 ,1,*
,1,*Development of efficient KASP molecular markers based on high throughput sequencing in maize
LU Hai-Yan1, ZHOU Ling1, LIN Feng1, WANG Rui2, WANG Feng-Ge2, ZHAO Han,1,*通讯作者:
收稿日期:2018-10-7接受日期:2019-01-19网络出版日期:2019-03-01
| 基金资助: |
Received:2018-10-7Accepted:2019-01-19Online:2019-03-01
| Fund supported: |
作者简介 About authors
E-mail:

摘要
关键词:
Abstract
Keywords:
PDF (2061KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
陆海燕, 周玲, 林峰, 王蕊, 王凤格, 赵涵. 基于高通量测序开发玉米高效KASP分子标记[J]. 作物学报, 2019, 45(6): 872-878. doi:10.3724/SP.J.1006.2019.83067
LU Hai-Yan, ZHOU Ling, LIN Feng, WANG Rui, WANG Feng-Ge, ZHAO Han.
玉米是重要的粮食及饲料作物, 由于具有较高的遗传多样性已成为重要模式作物[1]。其核苷酸多态性变化是所有作物中较高的, 不同材料基因组之间的差异达到1.42%, 大于人类与黑猩猩之间的差异(1.34%)[2]。丰富的基因组变异为开发具有高多态性分子标记提供了可能。根据变异序列的长短将基因组变异分为单核苷酸变异(SNP)及插入缺失(Indel 2~50 bp)和>50 bp的序列重排(SV)[2] 3种类型。基于Indel和SV开发的标记不仅检测通量低, 而且成本相对较高[3]。SNP标记具有分布广泛、遗传稳定性好且易于实现自动化等优点, 目前已迅速取代传统的分子标记[4,5]。
高通量测序技术的快速发展, 有效推动玉米基因组结构变异的研究。Jiao等[6]利用重测序技术对6个玉米自交系进行DNA结构变异分析, 发现中国玉米自交系中存在大量SNP位点的变异, 报道揭示了玉米基因组中超过100万个SNP。2018年, Sun等[7]报道了Mo17的从头基因组装配, 提供了又一个重要的玉米参考基因组序列。这一个参考基因组的产生为广泛比较玉米中种内基因组多样性提供了前所未有的机会, 通过比对B73和Mo17基因组, 发现了980多万个SNP位点、140多万的插入/缺失, 该研究为开发基于SNP位点的分子标记提供了理论依据和数据支撑。SNP是单核苷酸变异产生的多态性, 无法通过常规的PCR技术与凝胶电泳技术利用长度的差异来区分其多态性[8]。目前尽管开发了多种SNP基因分型方法, 包括定点测序技术、等位基因特异性PCR (AS-PCR)、裂解扩增多态性序列(CAPS)、温度开关PC (TS-PCR)和基因重测序[9,10,11], 但这些方法都存在低通量或高成本等局限性[12]。近些年, 高通量的SNP位点检测几乎都采用芯片技术。赵久然等[13]利用基于SNP位点的芯片技术揭示中国玉米育种种质的遗传多样性与群体遗传结构。王文斌等[14]利用含有2846个高质量SNP位点的芯片进行了31份骨干自交系群体遗传结构、遗传多样性分析。基于芯片技术的SNP基因分型平台的通量较高, 每张芯片包含几百到数百万的SNP位点[15], 但大多数基于芯片的基因分型平台用于样本量较多的全基因组扫描时成本较高[16]。
KASP (Kompetitive Allele Specific PCR), 即竞争性等位基因特异性PCR技术, 以其高度稳定性、准确性和低成本的特点, 已经被广泛应用于高通量SNP分型, 尤其在样本量大, SNP位点少时, KASP的应用特点最为显著[17]。Rasheed等[12]开发70个小麦KASP分子标记, 并成功验证了这些标记与品种亲本和双亲本群体的表型相关, 揭示了KASP分子标记在小麦育种中的潜在应用。国际玉米和小麦改良中心使用KASP, 每年产生超过一百万个数据点用于作物改良[18]。KASP在小到中等数量的标记应用中显示出了成本效益和可扩展的灵活性, 如在双亲本群体的质量控制分析、标记辅助的轮回选择、辅助回交和QTL精细定位等方面发挥重要作用[17]。KASP分子标记的开发需要综合考虑目的区段DNA的唯一变异, 多态性以及位点特异性等因素, 基于重测序数据可以筛选出目的区段100 bp内只有一个SNP位点的变异, 而通过高密度芯片筛选的位点无法保证唯一性。
本研究利用遗传来源不同的205份玉米自交系的全基因组重测序信息, 筛选出多态性高的二态性SNP位点, 基于这些位点以及侧翼序列信息开发KASP分子标记, 并利用其中46个代表玉米自交系进行验证, 获得了一套在染色体上均匀分布且多态性高的KASP分子标记。这些标记可以为玉米SNP基因分型研究提供重要的基础和分析方法, 同时为种质改良及杂交种组配提供DNA水平上的信息。
1 材料与方法
1.1 材料及全基因组测序数据
选用来自7个不同类群的205份玉米自交系, 其二代原始序列下载于NCBI (https://www.ncbi.nlm. nih.gov/)的3个SRA (Sequence Read Archive)数据库(PRJNA82843、SRP011907和PRJNA260788)。删除其中低测序深度的材料。利用生物信息学分析技术比对这些材料的序列, 选择最小等位基因频率(MAF)大于0.05的位点, 删除基因型缺失率超过20%的位点, 以哈迪温伯格平衡显著性阈值(HWE) P<0.001为标准再次过滤, 最终保留多态信息含量(PIC)>0.4并且是二态性的目的SNP位点[19,20]。从7个类群中, 分别抽出一定数量的代表自交系(详细信息见表1)为实验材料, 用于KASP验证。
1.2 标记开发和筛选
根据SNP位点和侧翼序列设计PCR扩增引物, 开发KASP分子标记。每个标记设计2条SNP特异性引物(F1/F2)和一条通用引物(R), F1尾部添加能够与FAM荧光结合的特异性序列, F2尾部添加能够与HEX荧光结合的特异性序列。KASP引物设计由mIndel软件包[21]完成, 引物由生工生物工程(上海)股份有限公司合成。对46份代表自交系的叶片进行取样, 按照上海浦迪植物基因组提取试剂盒操作步骤提取基因组DNA。KASP反应总体系为5 μL, 包含2×KASP Master Mix 2.5 μL、KASP Assay Mix (引物混合工作液) 0.07 μL、浓度为20 ng μL-1的模板DNA 2.43 μL。KASP反应程序为, 第一步: 94℃, 15 min; 第二步: 94℃, 20 s, 61~55℃, 1 min, 每个循环降低0.6℃, 共进行10个循环; 第三步: 94℃, 20 s, 55℃, 1 min, 共进行26个循环, PCR在购买自LGC公司型号为Hydrocycler 16的水浴PCR仪中进行。通过KASP荧光分析仪(LGC公司型号为PHERAstar plus)扫描分析PCR结果。根据基因型分型效果和在玉米基因组染色体上的分布等信息, 挑选有效的KASP标记。
Table 1
表1
表1实验材料详细信息
Table 1
| 类群 Group | 205份供试材料 205Experimental cultivars | 用于KASP验证的46份材料 46 cultivars for KASP | ||||
|---|---|---|---|---|---|---|
| 样本 大小 Size | 占总样本比例 Percentage (%) | 样本 大小 Size | 占总样本比例 Percentage (%) | 代表自交系 Representative inbred lines | ||
| 瑞德 Reid | 14 | 6.86 | 4 | 8.69 | B73, A632, 郑32, H84 B73, A632, Zheng 32, H84 | |
| 改良瑞德Improved Reid | 33 | 16.09 | 9 | 19.57 | 郑58, 478, 5003, 黄C, 1205A, 综3, 铁7922, K22 Zheng 58, 478, 5003, Huang C, 1205A, Zong 3, Tie 7922, K22 | |
| 热带 Tropic | 18 | 8.78 | 8 | 17.39 | 苏湾1611, 四路糯, CML162, DY206, CML52, Ki11, Ki3 Suwan 1611, Silunuo, CML162, DY206, CML52, Ki11, Ki3 | |
| PB | 22 | 10.73 | 7 | 15.22 | T877, 齐319, P138, 沈137, 沈135, Yu 87-1 T877, Qi 319, P138, Shen 137, Shen 135, Yu 87-1 | |
| 兰卡斯特 Lancaster | 50 | 24.39 | 6 | 13.04 | Mo17, OH43, LH51, LH61, LH54, 龙抗11 Mo17, OH43, LH51, LH61, LH54, Longkang 11 | |
| 四平头 Sipingtou | 45 | 21.95 | 9 | 19.57 | 黄早四, 昌7-2, S22, Huangyesi 3, Wu 126, 444, K12 Huangzaosi, Chang 7-2, S22, Huangyesi 3, Wu 126, 444, K12 | |
| 其他Other | 23 | 11.22 | 3 | 6.52 | F2, F7 | |
新窗口打开|下载CSV
1.3 数据分析
采用编写的Perl脚本和VCFtools软件计算最小等位基因频率(MAF)、多态信息含量(PIC)[20,22]。利用TASSELV5.0软件的邻接算法(N-J)[23]计算205份玉米自交系之间的遗传距离并构建聚类图。运用R软件的cor[17]函数包对205份多样性自交系的总SNP位点与实验验证筛选出的KASP标记位点进行遗传相关性分析。2 结果与分析
2.1 SNP位点的筛选和KASP分子标记的开发
利用205份玉米自交系全基因组测序数据开发SNP位点, 对总SNP位点进行过滤(缺失率<20%, MAF>0.05, PIC>0.4), 共筛选出1,660,336个二态性SNP位点, 这些位点均匀分布在玉米基因组的10条染色体上。采用Perl脚本进一步筛选出SNP位点前后各100 bp没有其他变异位点的SNP, 通过多窗口比对, 选择在不同材料之间具有保守型的SNP位点。经过引物软件设计, 开发出700个KASP分子标记。根据开发设计的700对KASP引物, 对B73、Mo17、昌7-2、郑58等46份玉米自交系进行基因型分型。获得了近490个高质量的分子标记引物, 这些标记位点可以准确清楚地将相同基因型的自交系聚类(聚类形式如图1), 同时阴性对照NTC (No Template Control)始终聚集在一起, 且不产生信号, 成功率达到70%。其他引物被淘汰的原因主要包括可以将相同基因型的样本聚类, 但是聚集略有分散, 或是同样可以分类但在一些基因型中的扩增效率不高等。
图1
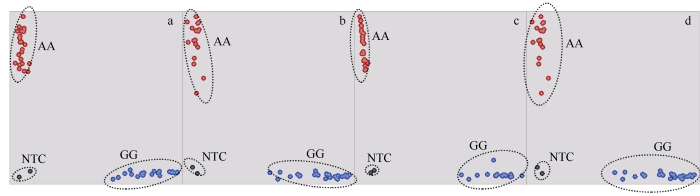 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1KASP标记基因分型图
a~d为编号不同的KASP标记的基因分型图; AA: 红色簇状是具有HEX型等位基因的品种; GG: 蓝色簇状是具有FAM型等位基因的品种; NTC: 是不含模板的空白对照。
Fig. 1Genotyping map of KASP markers
a-d represent genotyping map of different KASP markers; AA: red cluster is a variety with HEX allele; GG: blue cluster is a variety with FAM allele; NTC: blank control without template.
2.2 KASP分子标记的挑选和评价
为了进一步筛选出具有代表性的标记, 对鉴定出的490个标记进行过滤, 主要依据能够将HEX型和FAM型的基因型精确分型且MAF>0.40、PIC> 0.45, 以及在染色体上的位置分布均匀等信息, 最终筛选出202个位点(引物信息见附表), 这些位点覆盖在玉米基因组的10条染色体上, 平均间隔为8.74 Mb (图2-A)。利用Perl脚本对这些位点的等位基因频率等方面进行总体评价, 202个SNP位点PIC变化范围为0.458~0.469, 平均值为0.463, MAF变化范围为0.434~0.467, 平均值为0.451 (图2-B), 表明202个SNP位点具有高特异性和高稳定性。图2
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2202个SNP位点遗传多态性
A: 202个SNP位点在染色体上的物理位置, 右边数字是202个KASP标记的编号, 左边数字是202个KASP标记在染色体上物理位置, 单位为Mb; B: 202个SNP位点的最小等位基因频率分布(MAF)和多态信息含量分布(PIC)。
Fig. 2Genetic polymorphisms of 202 SNPs
A: the physical location on chromosome of 202 SNPs, the numbers on the right are 202 KASP markers ID, and the numbers on the left are the physical locations, the unit is Mb; B: minimum allele frequency (MAF) and polymorphic information content (PIC) of 202 SNPs.
2.3 KASP分子标记在品种鉴定中的应用
基于实验获得的202个KASP标记位点和重测序数据获得的1,660,336个二态性SNP位点, 分别对205份材料构建了系统发育树。图3显示, 基于两组不同数据构建的进化树均将205份自交系划分为7大类群, 分别是瑞德群(代表自交系B73)、改良瑞德群(代表自交系郑58)、热带群(代表自交系Ki11、Suwan 1611、Four-row)、PB群(代表自交系T877、Qi 319)、兰卡斯特群(代表自交系Mo17、OH43)、四平头群(代表自交系黄早四、昌7-2)、其他种质群(代表自交系F2、F7)。类群划分结果高度一致, 其中201份材料的划群结果相同, 4份材料的划群结果不同。图3-A显示, 205份自交系中用于KASP验证的46份代表自交系, 分别分布于各自的类群中, 46份自交系的类群划分与系谱来源相一致。表明202个KASP标记具有较好的重复性, 能够代替总SNP位点进行类群划分。另外, 利用R语言对KASP标记位点与重测序数据筛选的总SNP位点进行了遗传距离相似性分析, 两者之间的遗传距离相似性高达89.5%, 进一步表明202个KASP标记在鉴定玉米种质资源研究方面具有可靠性和代表性。图3
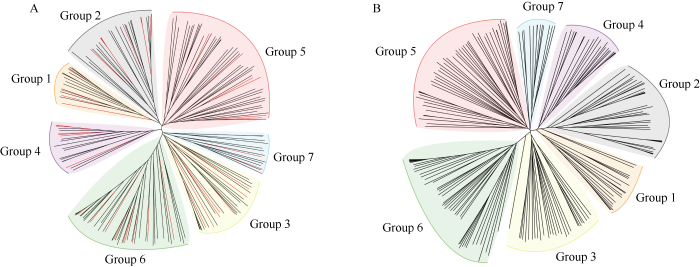 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3Neighbor-joining (N-J)树分析
A: 205份自交系基于202个位点的Neighbor-Joining (N-J)树分析, 红色线条代
Fig. 3Neighbor-joining (N-J) tree analysis
A: N-J tree of 205 inbred lines based on 202 SNPs and 46 experimental materials are represented by red lines; B: N-J tree of 205 inbred lines based on 1,660,336 SNPs; Groups 1-7 represent different groups of Reid, Improved Reid, Tropic, PB, Lancaster, Sipingtou, and other latitude.
3 讨论
玉米是一种经典的遗传模型, 是世界范围内的重要作物。玉米基因组在不同自交系间具有高度的遗传多样性。SNP标记具备二态性、等位基因性、高密度性、高代表性、高遗传稳定性等优点, 是构建生物高通量基因分型适合的标记。基于测序数据开发KASP标记不仅有大量SNP变异位点作为数据支撑, 而且能够筛选到唯一SNP位点变异的目的片段, 此外通过参数的设置和多窗口比对能够选择在不同材料之间具有保守性的位点。但是基于测序数据开发KASP标记的方法不仅要求基因组信息的准确性, 测序数据的分析和有效SNP位点的筛选等相关技术往往也需要配备生物信息学专业知识较强的科研人员, 因此目前关于高效KASP标记的开发研究较少。本研究基于205份自交系重测序数据, 过滤出1,660,336个SNP位点, 根据位点侧翼序列信息, 将其中700个位点开发为KASP标记。利用KASP平台验证, 获得了较高设计成功率, 主要原因是基于大量的重测序数据, 不适合用来设计KASP引物的位点在前期已经被过滤, 同时又对位点进行多态性评估, 这些步骤可以极大提高KASP引物设计效率。但是, 有些位点由于结构复杂, 测序深度浅, 或者序列信息存在错误, 可能导致一些引物设计失败。InDel (insertion-deletion)变异是同一位点不同大小片段的插入缺失, 变异结构比SNP变异复杂, 一些结构复杂的位点将来可以开发成基于InDel变异的KASP标记。
我们根据490个标记在玉米基因组染色体上的分布位置、多态信息含量等信息, 从中挑选出能对玉米品种特异性和真实性有效鉴定的202个KASP标记。205份材料的划群结果显示其中201份材料划群结果相同, 表明202个位点与总SNP位点的聚类分析结果高度一致, 且202个KASP标记的位点与总SNP位点的遗传距离相似性系数达到89.5%。本研究结果证明202个KASP标记位点能够代替总SNP位点应用在玉米种质资源鉴定等方面的研究。
KASP检测基因分型可以应用于分子辅助育种、种质资源鉴定、样本群体分析、性状基因的精细定位、全基因组关联分析等。何中虎等人[24]开发了一套基于KASP技术检测小麦功能基因的引物; 余四斌及其团队[25]基于KASP技术开发了用于水稻特殊营养物质、水稻产量等基因分型的引物。本研究开发的玉米高效KASP分子标记能够用于玉米群体多样性分析、群体结构分析、玉米种质资源的鉴定等研究, 大大减少了后期田间工作量, 可为提高玉米育种效率提供参考。
4 结论
基于高通量测序开发的202个KASP标记, 在染色体上分布均匀, 平均间隔8.74 Mb, PIC和MAF的平均值分别为0.463、0.451, 能成功区分玉米的杂种优势群。KASP标记将在玉米杂种优势群划分、连锁群构建、种质资源遗传背景确定等方面发挥作用。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1038/ng0914-1039URLPMID:22660547 [本文引用: 1]

The success of modern maize breeding has been demonstrated by remarkable increases in productivity over the last four decades. However, the underlying genetic changes correlated with these gains remain largely unknown. We report here the sequencing of 278 temperate maize inbred lines from different stages of breeding history, including deep resequencing of 4 lines with known pedigree information. The results show that modern breeding has introduced highly dynamic genetic changes into the maize genome. Artificial selection has affected thousands of targets, including genes and non-genic regions, leading to a reduction in nucleotide diversity and an increase in the proportion of rare alleles. Genetic changes during breeding happen rapidly, with extensive variation (SNPs, indels and copy-number variants (CNVs)) occurring, even within identity-by-descent regions. Our genome-wide assessment of genetic changes during modern maize breeding provides new strategies as well as practical targets for future crop breeding and biotechnology.
DOI:10.1038/s41588-018-0182-0URLPMID:30061735 [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1007/978-1-60327-411-1_26URL [本文引用: 1]
The increasing need for large-scale genotyping applications of single nucleotide polymorphisms ( SNPs ) in model and nonmodel organisms requires the development of low-cost technologies accessible to minimally equipped laboratories. The method presented here allows efficient discrimination of SNPs by allele-specific PCR in a single reaction with standard PCR conditions. A common reverse primer and two forward allele-specific primers with different tails amplify two allele-specific PCR products of different lengths, which are further separated by agarose gel electrophoresis. PCR specificity is improved by the introduction of a destabilizing mismatch within the 3鈥 end of the allele-specific primers. This is a simple and inexpensive method for SNP detection that does not require PCR optimization.
DOI:10.1021/jf047955bURLPMID:15769137 [本文引用: 1]
Grain hardness is one of the most important quality characteristics of cultivated bread wheat (Triticum aestivum L.) and has been reported to result from either a failure to express puroindoline a (Pina) or single-nucleotide mutations in puroindoline b (Pinb). Up to now, seven alleles from Pinb-D1a to Pinb-D1g were identified in bread wheat. Compared to the DNA coding region of Pinb-D1a (allele for softness), six single-nucleotide polymorphisms (SNPs) were detected in six alleles for Pinb-D1. In this study, we used pyrosequencing technology to develop two SNP assays for identification of the seven Pinb alleles and characterized SNP variations in the Pinb of 493 European wheat varieties. Of the three hardness alleles Pinb-D1b, Pinb-D1c, and Pinb-D1d detected in this study, Pinb-D1b was the most predominant hardness allele in European hard wheats. The hardness genotypes of partial German wheat varieties available confirmed the reliability and validation of the SNP assays developed for the Pinb locus. Therefore, pyrosequencing technology offers an efficient, precise, and reliable concept for high-throughout genotyping to assist selection of grain hardness genes in wheat quality breeding programs.
DOI:10.1186/1471-2164-10-580URLPMID:2795770 [本文引用: 1]
pAbstract/p pBackground/p pMany research and diagnostic applications rely upon the assay of individual single nucleotide polymorphisms (SNPs). Thus, methods to improve the speed and efficiency for single-marker SNP genotyping are highly desirable. Here, we describe the method of temperature-switch PCR (TSP), a biphasic four-primer PCR system with a universal primer design that permits amplification of the target locus in the first phase of thermal cycling before switching to the detection of the alleles. TSP can simplify assay design for a range of commonly used single-marker SNP genotyping methods, and reduce the requirement for individual assay optimization and operator expertise in the deployment of SNP assays./p pResults/p pWe demonstrate the utility of TSP for the rapid construction of robust and convenient endpoint SNP genotyping assays based on allele-specific PCR and high resolution melt analysis by generating a total of 11,232 data points. The TSP assays were performed under standardised reaction conditions, requiring minimal optimization of individual assays. High genotyping accuracy was verified by 100% concordance of TSP genotypes in a blinded study with an independent genotyping method./p pConclusion/p pTheoretically, TSP can be directly incorporated into the design of assays for most current single-marker SNP genotyping methods. TSP provides several technological advances for single-marker SNP genotyping including simplified assay design and development, increased assay specificity and genotyping accuracy, and opportunities for assay automation. By reducing the requirement for operator expertise, TSP provides opportunities to deploy a wider range of single-marker SNP genotyping methods in the laboratory. TSP has broad applications and can be deployed in any animal and plant species./p
DOI:10.1007/s00122-016-2743-xURLPMID:27306516 [本文引用: 2]
Key message We developed and validated a robust marker toolkit for high-throughput and cost-effective screening of a large number of functional genes in wheat.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.9787/PBB.2014.2.3.195URL [本文引用: 1]
Recent advances in next-generation sequencing (NGS) and single nucleotide polymorphism (SNP) genotyping promise to greatly accelerate crop improvement if properly deployed. High-throughput SNP genotyping offers a number of advantages over previous marker systems, including an abundance of markers, rapid processing of large populations, a variety of genotyping systems to meet different needs, and straightforward allele calling and database storage due to the bi-allelic nature of SNP markers. NGS technologies have enabled rapid whole genome sequencing, providing extensive SNP discovery pools to select informative markers for different sets of germplasm. Highly multiplexed fixed array platforms have enabled powerful approaches such as genome-wide association studies. On the other hand, routine deployment of trait-specific SNP markers requires flexible, low-cost systems for genotyping smaller numbers of SNPs across large breeding populations, using platforms such as Fluidigm’s Dynamic Arrays64, Douglas Scientific’s Array Tape64, and LGC’s automated systems for running KASP64 markers. At the same time, genotyping by sequencing (GBS) is rapidly becoming popular for low-cost high-density genome-wide scans through multiplexed sequencing. This review will discuss the range of options available to modern breeders for integrating SNP markers into their programs, whether by outsourcing to service providers or setting up in-house genotyping facilities, and will provide an example of SNP deployment for rice research and breeding as demonstrated by the Genotyping Services Lab at the International Rice Research Institute.
DOI:10.1111/pbi.12183URLPMID:4265271 [本文引用: 1]
SummaryHigh-density single nucleotide polymorphism (SNP) genotyping arrays are a powerful tool for studying genomic patterns of diversity, inferring ancestral relationships between individuals in populations and studying marker–trait associations in mapping experiments. We developed a genotyping array including about 9002000 gene-associated SNPs and used it to characterize genetic variation in allohexaploid and allotetraploid wheat populations. The array includes a significant fraction of common genome-wide distributed SNPs that are represented in populations of diverse geographical origin. We used density-based spatial clustering algorithms to enable high-throughput genotype calling in complex data sets obtained for polyploid wheat. We show that these model-free clustering algorithms provide accurate genotype calling in the presence of multiple clusters including clusters with low signal intensity resulting from significant sequence divergence at the target SNP site or gene deletions. Assays that detect low-intensity clusters can provide insight into the distribution of presence–absence variation (PAV) in wheat populations. A total of 4602977 SNPs from the wheat 90K array were genetically mapped using a combination of eight mapping populations. The developed array and cluster identification algorithms provide an opportunity to infer detailed haplotype structure in polyploid wheat and will serve as an invaluable resource for diversity studies and investigating the genetic basis of trait variation in wheat.
[本文引用: 3]
URL [本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 2]
[本文引用: 2]
DOI:10.1186/s12864-016-2614-5URLPMID:4832496 [本文引用: 1]
Rich in genetic information and cost-effective to genotype, the Insertion-Deletion (InDel) molecular marker system is an important tool for studies in genetics, genomics and for marker-assisted breeding. Advent of next-generation sequencing (NGS) revolutionized the speed and throughput of sequence data generation, and enabled genome-wide identification of insertion and deletion variation. However, current NGS-based InDel mining tools, such as Samtools, GATK and Atlas2, all rely on a reference genome for variant calling which hinders their application on unsequenced organisms and the output of short InDels compromised their use on gel-based genotyping platforms. To address these issues, an enhanced platform is needed to identify longer InDels and develop markers in absence of a reference genome. Here we present mInDel (multiple InDel), a next-generation variant calling tool specifically designed for InDel marker discovery. By taking in raw sequence reads and assembling them into contigsde novo, this software identifies InDel polymorphisms using a sliding window alignment from assembled contigs, rendering a unique advantage when a reference genome is unavailable. By providing an option of combining multiple discovered InDels as output, mInDel is amiable to gel-based genotyping platforms where markers with large polymorphisms are preferred. We demonstrated the usability and performance of this software through a case study using a set of maize NGS data, and experimentally validated the accuracy of markers generated from mInDel. mInDel is a novel and practical tool that enables rapid genome-wide InDel marker discovery. The features of being independent from a reference genome and the flexibility with downstream genotyping platforms will allow a broad range of applications across genetics research and plant breeding. The mInDel pipeline is freely available atwww.github.com/lyd0527/mInDel.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}