lowest, white is middle, and red is the highest.
3 讨论
3.1 复叶发育相关基因
与复叶发育相关的基因包括KNOX I、ARP、UFO/FIM等[27]。KNOX I (Class I KNOTTED-like homeobox)基因家族被认为是茎端分生组织(SAM)分化出叶原基和维持形态建成的关键基因, 叶原基形成初期(P0), KNOX I的表达会提高, 但在叶原基形成之后表达降低[28,29], 在单叶植物如玉米(Zea mays)中, 这种低表达是永久性的[30], 而在复叶植物如番茄和碎米荠(Cardamine hirsute L.)中, KNOX I基因的表达会再次升高[29,31]。在转基因的拟南芥中, KNOX I基因家族中任何一个基因的异位表达都会在引起叶边缘产生许多浅裂形成的小叶[32]。研究发现, KNOX I基因与复叶植物的形态建成有明显的关系, 在KNOX I基因参与复叶发育的物种中(如番茄), 如果增加KNOX I基因的表达, 可以产生明显的复叶重复结构单元[31,33]。ARP基因家族是在被子植物叶发育起着关键作用的另外一类基因, 它和KNOX I基因的相互拮抗作用共同决定叶的发生。KNOX I基因的功能是维持分生组织属性, 而ARP基因则参与侧生器官的发生和叶极性的建立等分化相关的发育过程[34,35,36]。另外, 相关研究表明, UFO/FIM在豌豆和百脉根中的直系同源基因STAMINA PISTILLOIDA (STP)和PROLIFERATING FLORAL ORGAN (PFO), 参与控制小叶原基的起始和形态发生[27,37-38]。关于大豆多小叶的基因定位研究较为深入的是Lf1和lf2基因, 在前期报道中, Jeong等[39]定位到了控制大豆五小叶的基因Lf1, 该基因编码一个假定AP2功能域, 虽然并未对该基因进行功能验证, 但有研究表明AP2转录因子参与调控玉米叶片发育, 影响玉米叶形。这说明影响植物叶片发育的基因除了KNOX I、ARP等研究比较深入的基因外, 还有其他基因有待发掘和验证。
Seversike等[6]在不同群体中验证了lf2基因与SSR标记Sat_272的连锁关系(LOD>4.0), 但并未得到定位区间。本研究通过ED和ΔSNP-index 2种方法进行关联分析, 将中黄622的多小叶相关基因定位在11号染色体末端, 经2种关联方法取交集后的关联区域大小3.36 Mb, 包含690个基因, 其中关联区域内部发生的SNP有6个, 分布在6个基因上。外显子发生SNP的基因有4个, 其中1个基因发生的变异为同义突变, 3个基因的变异导致氨基酸序列的变化。利用数据库(SoyBase)对比Sat_272 (物理位置为2,718,892至2,719,123)与本研究定位区间的物理位置发现, Sat_272在本研究的候选区间内。然而, 本研究定位的基因是否为多小叶基因lf2还有待进一步研究验证。
3.2 多小叶性状的遗传特性
Fehr[3]通过对三出复叶Hawkeye与七小叶突变体T255构建的群体的遗传分析发现, 小叶数目性状受1对基因控制, 控制正常的三出复叶的等位基因Lf2表现为完全显性, 但对T255和五小叶突变体T143构建的群体进行遗传分析发现, Lf2等位基因并不是在所有遗传背景中都是完全显性的。Seversike等[6]在七小叶大豆PI 548232和3个正常三出复叶大豆(Trill、MN1401、MN1801)构建的群体中再次证明lf2为控制七小叶的隐性基因, 而杂合单株整株为正常三出复叶或有一片复叶为四小叶。本研究中, 多小叶突变体的复叶有5到9个小叶, 控制多小叶性状的基因为1对不完全显性基因, 杂合单株表型介于突变体和野生型之间, 且表型并不稳定, 在105个F2杂合单株中有6个单株表现出与亲本相近的表型。这说明, 多小叶性状的表型不稳定, 特别是杂合单株在不同的环境和遗传背景下有着不同的表型。因此, 对多小叶全面的遗传分析需要在多年多点不同的材料之间进行。3.3 候选基因的功能
本研究定位区间内有6个候选基因(Glyma. 11G027100、Glyma.11G034100、Glyma.11G040200、Glyma.11G043100、Glyma.11G045200、Glyma. 11G083800)发生了SNP变异。Glyma.11G027100是II类KNOX亚家族(KNOX II)成员, 该基因在茎端分生组织(SAM)和叶片中均有着较高的表达。虽然在植物复叶发育过程中, I类KNOX亚家族(KNOX I)基因是SAM分化出叶原基和维持形态建成的关键基因, 但尚未有KNOX II基因与叶片发育相关的报道。Glyma.11G034100为一个编码亮氨酸-tRNA连接酶/亮氨酰-tRNA合成酶的基因, 其经典功能是催化合成亮氨酰-tRNA直接参与遗传信息的解码合成蛋白质[40]。Glyma.11G040200编码的无义转录物1的调节因子(UPF1、RENT1)参与调控无义介导的mRNA降解途径, 该途径能够识别并降解包括前终止的无义mRNA在内的异常mRNA[41,42], 并避免异常mRNA翻译成潜在有毒蛋白质, 从而对机体产生毒害效应。该途径的调控对基因的准确表达和机体细胞正常的生理活动起着重要作用[43,44,45]。Glyma.11G043100是一个AT-hook蛋白相关的基因, AT-hook蛋白可能在植物生长发育中起着重要作用, 相关研究表明, 拟南芥中AT-hook基因AHL27在过量表达时会延迟拟南芥的开花[46]; Glyma.11G045200基因的产物与核基质构成蛋白1相关, 核基质蛋白不仅是组成细胞核内部结构的支架, 而且同DNA的复制、RNA 的合成、激素的联接、基因表达的调节密切相关[47]。Glyma.11G083800为一个酰基激活酶1基因, 与过氧化氢酶体相关。这些基因对大豆复叶形成和发育的影响将在后续的工作中进行验证。4 结论
突变体中黄622的多小叶受1对不完全显性基因控制, 基于BSA-Seq技术将该基因定位在11号染色体上, 定位区间内亲本之间存在SNP的基因有6个, 为大豆多小叶基因图位克隆创造了条件。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
DOI:10.1111/j.1469-8137.2009.02854.xURLPMID:19413689 [本文引用: 1]

Climatic extremes can be as significant as averages in setting the conditions for successful organismal function and in determining the distribution of different forms. For lightweight, flexible structures such as leaves, even extremes lasting a few seconds can matter. The present review considers two extreme situations that may pose existential risks. Broad leaves heat rapidly when ambient air flows drop below c. 0.5 m s(-1). Devices implicated in minimizing heating include: reduction in size, lobing, and adjustments of orientation to improve convective cooling; low near-infrared absorptivity; and thickening for short-term heat storage. Different features become relevant when storm gusts threaten to tear leaves and uproot trees with leaf-level winds of 20 m s(-1) or more. Both individual leaves and clusters may curl into low-drag, stable cones and cylinders, facilitated by particular blade shapes, petioles that twist readily, and sufficient low-speed instability to initiate reconfiguration. While such factors may have implications in many areas, remarkably little relevant experimental work has addressed them.
[本文引用: 1]
[本文引用: 1]
DOI:10.2135/cropsci1972.0011183X001200020023xURL [本文引用: 2]
DOI:10.1007/s00122-017-2918-0URLPMID:28516383 [本文引用: 1]
KEY MESSAGE: A high-resolution genetic map that was constructed for the Lf1 -residing region will provide valuable information for map-based cloning and genetic improvement efforts in soybean. Changes in leaf architecture as photosynthesis factories remain a major challenge for the improvement of crop productivity. Unlike most soybeans, which have compound leaves comprising three leaflets, the soybean Lf1 mutant has a high frequency of compound leaves with five leaflets in a partially dominant manner. Here, we generated a fine genetic map to determine the genetic basis of this multifoliolate leaf trait. A five-leaflet variant Dusam was found in a recently collected landrace cultivar. Phenotypic data were collected from the F2 population of a cross between the Dusam and three-leaflet cultivar V94-5152. The mapping results generated using public markers indicated that the five-leaflet determining gene in Dusam is an allele of the previously studied Lf1 gene on chromosome 8. A high-resolution map delimited the genomic region controlling the leaflet number trait to a sequence length of 49 kb. AP2 domain-containing Glyma.08g281900 annotated in this 49 kb region appeared to be a strong candidate for the Lf1-encoding gene, as members of the AP2-type transcription factor family regulate lateral organ development. Dusam additionally exhibits visually distinct phenotypes for shattering and seed-coat cracking traits. However, the two traits were clearly unlinked to the Lf1 gene in our mapping population. Interestingly, the mapping results suggest that the Lf1 gene most likely exerts a pleiotropic effect on the number of seeds per pod. Thus, our results provide a strong foundation towards the cloning of this compound leaf development gene and marker-assisted selection of the seeds per pod trait.
DOI:10.2135/cropsci2003.2028URL [本文引用: 1]
DOI:10.1007/s00122-008-0759-6URL [本文引用: 3]
The seven-leaflet character of soybean [Glycine max L. (Merr.)] is a single recessive trait conditioned by the lf 2 gene. The lf 2 gene is located on linkage group (LG) 16 of the classical soybean genetic map, but it has not been placed on the molecular map. The objective of this research was to identify the location of the lf 2 gene on the soybean molecular map using simple sequence repeat (SSR) markers. A backcross breeding method was used to create three- and seven-leaflet near-isogenic lines in genetic backgrounds of ‘Traill’, ‘MN1401’, and ‘MN1801’. Eight mapping populations were derived from eight single heterozygous Lf 2 lf 2 plants. A total of 482 SSR markers that covered approximately every 10–20cM of all soybean molecular LG were used to screen the mapping populations for polymorphisms. For the 115 SSRs that were identified as polymorphic, possible linkage between the lf 2 gene and the polymorphic SSR markers was determined. One SSR marker from the LG B1, Sat_272, was linked (LOD>4.0) to the lf 2 gene in the Traill and MN1401 derived populations, with map distances ranging from 2.8 to 11.2cM. Two additional markers (a SSR, Sat_270 and a SNP, A588c) located on LG B1 were also polymorphic and were identified as linked to the lf 2 gene in one of the populations. This research was successful in mapping the lf 2 gene to LG B1 of the soybean molecular map and therefore, provides evidence that molecular LG B1 corresponds to classical LG 16.]]>
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1016/j.devcel.2010.06.004URLPMID:20643354 [本文引用: 1]
Division of the Arabidopsis zygote defines two fundamentally different developmental domains, the proembryo and suspensor. The resulting boundary separates domain-specific gene expression, and a signal originating from the proembryo instructs the suspensor to generate the root stem cell niche. While root induction is known to require the phytohormone auxin and the Auxin Response Factor MONOPTEROS, it has remained largely elusive how the two domains involved in this process are initially specified. Here, we show that the GATA factor HANABA TARANU (HAN) is required to position the inductive proembryo boundary. Mutations in HAN cause a coordinated apical shift of gene expression patterns, revealing that HAN regulates transcription in the basal proembryo. Key auxin transporters are affected as early as the 8 cell stage, resulting in apical redistribution of auxin. Remarkably, han embryos eventually organize a root independent of MONOPTEROS and the suspensor around a new boundary marked by the auxin maximum.
[本文引用: 1]
DOI:10.1126/science.1248384URLPMID:24531971 [本文引用: 1]
In this work, we investigate morphological differences between Arabidopsis thaliana, which has simple leaves, and its relative Cardamine hirsuta, which has dissected leaves comprising distinct leaflets. With the use of genetics, interspecific gene transfers, and time-lapse imaging, we show that leaflet development requires the REDUCED COMPLEXITY (RCO) homeodomain protein. RCO functions specifically in leaves, where it sculpts developing leaflets by repressing growth at their flanks. RCO evolved in the Brassicaceae family through gene duplication and was lost in A. thaliana, contributing to leaf simplification in this species. Species-specific RCO action with respect to its paralog results from its distinct gene expression pattern in the leaf base. Thus, regulatory evolution coupled with gene duplication and loss generated leaf shape diversity by modifying local growth patterns during organogenesis.
DOI:10.1371/journal.pone.0146492URLPMID:26745275 [本文引用: 2]
Cytokinesis in plants involves the formation of unique cellular structures such as the phragmoplast and the cell plate, both of which are required to divide the cell after nuclear division. In order to isolate genes that are involved in de novo cell wall formation, we performed a large-scale, microscope-based screen for Arabidopsis mutants that severely impair cytokinesis in the embryo. We recovered 35 mutations that form abnormally enlarged cells with multiple, often polyploid nuclei and incomplete cell walls. These mutants represent seven genes, four of which have previously been implicated in phragmoplast or cell plate function. Mutations in two loci show strongly reduced transmission through the haploid gametophytic generation. Molecular cloning of both corresponding genes reveals that one is represented by hypomorphic alleles of the kinesin-5 gene RADIALLY SWOLLEN 7 (homologous to tobacco kinesin-related protein TKRP125), and that the other gene corresponds to the Arabidopsis FUSED ortholog TWO-IN-ONE (originally identified based on its function in pollen development). No mutations that completely abolish the formation of cross walls in diploid cells were found. Our results support the idea that cytokinesis in the diploid and haploid generations involve similar mechanisms.
DOI:10.1038/nbt.2095URLPMID:22267009 [本文引用: 1]
The majority of agronomic traits are controlled by multiple genes that cause minor phenotypic effects, making the identification of these genes difficult. Here we introduce MutMap, a method based on whole-genome resequencing of pooled DNA from a segregating population of plants that show a useful phenotype. In MutMap, a mutant is crossed directly to the original wild-type line and then selfed, allowing unequivocal segregation in second filial generation (F(2)) progeny of subtle phenotypic differences. This approach is particularly amenable to crop species because it minimizes the number of genetic crosses (n = 1 or 0) and mutant F(2) progeny that are required. We applied MutMap to seven mutants of a Japanese elite rice cultivar and identified the unique genomic positions most probable to harbor mutations causing pale green leaves and semidwarfism, an agronomically relevant trait. These results show that MutMap can accelerate the genetic improvement of rice and other crop plants.
DOI:10.1111/tpj.12105URL [本文引用: 1]
The majority of agronomically important crop traits are quantitative, meaning that they are controlled by multiple genes each with a small effect (quantitative trait loci, QTLs). Mapping and isolation of QTLs is important for efficient crop breeding by marker-assisted selection (MAS) and for a better understanding of the molecular mechanisms underlying the traits. However, since it requires the development and selection of DNA markers for linkage analysis, QTL analysis has been time-consuming and labor-intensive. Here we report the rapid identification of plant QTLs by whole-genome resequencing of DNAs from two populations each composed of 2050 individuals showing extreme opposite trait values for a given phenotype in a segregating progeny. We propose to name this approach QTL-seq as applied to plant species. We applied QTL-seq to rice recombinant inbred lines and F2 populations and successfully identified QTLs for important agronomic traits, such as partial resistance to the fungal rice blast disease and seedling vigor. Simulation study showed that QTL-seq is able to detect QTLs over wide ranges of experimental variables, and the method can be generally applied in population genomics studies to rapidly identify genomic regions that underwent artificial or natural selective sweeps.
DOI:10.1016/j.molp.2018.12.018URLPMID:30597214 [本文引用: 1]
Deciphering the genetic mechanisms underlying agronomic traits is of great importance for crop improvement. Most of these traits are controlled by multiple quantitative trait loci (QTLs), and identifying the underlying genes by conventional QTL fine-mapping is time-consuming and labor-intensive. Here, we devised a new method, named quantitative trait gene sequencing (QTG-seq), to accelerate QTL fine-mapping. QTG-seq combines QTL partitioning to convert a quantitative trait into a near-qualitative trait, sequencing of bulked segregant pools from a large segregating population, and the use of a robust new algorithm for identifying candidate genes. Using QTG-seq, we fine-mapped a plant-height QTL in maize (Zea mays L.), qPH7, to a 300-kb genomic interval and verified that a gene encoding an NF-YC transcription factor was the functional gene. Functional analysis suggested that qPH7-encoding protein might influence plant height by interacting with a CO-like protein and an AP2 domain-containing protein. Selection footprint analysis indicated that qPH7 was subject to strong selection during maize improvement. In summary, QTG-seq provides an efficient method for QTL fine-mapping in the era of
[本文引用: 1]
DOI:10.1186/s12864-015-2344-0URLPMID:26739042 [本文引用: 1]
BACKGROUND: A landmark in soybean research, Glyma1.01, the first whole genome sequence of variety Williams 82 (Glycine max L. Merr.) was completed in 2010 and is widely used. However, because the assembly was primarily built based on the linkage maps constructed with a limited number of markers and recombinant inbred lines (RILs), the assembled sequence, especially in some genomic regions with sparse numbers of anchoring markers, needs to be improved. Molecular markers are being used by researchers in the soybean community, however, with the updating of the Glyma1.01 build based on the high-resolution linkage maps resulting from this research, the genome positions of these markers need to be mapped. RESULTS: Two high density genetic linkage maps were constructed based on 21,478 single nucleotide polymorphism loci mapped in the Williams 82 x G. soja (Sieb. & Zucc.) PI479752 population with 1083 RILs and 11,922 loci mapped in the Essex x Williams 82 population with 922 RILs. There were 37 regions or single markers where marker order in the two populations was in agreement but was not consistent with the physical position in the Glyma1.01 build. In addition, 28 previously unanchored scaffolds were positioned. Map data were used to identify false joins in the Glyma1.01 assembly and the corresponding scaffolds were broken and reassembled to the new assembly, Wm82.a2.v1. Based upon the plots of the genetic on physical distance of the loci, the euchromatic and heterochromatic regions along each chromosome in the new assembly were delimited. Genomic positions of the commonly used markers contained in BARCSOYSSR_1.0 database and the SoySNP50K BeadChip were updated based upon the Wm82.a2.v1 assembly. CONCLUSIONS: The information will facilitate the study of recombination hot spots in the soybean genome, identification of genes or quantitative trait loci controlling yield, seed quality and resistance to biotic or abiotic stresses as well as other genetic or genomic research.
DOI:10.1101/gr.107524.110URLPMID:20644199 [本文引用: 1]
Next-generation DNA sequencing (NGS) projects, such as the 1000 Genomes Project, are already revolutionizing our understanding of genetic variation among individuals. However, the massive data sets generated by NGS--the 1000 Genome pilot alone includes nearly five terabases--make writing feature-rich, efficient, and robust analysis tools difficult for even computationally sophisticated individuals. Indeed, many professionals are limited in the scope and the ease with which they can answer scientific questions by the complexity of accessing and manipulating the data produced by these machines. Here, we discuss our Genome Analysis Toolkit (GATK), a structured programming framework designed to ease the development of efficient and robust analysis tools for next-generation DNA sequencers using the functional programming philosophy of MapReduce. The GATK provides a small but rich set of data access patterns that encompass the majority of analysis tool needs. Separating specific analysis calculations from common data management infrastructure enables us to optimize the GATK framework for correctness, stability, and CPU and memory efficiency and to enable distributed and shared memory parallelization. We highlight the capabilities of the GATK by describing the implementation and application of robust, scale-tolerant tools like coverage calculators and single nucleotide polymorphism (SNP) calling. We conclude that the GATK programming framework enables developers and analysts to quickly and easily write efficient and robust NGS tools, many of which have already been incorporated into large-scale sequencing projects like the 1000 Genomes Project and The Cancer Genome Atlas.
DOI:10.4161/fly.19695URL [本文引用: 1]
We describe a new computer program, SnpEff, for rapidly categorizing the effects of variants in genome sequences. Once a genome is sequenced, SnpEff annotates variants based on their genomic locations and predicts coding effects. Annotated genomic locations include intronic, untranslated region, upstream, downstream, splice site, or intergenic regions. Coding effects such as synonymous or non-synonymous amino acid replacement, start codon gains or losses, stop codon gains or losses, or frame shifts can be predicted. Here the use of SnpEff is illustrated by annotating similar to 356,660 candidate SNPs in similar to 117 Mb unique sequences, representing a substitution rate of similar to 1/305 nucleotides, between the Drosophila melanogaster w(1118); iso-2; iso-3 strain and the reference y(1); cn(1) bw(1) sp(1) strain. We show that similar to 15,842 SNPs are synonymous and similar to 4,467 SNPs are non-synonymous (N/S similar to 0.28). The remaining SNPs are in other categories, such as stop codon gains (38 SNPs), stop codon losses (8 SNPs), and start codon gains (297 SNPs) in the 5'UTR. We found, as expected, that the SNP frequency is proportional to the recombination frequency (i.e., highest in the middle of chromosome arms). We also found that start-gain or stop-lost SNPs in Drosophila melanogaster often result in additions of N-terminal or C-terminal amino acids that are conserved in other Drosophila species. It appears that the 5' and 3'UTRs are reservoirs for genetic variations that changes the termini of proteins during evolution of the Drosophila genus. As genome sequencing is becoming inexpensive and routine, SnpEff enables rapid analyses of whole-genome sequencing data to be performed by an individual laboratory.
DOI:10.1101/gr.146936.112URL [本文引用: 4]
Forward genetic screens in model organisms are vital for identifying novel genes essential for developmental or disease processes. One drawback of these screens is the labor-intensive and sometimes inconclusive process of mapping the causative mutation. To leverage high-throughput techniques to improve this mapping process, we have developed a Mutation Mapping Analysis Pipeline for Pooled RNA-seq (MMAPPR) that works without parental strain information or requiring a preexisting SNP map of the organism, and adapts to differential recombination frequencies across the genome. MMAPPR accommodates the considerable amount of noise in RNA-seq data sets, calculates allelic frequency by Euclidean distance followed by Loess regression analysis, identifies the region where the mutation lies, and generates a list of putative coding region mutations in the linked genomic segment. MMAPPR can exploit RNA-seq data sets from isolated tissues or whole organisms that are used for gene expression and transcriptome analysis in novel mutants. We tested MMAPPR on two known mutant lines in zebrafish, nkx2.5 and tbx1, and used it to map two novel ENU-induced cardiovascular mutants, with mutations found in the ctr9 and cds2 genes. MMAPPR can be directly applied to other model organisms, such as Drosophila and Caenorhabditis elegans, that are amenable to both forward genetic screens and pooled RNA-seq experiments. Thus, MMAPPR is a rapid, cost-efficient, and highly automated pipeline, available to perform mutant mapping in any organism with a well-assembled genome.
DOI:10.1371/journal.pone.0068529URLPMID:23874658 [本文引用: 2]
Advances in genome sequencing technologies have enabled researchers and breeders to rapidly associate phenotypic variation to genome sequence differences. We recently took advantage of next-generation sequencing technology to develop MutMap, a method that allows rapid identification of causal nucleotide changes of rice mutants by whole genome resequencing of pooled DNA of mutant F2 progeny derived from crosses made between candidate mutants and the parental line. Here we describe MutMap+, a versatile extension of MutMap, that identifies causal mutations by comparing SNP frequencies of bulked DNA of mutant and wild-type progeny of M3 generation derived from selfing of an M2 heterozygous individual. Notably, MutMap+ does not necessitate artificial crossing between mutants and the wild-type parental line. This method is therefore suitable for identifying mutations that cause early development lethality, sterility, or generally hamper crossing. Furthermore, MutMap+ is potentially useful for gene isolation in crops that are recalcitrant to artificial crosses.
DOI:10.1093/nar/25.17.3389URLPMID:9254694 [本文引用: 1]
The BLAST programs are widely used tools for searching protein and DNA databases for sequence similarities. For protein comparisons, a variety of definitional, algorithmic and statistical refinements described here permits the execution time of the BLAST programs to be decreased substantially while enhancing their sensitivity to weak similarities. A new criterion for triggering the extension of word hits, combined with a new heuristic for generating gapped alignments, yields a gapped BLAST program that runs at approximately three times the speed of the original. In addition, a method is introduced for automatically combining statistically significant alignments produced by BLAST into a position-specific score matrix, and searching the database using this matrix. The resulting Position-Specific Iterated BLAST (PSI-BLAST) program runs at approximately the same speed per iteration as gapped BLAST, but in many cases is much more sensitive to weak but biologically relevant sequence similarities. PSI-BLAST is used to uncover several new and interesting members of the BRCT superfamily.
[本文引用: 1]
DOI:10.1038/75556URLPMID:10802651 [本文引用: 1]
DOI:10.1093/nar/gkh063URLPMID:14681412 [本文引用: 1]
A grand challenge in the post-genomic era is a complete computer representation of the cell and the organism, which will enable computational prediction of higher-level complexity of cellular processes and organism behavior from genomic information. Toward this end we have been developing a knowledge-based approach for network prediction, which is to predict, given a complete set of genes in the genome, the protein interaction networks that are responsible for various cellular processes. KEGG at http://www.genome.ad.jp/kegg/ is the reference knowledge base that integrates current knowledge on molecular interaction networks such as pathways and complexes (PATHWAY database), information about genes and proteins generated by genome projects (GENES/SSDB/KO databases) and information about biochemical compounds and reactions (COMPOUND/GLYCAN/REACTION databases). These three types of database actually represent three graph objects, called the protein network, the gene universe and the chemical universe. New efforts are being made to abstract knowledge, both computationally and manually, about ortholog clusters in the KO (KEGG Orthology) database, and to collect and analyze carbohydrate structures in the GLYCAN database.
DOI:10.1093/nar/28.1.33URLPMID:10592175 [本文引用: 1]
Rational classification of proteins encoded in sequenced genomes is critical for making the genome sequences maximally useful for functional and evolutionary studies. The database of Clusters of Orthologous Groups of proteins (COGs) is an attempt on a phylogenetic classification of the proteins encoded in 21 complete genomes of bacteria, archaea and eukaryotes (http://www. ncbi.nlm. nih.gov/COG). The COGs were constructed by applying the criterion of consistency of genome-specific best hits to the results of an exhaustive comparison of all protein sequences from these genomes. The database comprises 2091 COGs that include 56-83% of the gene products from each of the complete bacterial and archaeal genomes and approximately 35% of those from the yeast Saccharomyces cerevisiae genome. The COG database is accompanied by the COGNITOR program that is used to fit new proteins into the COGs and can be applied to functional and phylogenetic annotation of newly sequenced genomes.
[本文引用: 2]
[本文引用: 2]
DOI:10.1038/379066a0URL [本文引用: 1]
DOI:10.1126/science.1070343URLPMID:12052958 [本文引用: 2]
KNOTTEDI-like homeobox (KNOXI) genes regulate development of the leaf from the shoot apical meristem (SAM) and may regulate leaf form. We examined KNOXI expression in SAMs of various vascular plants and found that KNOXI expression correlated with complex leaf primordia. However, complex primordia may mature into simple leaves. Therefore, not all simple leaves develop similarly, and final leaf morphology may not be an adequate predictor of homology.
DOI:10.1105/tpc.6.12.1859URLPMID:7866029 [本文引用: 1]
The homeobox gene knotted1 (kn1) was first isolated by transposon tagging a dominant leaf mutant in maize. Related maize genes, isolated by virtue of sequence conservation within the homeobox, fall into two classes based on sequence similarity and expression patterns. Here, we report the characterization of two genes, KNAT1 and KNAT2 (for knotted-like from Arabidopsis thaliana) that were cloned from Arabidopsis using the kn1 homeobox as a heterologous probe. The homeodomains of KNAT1 and KNAT2 are very similar to the homeodomains of proteins encoded by class 1 maize genes, ranging from 78 to 95% amino acid identity. Overall, the deduced KNAT1 and KNAT2 proteins share amino acid identities of 53 and 40%, respectively, with the KN1 protein. Intron positions are also fairly well conserved among KNAT1, KNAT2, and kn1. Based on in situ hybridization analysis, the expression pattern of KNAT1 during vegetative development is similar to that of class 1 maize genes. In the shoot apex, KNAT1 transcript is localized primarily to the shoot apical meristem; down-regulation of expression occurs as leaf primordia are initiated. In contrast to the expression of class 1 maize genes in floral and inflorescence meristems, the expression of KNAT1 in the shoot meristem decreases during the floral transition and is restricted to the cortex of the inflorescence stem. Transgenic Arabidopsis plants carrying the KNAT1 cDNA and the kn1 cDNA fused to the cauliflower mosaic virus 35S promoter were generated. Misexpression of KNAT1 and kn1 resulted in highly abnormal leaf morphology that included severely lobed leaves. The expression pattern of KNAT1 in the shoot meristem combined with the results of transgenic overexpression experiments supports the hypothesis that class 1 kn1-like genes play a role in morphogenesis.
DOI:10.1038/ng1835URLPMID:16823378 [本文引用: 2]
A key question in biology is how differences in gene function or regulation produce new morphologies during evolution. Here we investigate the genetic basis for differences in leaf form between two closely related plant species, Arabidopsis thaliana and Cardamine hirsuta. We report that in C. hirsuta, class I KNOTTED1-like homeobox (KNOX) proteins are required in the leaf to delay cellular differentiation and produce a dissected leaf form, in contrast to A. thaliana, in which KNOX exclusion from leaves results in a simple leaf form. These differences in KNOX expression arise through changes in the activity of upstream gene regulatory sequences. The function of ASYMMETRIC LEAVES1/ROUGHSHEATH2/PHANTASTICA (ARP) proteins to repress KNOX expression is conserved between the two species, but in C. hirsuta the ARP-KNOX regulatory module controls new developmental processes in the leaf. Thus, evolutionary tinkering with KNOX regulation, constrained by ARP function, may have produced diverse leaf forms by modulating growth and differentiation patterns in developing leaf primordia.
DOI:10.1105/tpc.109.068148URLPMID:19820191 [本文引用: 1]
Class 1 KNOTTED1-LIKE HOMEOBOX (KNOXI) genes encode transcription factors that are expressed in the shoot apical meristem (SAM) and are essential for SAM maintenance. In some species with compound leaves, including tomato (Solanum lycopersicum), KNOXI genes are also expressed during leaf development and affect leaf morphology. To dissect the role of KNOXI proteins in leaf patterning, we expressed in tomato leaves a fusion of the tomato KNOXI gene Tkn2 with a sequence encoding a repressor domain, expected to repress common targets of tomato KNOXI proteins. This resulted in the formation of small, narrow, and simple leaves due to accelerated differentiation. Overexpression of the wild-type form of Tkn1 or Tkn2 in young leaves also resulted in narrow and simple leaves, but in this case, leaf development was blocked at the initiation stage. Expression of Tkn1 or Tkn2 during a series of spatial and temporal windows in leaf development identified leaf initiation and primary morphogenesis as specific developmental contexts at which the tomato leaf is responsive to KNOXI activity. Arabidopsis thaliana leaves responded to overexpression of Arabidopsis or tomato KNOXI genes during the morphogenetic stage but were largely insensitive to their overexpression during leaf initiation. These results imply that KNOXI proteins act at specific stages within the compound-leaf development program to delay maturation and enable leaflet formation, rather than set the compound leaf route.
DOI:10.1016/s0092-8674(00)81051-xURLPMID:8625411 [本文引用: 1]
The most distinctive morphogenetic feature of leaves is their being either simple or compound. To study the basis for this dichotomy, we have exploited the maize homeobox-containing Knotted-1 (Kn1) gene in conjunction with mutations that alter the tomato compound leaf. We show that misexpression of Kn1 confers different phenotypes on simple and compound leaves. Up to 2000 leaflets, organized in compound reiterated units, are formed in tomato leaves expressing Kn1. In contrast, Kn1 induces leaf malformations but fails to elicit leaf ramification in plants with inherent simple leaves such as Arabidopsis or in tomato mutant plants with simple leaves. Moreover, the tomato Kn1 ortholog, unlike that of Arabidopsis, is expressed in the leaf primordia. Presumably, the two alternative leaf forms are conditioned by different developmental programs in the primary appendage that is common to all types of leaves.
DOI:10.1038/35050091URLPMID:11140682 [本文引用: 1]
Meristem function in plants requires both the maintenance of stem cells and the specification of founder cells from which lateral organs arise. Lateral organs are patterned along proximodistal, dorsoventral and mediolateral axes. Here we show that the Arabidopsis mutant asymmetric leaves1 (as1) disrupts this process. AS1 encodes a myb domain protein, closely related to PHANTASTICA in Antirrhinum and ROUGH SHEATH2 in maize, both of which negatively regulate knotted-class homeobox genes. AS1 negatively regulates the homeobox genes KNAT1 and KNAT2 and is, in turn, negatively regulated by the meristematic homeobox gene SHOOT MERISTEMLESS. This genetic pathway defines a mechanism for differentiating between stem cells and organ founder cells within the shoot apical meristem and demonstrates that genes expressed in organ primordia interact with meristematic genes to regulate shoot morphogenesis.
DOI:10.1016/s0092-8674(00)81439-7URLPMID:9630222 [本文引用: 1]
The organs of a higher plant show two fundamental axes of asymmetry: proximodistal and dorsoventral. Dorsoventrality in leaves, bracts, and petal lobes of Antirrhinum majus requires activity of the PHANTASTICA (PHAN) gene. Conditional mutants revealed that PHAN is also required for earlier elaboration of the proximodistal axis. PHAN was isolated and shown to encode a MYB transcription factor homolog. PHAN mRNA is first detected in organ initials before primordium initiation. The structure and expression pattern of PHAN, together with its requirement in two key features of organ development, are consistent with a role in specifying lateral organ identity as distinct from that of the stem or meristem. PHAN also appears to maintain meristem activity in a non-cell-autonomous manner.
DOI:10.1242/dev.00655URLPMID:12900456 [本文引用: 1]
Recent work on species with simple leaves suggests that the juxtaposition of abaxial (lower) and adaxial (upper) cell fates (dorsiventrality) in leaf primordia is necessary for lamina outgrowth. However, how leaf dorsiventral symmetry affects leaflet formation in species with compound leaves is largely unknown. In four non-allelic dorsiventrality-defective mutants in tomato, wiry, wiry3, wiry4 and wiry6, partial or complete loss of ab-adaxiality was observed in leaves as well as in lateral organs in the flower, and the number of leaflets in leaves was reduced significantly. Morphological analyses and expression patterns of molecular markers for ab-adaxiality [LePHANTASTICA (LePHAN) and LeYABBY B (LeYAB B)] indicated that ab-adaxial cell fates were altered in mutant leaves. Reduction in expression of both LeT6 (a tomato KNOX gene) and LePHAN during post-primordial leaf development was correlated with a reduction in leaflet formation in the wiry mutants. LePHAN expression in LeT6 overexpression mutants suggests that LeT6 is a negative regulator of LePHAN. KNOX expression is known to be correlated with leaflet formation and we show that LeT6 requires LePHAN activity to form leaflets. These phenotypes and gene expression patterns suggest that the abaxial and adaxial domains of leaf primordia are important for leaflet primordia formation, and thus also important for compound leaf development. Furthermore, the regulatory relationship between LePHAN and KNOX genes is different from that proposed for simple-leafed species. We propose that this change in the regulatory relationship between KNOX genes and LePHAN plays a role in compound leaf development and is an important feature that distinguishes simple leaves from compound leaves.
DOI:10.1105/tpc.13.1.31URLPMID:11158527 [本文引用: 1]
Isolation and characterization of two severe alleles at the Stamina pistilloida (Stp) locus reveals that Stp is involved in a wide range of developmental processes in the garden pea. The most severe allele, stp-4, results in flowers consisting almost entirely of sepals and carpels. Production of ectopic secondary flowers in stp-4 plants suggests that Stp is involved in specifying floral meristem identity in pea. The stp mutations also reduce the complexity of the compound pea leaf, and primary inflorescences often terminate prematurely in an aberrant sepaloid flower. In addition, stp mutants were shorter than their wild-type siblings due to a reduction in cell number in their internodes. Fewer cells were also found in the epidermis of the leaf rachis of stp mutants. Examination of the effects of stp-4 in double mutant combinations with af, tl, det, and veg2-2-mutations known to influence leaf, inflorescence, and flower development in pea-suggests that Stp function is independent of these genes. A synergistic interaction between weak mutant alleles at Stp and Uni indicated that these two genes act together, possibly to regulate primordial growth. Molecular analysis revealed that Stp is the pea homolog of the Antirrhinum gene Fimbriata (Fim) and of UNUSUAL FLORAL ORGANS (UFO) from Arabidopsis. Differences between Fim/UFO and Stp mutant phenotypes and expression patterns suggest that expansion of Stp activity into the leaf was an important step during evolution of the compound leaf in the garden pea.
DOI:10.1104/pp.104.054288URLPMID:15824286 [本文引用: 1]
Floral patterning in Papilionoideae plants, such as pea (Pisum sativum) and Medicago truncatula, is unique in terms of floral organ number, arrangement, and initiation timing as compared to other well-studied eudicots. To investigate the molecular mechanisms involved in the floral patterning in legumes, we have analyzed two mutants, proliferating floral meristem and proliferating floral organ-2 (pfo-2), obtained by ethyl methanesulfonate mutagenesis of Lotus japonicus. These two mutants showed similar phenotypes, with indeterminate floral structures and altered floral organ identities. We have demonstrated that loss of function of LjLFY and LjUFO/Pfo is likely to be responsible for these mutant phenotypes, respectively. To dissect the regulatory network controlling the floral patterning, we cloned homologs of the ABC function genes, which control floral organ identity in Arabidopsis (Arabidopsis thaliana). We found that some of the B and C function genes were duplicated. RNA in situ hybridization showed that the C function genes were expressed transiently in the carpel, continuously in stamens, and showed complementarity with the A function genes in the heterogeneous whorl. In proliferating floral meristem and pfo-2 mutants, all B function genes were down-regulated and the expression patterns of the A and C function genes were drastically altered. We conclude that LjLFY and LjUFO/Pfo are required for the activation of B function genes and function together in the recruitment and determination of petals and stamens. Our findings suggest that gene duplication, change in expression pattern, gain or loss of functional domains, and alteration of key gene functions all contribute to the divergence of floral patterning in L. japonicus.
DOI:10.1371/journal.pone.0037040URLPMID:22649507 [本文引用: 1]
BACKGROUND: Plant height is an important agronomic trait that affects yield and tolerance to certain abiotic stresses. Understanding the genetic control of plant height is important for elucidating the regulation of maize development and has practical implications for trait improvement in plant breeding. METHODOLOGY/PRINCIPAL FINDINGS: In this study, two independent, semi-dwarf maize EMS mutants, referred to as dwarf & irregular leaf (dil1), were isolated and confirmed to be allelic. In comparison to wild type plants, the mutant plants have shorter internodes, shorter, wider and wrinkled leaves, as well as smaller leaf angles. Cytological analysis indicated that the leaf epidermal cells and internode parenchyma cells are irregular in shape and are arranged in a more random fashion, and the mutants have disrupted leaf epidermal patterning. In addition, parenchyma cells in the dil1 mutants are significantly smaller than those in wild-type plants. The dil1 mutation was mapped on the long arm of chromosome 6 and a candidate gene, annotated as an AP2 transcription factor-like, was identified through positional cloning. Point mutations near exon-intron junctions were identified in both dil1 alleles, resulting in mis-spliced variants. CONCLUSION: An AP2 transcription factor-like gene involved in stalk and leaf development in maize has been identified. Mutations near exon-intron junctions of the AP2 gene give mis-spliced transcript variants, which result in shorter internodes and wrinkled leaves.
DOI:10.1146/annurev.biochem.69.1.617URLPMID:10966471 [本文引用: 1]
Aminoacyl-tRNAs are substrates for translation and are pivotal in determining how the genetic code is interpreted as amino acids. The function of aminoacyl-tRNA synthesis is to precisely match amino acids with tRNAs containing the corresponding anticodon. This is primarily achieved by the direct attachment of an amino acid to the corresponding tRNA by an aminoacyl-tRNA synthetase, although intrinsic proofreading and extrinsic editing are also essential in several cases. Recent studies of aminoacyl-tRNA synthesis, mainly prompted by the advent of whole genome sequencing and the availability of a vast body of structural data, have led to an expanded and more detailed picture of how aminoacyl-tRNAs are synthesized. This article reviews current knowledge of the biochemical, structural, and evolutionary facets of aminoacyl-tRNA synthesis.
DOI:10.1007/s00018-015-2017-9URLPMID:26283621 [本文引用: 1]
Nonsense-mediated mRNA decay (NMD) is a translation-dependent, multistep process that degrades irregular or faulty messenger RNAs (mRNAs). NMD mainly targets mRNAs with a truncated open reading frame (ORF) due to premature termination codons (PTCs). In addition, NMD also regulates the expression of different types of endogenous mRNA substrates. A multitude of factors are involved in the tight regulation of the NMD mechanism. In this review, we focus on the molecular mechanism of mammalian NMD. Based on the published data, we discuss the involvement of translation termination in NMD initiation. Furthermore, we provide a detailed overview of the core NMD machinery, as well as several peripheral NMD factors, and discuss their function. Finally, we present an overview of diseases associated with NMD factor mutations and summarize the current state of treatment for genetic disorders caused by nonsense mutations.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/gtc.12033URL [本文引用: 1]
SMG-1, a member of the PIKK (phosphoinositide 3-kinase-related kinase) family, plays a critical role in the mRNA quality control system known as nonsense-mediated mRNA decay (NMD). NMD protects cells from the accumulation of aberrant mRNAs with premature termination codons (PTCs) which encode nonfunctional or potentially harmful truncated proteins. SMG-1 directly phosphorylates Upf1 helicase, another key component of NMD, upon recognition of PTC on postspliced mRNA during the initial round of translation. Phosphorylated-Upf1 recruits the SMG-5/SMG-7 complex to induce ribosome dissociation and decapping-mediated decay. Phospho-Upf1 also recruits the SMG-6 endonuclease which might be involved in endo-cleavage. Upf1 ATPase/helicase activities are likely required for the activation of other mRNA decay enzymes and the mRNA-protein complex dissociation to complete NMD. At present, a variety of tools are available that can specifically suppress NMD, and it has become possible to examine the contribution of NMD in a variety of physiological and pathological conditions.
DOI:10.1073/pnas.1501558112URLPMID:25918386 [本文引用: 1]
Bacterial strains carrying nonsense suppressor tRNA genes played a crucial role in early work on bacterial and bacterial viral genetics. In eukaryotes as well, suppressor tRNAs have played important roles in the genetic analysis of yeast and worms. Surprisingly, little is known about genetic suppression in archaea, and there has been no characterization of suppressor tRNAs or identification of nonsense mutations in any of the archaeal genes. Here, we show, using the beta-gal gene as a reporter, that amber, ochre, and opal suppressors derived from the serine and tyrosine tRNAs of the archaeon Haloferax volcanii are active in suppression of their corresponding stop codons. Using a promoter for tRNA expression regulated by tryptophan, we also show inducible and regulatable suppression of all three stop codons in H. volcanii. Additionally, transformation of a DeltapyrE2 H. volcanii strain with plasmids carrying the genes for a pyrE2 amber mutant and the serine amber suppressor tRNA yielded transformants that grow on agar plates lacking uracil. Thus, an auxotrophic amber mutation in the pyrE2 gene can be complemented by expression of the amber suppressor tRNA. These results pave the way for generating archaeal strains carrying inducible suppressor tRNA genes on the chromosome and their use in archaeal and archaeviral genetics. We also provide possible explanations for why suppressor tRNAs have not been identified in archaea.
[本文引用: 1]
[本文引用: 1]
DOI:10.1002/jcb.240470402URLPMID:1795013 [本文引用: 1]
The organization of DNA within the nucleus has been demonstrated to be both cell and tissue specific and is arranged in a non-random fashion in both sperm and somatic cells. Nuclear structure has a pivotal role in this three-dimensional organization of DNA and RNA and contributes as well to forming fixed organizing sites for nuclear functions, such as DNA replication, transcription, and RNA processing. In sperm, DNA is also organized in a specific fashion by the nuclear matrix and DNA-protamine interactions. Within somatic cells, the nuclear matrix provides a three-dimensional framework for the tissue specific regulation of genes by directed interaction with transcriptional activators. This differential organization of the DNA by the nuclear matrix, in a tissue specific manner, contributes to tissue specific gene expression. The nuclear matrix is the first link from the DNA to the entire tissue matrix system and provides a direct structural linkage to the cytomatrix and extracellular matrix. In summary, the tissue matrix serves as a dynamic structural framework for the cell which interacts to organize and process spatial and temporal information to coordinate cellular functions and gene expression. The tissue matrix provides a structural system for integrating form and function.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}