 ,1,*, 杨梅,2,*, 殷鹏程2, 周玉乾1, 何海军1, 邱法展,2,*
,1,*, 杨梅,2,*, 殷鹏程2, 周玉乾1, 何海军1, 邱法展,2,*Fine Mapping and Genetic Effect Analysis of a Major QTL qPH3.2 Associated with Plant Height in Maize (Zea mays L.)
LIU Zhong-Xiang,1,*, YANG Mei,2,*, YIN Peng-Cheng2, ZHOU Yu-Qian1, HE Hai-Jun1, QIU Fa-Zhan,2,*通讯作者:
第一联系人:
收稿日期:2018-01-8接受日期:2018-04-11网络出版日期:2018-05-14
| 基金资助: |
Received:2018-01-8Accepted:2018-04-11Online:2018-05-14
| Fund supported: |

摘要
关键词:
Abstract
Keywords:
PDF (3669KB)元数据多维度评价相关文章导出EndNote|Ris|Bibtex收藏本文
本文引用格式
刘忠祥, 杨梅, 殷鹏程, 周玉乾, 何海军, 邱法展. 玉米株高主效QTL qPH3.2精细定位及遗传效应分析[J]. 作物学报, 2018, 44(9): 1357-1366. doi:10.3724/SP.J.1006.2018.01357
LIU Zhong-Xiang, YANG Mei, YIN Peng-Cheng, ZHOU Yu-Qian, HE Hai-Jun, QIU Fa-Zhan.
在玉米诸多株型构成中, 茎秆高度(株高)和穗位高度(穗位高)尤为重要, 这两个因素均与产量密切相关, 因此解析玉米株高的遗传基础, 对玉米株型育种乃至产量提高具有重要意义。在水稻、玉米、高粱、小麦等作物中, 株高主要是由节间数目和节间长度决定[1,2]。迄今为止, 很多研究表明玉米株高与穗上节间数目显著相关, 穗位高与穗下节间数目密切相关, 穗上节间数受环境因素影响不大[3,4,5,6,7]。而节间长度的变化是影响株高的又一关键因素, 大量研究发现细胞数目和细胞大小是影响玉米节间长度的关键因子, 即节间细胞数增多或减少及节间细胞纵向长度都影响株高性状[8,9,10,11,12,13]。
株高是数量性状, 在玉米中, 通常一个群体内就可同时检测到多个株高QTL, 并且不同群体中检测到的QTL也不相同[14]。迄今为止, 利用不同的遗传群体在玉米中已定位了大量的株高主效QTL [4,14-27], 但是图位克隆的案例很少, 已报道克隆了跟GA 合成相关的基因ZmGA3ox2、D3和AN1[28,29,30]; 其他已克隆的近30个玉米矮生基因多是通过突变体克隆的方式获得, 如Br2基因通过转座子标签方式获得, Br2隐性基因可以显著降低株高, Br2突变体是通过减小节间长度来降低株高, 它的节间数目和叶片大小没有变化[31]; 还有的通过GA敏感型突变体获得株高相关基因, 在玉米中已经鉴定了5个即dwarf1 (d1)、dwarf2 (d2)、dwarf3 (d3)、dwarf5 (d5)和another ear 1 (an1) [32,33], 玉米Dwarf8基因的3个突变体d8-1、d8-2023和d8-MP1分别在DELLA区、VHYNP区以及同时在DELLA和VHYNP区发生氨基酸不等数目的缺失导致GA信号传导受阻而产生矮秆半矮秆表型[34], Dwarf9与Dwarf8相似[35]。Vanishing tassel2 (vt2)突变体表现出株高降低的特征[36]。除了上述基因从激素方面调节株高外, 还存在一些其他类型的基因调控株高, 如玉米的矮秆突变体d2003, 在基因Viviparous8 (Vp8)的编码区插入一个碱基从而使翻译提前终止, 表现出节间数目增加, 节间长度缩短[11]。
玉米株高与产量高度相关, 适当降低株高, 增加种植密度, 提高抗倒伏能力从而增加产量; 增加株高也可以增加玉米生物学产量, 提高玉米在生物能源和青贮饲料中的应用价值。迄今为止, 图位克隆的玉米株高QTL或基因很少, 因此利用特殊的材料进一步寻找新的株高主效QTL并进行遗传剖析, 不仅有利于阐明玉米株高形成的遗传基础, 也对玉米株高的遗传改良及育种新材料的创制具有重要的应用价值, 同时为克隆株高QTL奠定扎实基础。本研究利用构建的分离群体, 共定位到5个与株高相关的主效QTL位点, qPH3-1、qPH3-2、qPH3.3、qPH9-1和qPH9-2, 分别可以解释3.30%~22.22%的表型变异。在此基础上, 针对多年多环境共同检测到且解释表型变异最大的主效QTL qPH3.2进一步利用剩余杂合系及目标区段跨叠系开展重定位及精细定位工作, 为图位克隆该QTL/基因奠定基础。
1 材料与方法
1.1 试验材料
2个株高差异显著的高代回交重组自交系(BCRIL) W1 (高)和W2 (矮), 株高相差约20 cm (图1)。定位所用F2群体及目标区段跨叠系群体均由W1和W2杂交及自交构建所得。1.2 试验方法
1.2.1 群体材料田间种植方式 2013年春于湖北武汉(30.35°N, 114.17°E)种植包含465个单株的F2群体, 自交构建相应的F2:3家系, 考察F2群体的株高。于2014年春、2014年夏季分别于湖北黄冈(30.44°N, 114.87°E)和山东潍坊(35.32°N, 118°E)种植F2:3家系, 按随机区组设计, 2次重复, 每个家系1行, 行长3.00 m, 株距0.25 m, 行距0.50 m。每次重复起始种植亲本W1、W2以及F1各1行作为对照, 田间管理与当地大田管理相同。成熟期对每个家系的株高和穗位高进行考察, 株高是从地面至雄穗顶端的高度, 穗位高是从地面到最上部果穗着生节的高度, 对所有单株开放授粉。同时2014年春在湖北武汉种植包含200个单株的BC1群体, 成熟期考察株高与穗位高。将4个环境分别命名为2013WH (2013年湖北武汉)、2014WH (2014年湖北武汉)、2014HG (2014年湖北黄冈)和2014SD (2014年山东潍坊)。根据初定位结果, 以W2为背景, 利用MAS方法回交获得BC2F2, 并筛选重组单株。2015年于海南自交所有类型的重组单株, 构建次级跨叠系, 自交后代于2016年分别种植于湖北武汉、湖北黄冈、山东潍坊3个环境, 每个环境3次重复, 开放授粉, 种植方式及管理同上述F2群体相同。
1.2.2 外源GA处理W1和W2的时期及方法
2016年将W1和W2种植于网室, 在二叶一心期开始使用移液枪将100 μL GA溶液(浓度为100 μg mL-1)注入植株顶端, 一周处理2次, 选用空白对照, 各处理40株, 连续处理至抽雄散粉期。
1.2.3 节间细胞数目观察 在W1和W2株高差异明显的拔节期, 各选取穗上部第3节间为样本, 使用双刃剃须刀对所取材料沿着纵向快速切割, 制作徒手切片, 将削下的切片组织悬浮于水中, 选取薄的具有单层细胞的切片, 采用配置Olympus DP72照相机的Olympus 1X71荧光倒置显微镜(Olympus, Tokyo, Japan)观察照相。观察统计每个材料10张切片距表皮组织5~10层的细胞大小, 采用t测验检验2个材料间细胞大小的差异。
1.3 基因型鉴定、标记开发、数据统计分析及QTL定位
用CTAB法抽提所有材料的DNA; 常规PCR扩增程序分析基因型; 参考B73基因组序列, 根据InDel的侧翼序列设计InDel标记。参考B73序列, 利用在线工具Primer (http://122.205.95.20/cgibin/primer3 plus/primer3plus.cgi)设计SSR标记, 在150~400 bp的扩增序列范围与玉米基因序列进行Blast (http:// 122.205.95.20/blast/blast.html), 以确保所设计的引物扩增的特异性。用SPSS17.0软件对各性状进行描述性统计分析、相关性分析和方差分析; 用软件MapMaker/EXP V3.0[37,38]构建遗传连锁图, 获得各标记间的遗传距离。利用Windows QTL Cartographer 2.5软件, 复合区间作图法(CIM)确定QTL的阈值。
2 结果与分析
2.1 W1与W2农艺性状
2.1.1 W1与W2在多个环境中植株表型性状2015年和2016年在3个环境中各种植80株W1和W2开展株高表型调查, 发现株高差异在不同环境中能稳定遗传。2015年湖北武汉W1株高为148.7±3.9 cm, W2为123.5±4.8 cm; 2016年湖北黄冈W1株高为140.3±3.4 cm, W2为129.1±4.0 cm; 2016年山东W1株高为162.8±3.9 cm, W2为136.4±4.0 cm。2年3点试验中株高差异都达到了极显著水平(图1)。
图1
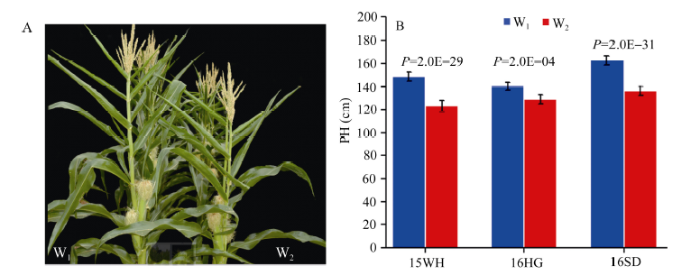 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图1RIL系W1和W2的株高比较
A: 亲本W1和W2的株高表型; B: 亲本在湖北武汉(15WH)、湖北黄冈(16HG)、山东潍坊(16SD)的株高表型差异分析。
Fig. 1Comparison of plant height (PH) between W1 and W2
A: The plant height (PH) between W1 and W2; B: The difference analysis of W1 and W2 in Wuhan of Hubei (15WH), Huanggang of Hubei (16HG), and Weifang of Shandong (16SD), respectively.
2.1.2 W1与W2节间细胞学观察 W1和W2节间数目相同, 穗上和穗下均为6个节间, 表明节间长度差异是引起2个材料株高差异的主要原因; 同时W1和W2节间长度的差异主要集中在穗上部节间(图2); 因此选择穗上第3节间作为取样材料观察节间细胞数目和大小, 结果W1的节间细胞长度显著大于W2 (P<0.01), 表明细胞长度的增加导致W1的节间长于W2 (图3)。
图2
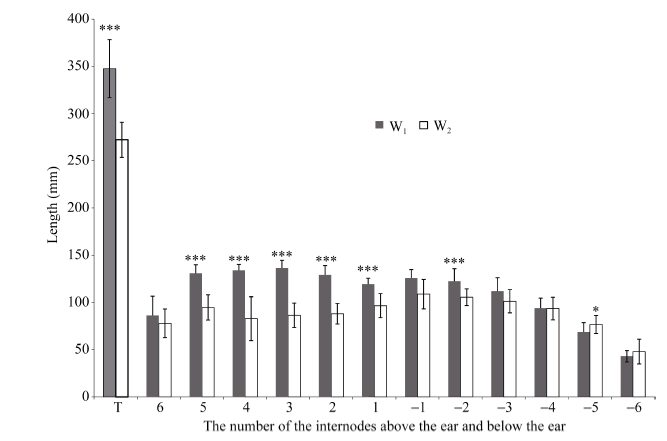 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图2W1和W2的节间长度比较
*、**、***分别表示在P<0.05, 0.01, 0.001概率水平下差异极显著, 误差线代表SD, nW1=30, nW2=49。穗上节间用1、2、3、4、5、6表示, 穗下节间用-1、-2、-3、-4、-5、-6表示, 雄穗用T表示。
Fig. 2Comparison of internode length between W1 and W2
*, **, *** indicate significant difference at the 0.05, 0.01, and 0.001 probability levels, respectively. Error bars represent the SD of the mean (nW1=30, nW2=49). The internodes above the ear are labeled as 1, 2, 3, 4, 5, and 6. The internodes below the ear are labeled as -1, -2, -3, -4, -5, and -6. The tassel is labeled as T.
图3
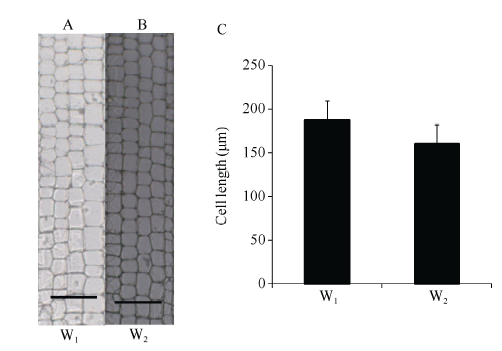 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图3节间细胞形态学观察
A和B分别是W1和W2的徒手切片(比例尺: 500 μm); C为W1和W2节间细胞长度比较。
Fig. 3Cell morphological observation of stem
A and B are hand-cut longitudinal section of W1 and W2 (scale bar: 500 μm); C: the length of internode cells in W1 and W2.
2.1.3 W1和W2对外源GA响应分析 玉米中已克隆的几个株高相关基因都涉及到GA合成或信号传导途径。从二叶一心期开始使用100 μg mL-1外源GA处理W1与W2发现其株高显著高于相应的对照, 说明W1和W2对GA信号响应的途径均正常(图4和表1)。
Table 1
表1
表1外源GA处理株高结果
Table 1
| 对照CK | GA | |
|---|---|---|
| W1 | 149.33±3.39 | 161.83±6.99*** |
| W2 | 143.78±3.60 | 156.41±6.49*** |
新窗口打开|下载CSV
图4
 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图4外源GA处理对W1和W2株高的效应
A和B分别为W1的处理与对照; C和D分别为W2的处理与对照。
Fig. 4Effect of exogenous GA on the plant height of W1 and W2
A and B are the treatment and control of W1, respectively; C and D are the treatment and control of W2, respectively.
2.2 F2:3家系表型数据分析及株高QTL初定位
2.2.1 F2:3家系表型数据分析 不同环境条件下株高和穗位高有一定差异, 湖北黄冈环境株高的均值低于湖北武汉和山东潍坊2个环境的均值。2014HG和2014SD的群体株高变异系数无明显差异, 穗位高变异系数差异较大, 株高的变异系数显著小于穗位高(表2)。Table 2
表2
表2F2:3群体株高及穗位高的表型统计
Table 2
| 性状 Trait | 环境 Environment | 均值 Average (cm) | 范围 Range (cm) | 变异系数 CV (%) | 偏度Skewness | 峰度 Kurtosis | P值 P-value |
|---|---|---|---|---|---|---|---|
| 株高PH | 2013WH | 165.47 | 116.50-191.00 | 6.76 | -0.379 | 0.527 | 0.19ns |
| 2014HG | 127.60 | 104.92-160.57 | 6.23 | 0.163 | 0.335 | 0.32ns | |
| 2014SD | 162.71 | 128.41-192.81 | 5.44 | 0.042 | 0.337 | 0.92ns | |
| 穗位高EH | 2014HG | 42.77 | 22.96-65.79 | 12.50 | 0.319 | 0.844 | 0.85ns |
| 2014SD | 60.94 | 42.86-78.59 | 8.80 | 0.168 | 0.113 | 0.98ns |
新窗口打开|下载CSV
在2014HG和2014SD环境中, 家系间株高和穗位高差异达极显著水平, 表明家系间存在显著的遗传变异, 适宜进行QTL定位。株高和穗位高在2个环境中的广义遗传力较高, 均在0.80以上, 表明这2个性状主要受遗传因素影响(表3)。
Table 3
表3
表3F2:3家系株高和穗位高遗传力分析
Table 3
| 性状 Trait | 环境 Environment | 遗传方差 Genetic variance | 剩余方差 Residual variance | 遗传力 Heritability (%) |
|---|---|---|---|---|
| 株高PH | 2014HG | 100.08*** | 38.03 | 83.04 |
| 2014SD | 157.54*** | 46.49 | 87.14 | |
| 穗位高EH | 2014HG | 44.53*** | 15.74 | 84.98 |
| 2014SD | 55.84*** | 22.10 | 80.84 |
新窗口打开|下载CSV
2.2.2 株高QTL定位 从1052对SSR标记中共筛选到86对标记在双亲W1和W2间具有多态性, 集中在第3、第4、第5和第9共4条染色体上, 多态性比例分别为20.37% (22/108)、16.82% (18/107)、14.81% (16/108)和13.08% (14/107), 说明导致W1和W2株高差异的重组片段可能位于这4条染色体上。利用F2群体及F2:3家系株高数据分别在2013WH、2014HG、2014SD 3个环境条件下开展株高QTL定位, 均在第3染色体上扫描到1个主效QTL (标记C42-P17间), 可解释8.47%~22.22%的表型变异, 来自W1的等位基因起增效作用, 表现为加性和部分显性效应, 命名为qPH3.2 (表4)。
Table 4
表4
表43个环境下定位到的控制玉米株高(PH, cm)的主效QTL (qPH3.2)
Table 4
| 环境 Environment | 范围 Range (cM) | 相邻标记 Flanking markers | 峰值标记 Peak position (cM) | bin | LOD | 加性效应 A | 显性效应 D | D/A | 基因作用 G | 贡献率PVE% |
|---|---|---|---|---|---|---|---|---|---|---|
| 2013WH | 59.09-81.46 | bnlg602-SSR5 | 73.51 | 3.04-3.05 | 4.98 | 4.42 | 0.35 | 0.08 | A | 8.47 |
| 2014HG | 59.09-81.46 | bnlg602-SSR5 | 75.51 | 3.04-3.05 | 13.85 | 4.29 | 1.11 | 0.26 | PD | 22.22 |
| 2014SD | 71.46-102.9 | SSR4-umc2266 | 87.01 | 3.05-3.06 | 12.15 | 4.37 | 1.76 | 0.40 | PD | 14.21 |
新窗口打开|下载CSV
2.3 qPH3.2的遗传剖析及精细定位
2.3.1 qPH3.2的遗传剖析 主效QTL qPH3.2被定位在标记C42-P17间, 在BC2F1群体中利用区间标记寻找到2个剩余杂合单株(RHL、Y36和Y113)并自交, 它覆盖了qPH3.2的整个区间; 利用开发的多态性标记与已有标记对2个剩余杂合系Y36和Y113自交后代进行单标记分析, 发现在标记Y91和YH-55间, 存在一个断点即在标记YH230处存在断点, 均为背景片段; 而且Y36家系和Y113家系单标记分析差异均达到显著水平(表5和表6), 证实了qPH3.2定位结果的真实性; 进而根据单标记分析的结果将QTL qPH3.2分解为2个主效QTL分别命名为qPH3.2.1和qPH3.2.2 (图5-A和图6-A)。Table 5
表5
表5RHL (Y36)的单标记分析
Table 5
| 标记 Marker | P值 P-value | AA qPH3.2 | aaW2 | |||
|---|---|---|---|---|---|---|
| 均值±标准差 Mean±SD | 群体 Size of population | 均值±标准差 Mean±SD | 个体数 Number of individual | |||
| C42 | 6E-04 | 154.2±4.2 | 20 | 147.1±4.8 | 24 | |
| Y45 | 4E-09 | 155.8±5.1 | 19 | 141.9±6.1 | 21 | |
| Y72 | 1E-09 | 155.3±6.0 | 23 | 141.5±6.3 | 25 | |
| YH196 | 4E-10 | 155.4±5.6 | 25 | 141.4±5.9 | 22 | |
| Y91 | 2E-06 | 152.7±5.7 | 22 | 141.7±6.6 | 20 | |
新窗口打开|下载CSV
Table 6
表6
表6RHL (Y113)的单标记分析
Table 6
| 标记 Marker | P值 P-value | AA qPH3.2 | aaW2 | |||
|---|---|---|---|---|---|---|
| 均值±标准差 Mean±SD | 群体 Size of population | 均值±标准差 Mean±SD | 个体数 Number of individual | |||
| YH55 | 0.03 | 152.1±3.5 | 12 | 149.0±2.8 | 13 | |
| YH61 | 0.03 | 153.5±4.4 | 24 | 150.8±4.4 | 26 | |
| P17 | 4E-05 | 155.1±3.2 | 14 | 147.7±4.0 | 13 | |
| YH112 | 0.04 | 155.5±3.2 | 14 | 151.8±3.2 | 12 | |
| YH121 | 3E-05 | 157.2±3.1 | 21 | 151.4±3.5 | 22 | |
新窗口打开|下载CSV
图5
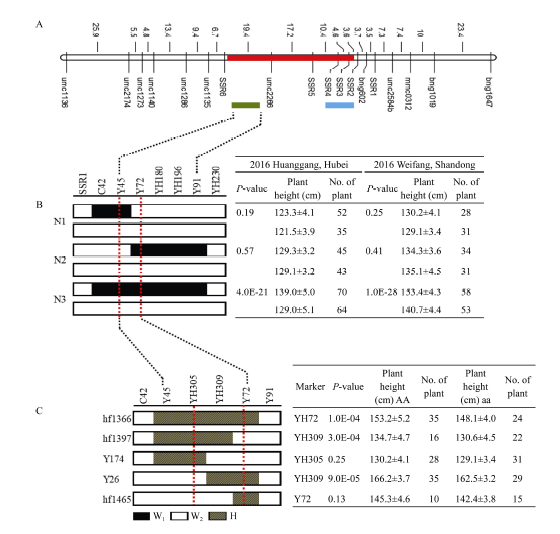 新窗口打开|下载原图ZIP|生成PPT
新窗口打开|下载原图ZIP|生成PPT图5qPH3.2.1的精细定位
A: 红色区域为qPH3.2初定位区间, 绿色和蓝色分别是qPH3.2.1和qPH3.2.2重定位区间; B: 利用3个跨叠系N1、N2和N3定位qPH3.2.1; C: qPH3.2.1精细定位结果; N代表评估表型的单株数目。AA表示携带qPH3.2.1位点的等位基因; aa是以W2为背景的等位基因。
Fig. 5Fine mapping of qPH3.2.1
A: the pre-mapping result of qPH3.2 located on red bar, green and blue bars indicate the remapping results of qPH3.2.1 and qPH3.2.2, respectively; B: the mapping results of qPH3.2.1 using N1, N2, and N3 three overlapping lines; C: the fine mapping results of qPH3.2.1. N indicates the number of the evaluated individuals in each line. AA means the allele derived from qPH3.2.1; aa indicates the allele derived from genetic background W2.
图6
 新窗口打开|下载原图ZIP|生成PPT
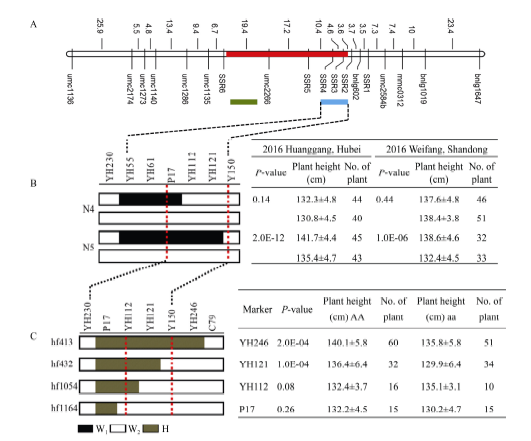
新窗口打开|下载原图ZIP|生成PPT图6qPH3.2.2的精细定位
A: 红色区域为qPH3.2初定位区间, 绿色和蓝色分别是qPH3.2.1和qPH3.2.2重定位区间; B: 利用2个跨叠系N4和N5定位qPH3.2.2; C: qPH3.2.2精细定位结果。N代表评估表型的单株数目。AA表示携带qPH3.2.2位点的等位基因; aa是以W2为背景的等位基因。
Fig. 6Fine mapping of qPH3.2.2
A: the pre-mapping result of qPH3.2 located on red bar, green and blue bars indicate the remapping results of qPH3.2.1 and qPH3.2.2, respectively; B: the mapping results of qPH3.2.2 using N4 and N5 overlapping lines; C: the fine mapping results of qPH3.2.2; N indicates the number of the evaluated individuals in each line. AA means the allele derived from qPH3.2.2; aa indicates the allele derived from genetic background W2.
2.3.2 qPH3.2.1和qPH3.2.2的精细定位 为了进一步证实剩余杂合系Y36的分析结果, 2016年在湖北黄冈、山东潍坊播种标记C42-Y91间呈阶梯覆盖的3种重组类型N1、N2和N3其自交后代中携带目标QTL区段的纯系定义为纯系A, 反之则定义为纯系B, 其余为杂合交换单株。表型数据统计分析发现, N3的子代纯系株高均值比N1和N2子代纯系株高均值高, 湖北黄冈环境中, N3比N1均值高约11.6 cm, 比N2子代株高平均值高约5.1 cm; 在山东环境中, N3子代纯系平均值比N1子代纯系高17.6 cm, 比N2高12.5 cm。可以推测重组类型N3携带影响株高的导入片段。
对3种重组类型子代纯系进行t测验进一步证明了2015年海南Y36衍生的剩余杂合系的分析结果(图5-B)。N1和N2子代纯系内作t测验分析表明N1和N2子代中2种纯系的株高均值均无明显差异。N3的子代纯系之间差异极显著(湖北黄冈: P = 4E-21、山东潍坊: P = 1E-28), 结合基因型及表型分析发现携带qPH3.2.1的纯系株高均值显著高于不携带qPH3.2.1的纯系株高均值。综合分析上述结果, 可将qPH3.2.1定位在标记Y45-Y72之间, 遗传距离为7.9 cM, 且该位点的加性效应值为5.0, 起增效作用; 进一步在目标区段内开发标记, 并新增多态性标记YH305和YH309, 重新筛选到5种类型的跨叠系, 编号分别为hf1366、hf1397、hf1465、Y174和Y26。利用这5种跨叠类型子代测验将qPH3.2.1定位在标记YH305-Y72间2 Mb范围内(图5-C)。
利用同样的方法, 借助qPH3.2.2目标区段筛选的4个重组类型(hf413、hf432、hf1054和hf1164)构建的跨叠系结合新开发的标记, 将其定位在标记YH112与Y150间1.6 Mb范围内(图6-B和图6-C); qPH3.2.2位点的加性效应值为1.3, 同样对株高起增效作用。
3 讨论
本试验所用材料W1和W2为高代回交重组自交系, 由多态性标记分析结果可知, 二者株高的差异是由少数重组片段导致的, 这样就为我们开展株高QTL定位提供了优良的材料。另外, 筛选的RHL相当于NIL形成的F1, 对于同一个QTL, 同一个株系它们的背景完全一致, 株系之间存在着一定的差异; 因此, 本实验中针对遗传剖析出的2个QTL qPH3.2.1和qPH3.2.2构建的RHL, 在作单标记分析或t测验时, 采取的是系内比较方法, 保证了定位的准确性; 另外, 同一染色体或不同染色体间QTL可以利用剩余杂合系分析不同QTL之间的相互作用。本试验精细定位的2个QTL对株高表型的调控起增效作用, 后期可以开展两位点互作研究。株高是产量性状的重要构成因子, 产量相关性状QTL的稳定性主要受遗传背景、遗传力和环境三方面的影响。环境与QTL的互作对数量性状的研究具有重要影响。对同一位点的主效QTL在不同环境下进行分析, 可以阐述QTL与环境的互作关系, 但是对于微效QTL, 在不同的环境中可能检出的效率不同。本研究的材料是利用纯系做子代验证, 在山东潍坊和湖北黄冈的纯系分析结果发现, 相同材料在不同环境中表型不一致, 在湖北黄冈的株高表型均值比山东对应的纯系株高均值低, 但是携带QTL位点的纯系和背景t测验都达到显著水平, 证明QTL qPH3.2.1和qPH3.2.2虽在不同环境受到不同程度的影响, 但却能稳定遗传; 另外, 本试验所用材料在不同环境中受到了不同程度的影响, 但株高的遗传力在不同环境中都很高, 在海南、湖北武汉、山东潍坊、湖北黄冈都能稳定地定位到qPH3.2.1和qPH3.2.2, 证明这2个QTL能够在不同环境中稳定遗传, 真实存在, 即环境稳定性QTL, 此种类型的位点将为玉米株高的遗传改良提供重要的QTL或筛选标记[39]。
株高差异影响因素主要是节间数目和节间长度, 节间数目又分为穗上节间数目和穗下节间数目, 其中节间长度又分为穗上和穗下, 主要是穗上节间的差异。穗上节间数是一个稳定的植物学性状, 表现加性效应遗传, 受环境影响较小, 因此穗上节间数目既是提升株高的重要元素也是研究玉米抗倒伏能力的一个理想性状[1,4-7]。很多研究表明可以通过增加节间细胞数来增加节间长度, 如水稻中CYP94C2b、COI1和Ghd7 3个基因及玉米中d2003突变基因等都是通过增加纵向细胞数目来增加植株节间长度[8-9,11]; 另外, 增加纵向节间细胞长度也会增加节间长度, 如水稻eui突变基因和玉米AP2基因均是通过调控细胞体积来影响节间长度的[12,13]; 本研究中W1和W2之间穗上和穗下节间数目相同, 而穗上节间长度差异是引起株高变化的主要原因。通过节间细胞形态观察分析, 推测引起节间长度差异的原因可能是细胞纵向长度的改变。因此, 后期工
作中, 可以结合上述结果或结论有针对性地筛选关键候选基因, 开展功能研究。
4 结论
精细定位了2个控制玉米株高的主效QTL qPH3.2.1和qPH3.2.2。这2个QTL真实可靠, 且贡献率高, 后期将更容易进行精细定位和图位克隆, 也将为玉米株高性状的遗传改良提供重要的基础材料和选择位点。参考文献 原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
[本文引用: 2]
[本文引用: 1]
URL [本文引用: 1]

Four inbred lines derived from Tangsipingtou heterotic group and twenty inbred lines selected from the US hybrids used as materials,this paper aims to study the relationship between the internode number above ear and ear height,plant height,as well as grain yield by means of NC design.The results showed that plant height had significantly positive correlation with plot yield,and was a key factor to grain yield with the largest path coefficient of 2.753 1.The internode number above ear had significantly negative correlation to ear height and the ratio of ear height to plant height.The increase of the ratio of ear height to plant height might have dominant effect on plot yield with a low path coefficient of-2.242 9,providing feasibility of improving plant lodging resistance by decreasing the ear height relying on increasing internode number above ear.
DOI:10.1186/1471-2229-11-4URLPMID:21211047 [本文引用: 3]
pAbstract/p pBackground/p pCollections of nearly isogenic lines where each line carries a delimited portion of a donor source genome into a common recipient genetic background are known as introgression libraries and have already shown to be instrumental for the dissection of quantitative traits. By means of marker-assisted backcrossing, we have produced an introgression library using the extremely early-flowering maize (itZea mays /itL.) variety Gasp Flint and the elite line B73 as donor and recipient genotypes, respectively, and utilized this collection to investigate the genetic basis of flowering time and related traits of adaptive and agronomic importance in maize./p pResults/p pThe collection includes 75 lines with an average Gasp Flint introgression length of 43.1 cM. The collection was evaluated for flowering time, internode length, number of ears, number of nodes (phytomeres), number of nodes above the ear, number and proportion of nodes below the ear and plant height. Five QTLs for flowering time were mapped, all corresponding to major QTLs for number of nodes. Three additional QTLs for number of nodes were mapped. Besides flowering time, the QTLs for number of nodes drove phenotypic variation for plant height and number of nodes below and above the top ear, but not for internode length. A number of apparently Mendelian-inherited phenotypes were also observed./p pConclusions/p pWhile the inheritance of flowering time was dominated by the well-known QTL itVgt1/it, a number of other important flowering time QTLs were identified and, thanks to the type of plant material here utilized, immediately isogenized and made available for fine mapping. At each flowering time QTL, early flowering correlated with fewer vegetative phytomeres, indicating the latter as a key developmental strategy to adapt the maize crop from the original tropical environment to the northern border of the temperate zone (southern Canada), where Gasp Flint was originally cultivated. Because of the trait differences between the two parental genotypes, this collection will serve as a permanent source of nearly isogenic materials for multiple studies of QTL analysis and cloning./p
URL [本文引用: 1]
[Objective] Relevancy of plant traits of different maize varieties in Jiangsu was analyzed,and impact of environmental factors on plant traits explored. [Method]Relevancy test of maize plant traits was conducted in Dafeng,Yancheng City,Jiangsu Province by adopting randomized block test,and sowing density was 67 500 plants / hm2; impact test of environmental factors on maize plant traits was conducted in Shuyang County( Suqian City),Ganyu County( Lianyungang City),Fengxian County( Xuzhou City),sowing density was 67 500 plants / hm2. [Result] Plant traits of different varieties were compared,below-ear internode number,above-ear internode number,aboveground node number,plant height,ear height of different varieties varied greatly; growth season showed extremely significant positive correlation with aboveground node number and below-ear internode number,plant height showed significant positive correlation with total node number,ear height showed significant positive correlation with below-ear internode number. By studying plant traits and environmental factors in the 3 locations,it was found that above-ear internode number was less impacted by environment,accumulated temperature showed extremely significant positive correlation with plant height and aboveground total node number,accumulated temperature and ear height showed significant positive correlation; precipitation showed positive correlation with ear height,above-ear internode,below-ear internode,aboveground total node number,and significant positive correlation with plant height. [Conclusion] The study provides references for the new varieties breeding and high-yield cultivation of maize in Jiangsu.
DOI:10.1002/j.1537-2197.1992.tb14557.xURL [本文引用: 1]
Shoot growth and histogenesis were followed in five unrelated tree taxa possessing inherently diverse patterns of shoot development. Following the resumption of growth in spring, each species differs quantitatively in the number of internodes elongating contemporaneously, in rates and duration of internodal elongation and seasonal periodicity of shoot growth. The basic pattern of internode elongation and histogenesis is qualitatively similar in each of the dicotyledonous species observed irrespective of growth habit or final form of the shoot produced. During the initial phase of internode development, growth is essentially uniform throughout young internodes, corresponding to an active period of cell division during which time pith cells increase in size to about one-third their final length. Subsequently, the pattern of cell division shifts progressively upward concomitant with increased elongation and maturation of pith cells in the basal portion of developing internodes. Thereafter, a wave of cell division accompanied by cell elongation continues to proceed acropetally until growth finally ceases in the distal portion of each internode. As long as internode elongation continues, frequently at distances 15-20 cm below the shoot apex, cell divisions still occur in the distal growing portion. As successive portions of each internode mature acropetally, final length of pith cells becomes relatively uniform throughout the internode. During the process of internode growth and development, cell lengths increase only two- to threefold, whereas cell numbers increase ten- to 30-fold, indicating the dominant role of cell division and increases in cell number to final internode length. Morphological patterns of shoot expression associated with differences in internode lengths along the axis of either preformed or neoformed shoots, as well as sylleptic branches, are due to differences in cell number rather than final cell length. Significant variations in final internode lengths along the axis of episodic shoots, caused by either endogenous or exogenous factors, are also attributed to differences in cell number.
[本文引用: 2]
[本文引用: 2]
[本文引用: 2]
DOI:10.1038/ng.143URLPMID:18454147 [本文引用: 1]
Yield potential,plant height and heading date are three classes of traits that determine the productivity of many crop plants. Here we showed that the quantitative trait locus (QTL) Ghd7,isolated from an elite rice hybrid and encoding a CCT domain protein,had major effects on an array of traits in rice,including number of grains per panicle,plant height and heading date. Enhanced expression of Ghd7 under long-day conditions delays heading and increases plant height and panicle size. Natural mutants with reduced function enable rice to be cultivated in temperate and cooler regions. Thus,Ghd7 has played crucial roles for increasing productivity and adaptability of rice globally.
DOI:10.1007/s11103-013-0129-xURLPMID:24214124 [本文引用: 3]
The d2003 is a natural dwarf mutant from maize inbred line K36 and has less than one-third of K36 plant height with severely shortened internodes. In this study, we reported the cloning of d2003 gene using positional cloning. The results showed that there was a single-base insertion in the coding region of Viviparous8 (VP8) in d2003 mutant, which resulted in a premature stop codon. Further genetic allelism tests confirmed that d2003 mutation is a novel allele of VP8. VP8 is mainly expressed in the stem apex, young leaves, and developing vascular tissues, and its expression levels in nodes are significantly higher than that in internodes at 12-leaf stage. Subcellular localization demonstrated that the VP8 protein is localized to the endoplasmic reticulum and the N-terminal 26 amino acids (aa) of VP8 protein are essential to its localization in ER. Further transgenic experiments showed that lack of the 26 aa leads to loss of VP8 function in Arabidopsis amp1 phenotype rescue. These results strongly suggested that the N-terminal 26 aa is critical for VP8 protein localization, and the correct protein localization of VP8 in ER is necessary for its function.
[本文引用: 2]
[本文引用: 2]
DOI:10.2135/cropsci1995.0011183X003500060004xURL [本文引用: 2]
The inheritance of quantitative traits is not well understood. A study was conducted to determine the number and chromosomal locations of quantitative trait loci (QTL) controlling male anthesis date ; plant and ear height ; kernel weight ; and kernel protein, oil, and starch concentration in maize (Zea mays L.). Two hundred S
DOI:10.1007/s11032-005-5679-4URL
The vast majority of reported QTL mapping for maize ( Zea mays L.) traits are from temperate germplasm and, also, QTL by environment interaction (QTL E) has not been thoroughly evaluated and analyzed in most of these papers. The maize growing areas in tropical regions are more prone to environmental variability than in temperate areas, and, therefore, genotype by environment interaction is of great concern for maize breeders. The objectives of this study were to map QTL and to test their interaction with environments for several traits in a tropical maize population. Two-hundred and fifty-six F 2:3 families evaluated in five environments, a genetic map with 139 microsatellites markers, and the multiple-environment joint analysis (mCIM) were used to map QTL and to test QTL E interaction. Sixteen, eight, six, six, nine, and two QTL were mapped for grain yield, ears per plant, plant lodging, plant height, ear height, and number of leaves, respectively. Most of these QTL interacted significantly with environments, most of them displayed overdominance for all traits, and genetic correlated traits had a low number of QTL mapped in the same genomic regions. Few of the QTL mapped had already been reported in both temperate and tropical germplasm. The low number of stable QTL across environments imposes additional challenges to design marker-assisted selection in tropical areas, unless the breeding programs could be directed towards specific target areas.
[本文引用: 1]
[本文引用: 1]
DOI:10.1111/tpj.12038URLPMID:23020630 [本文引用: 1]
Maize plant height is closely associated with biomass, lodging resistance and grain yield. Determining the genetic basis of plant height by characterizing and cloning plant height genes will guide the genetic improvement of crops. In this study, a quantitative trait locus (QTL) for plant height, qPH3.1, was identified on chromosome3 using populations derived from a cross between Zong3 and its chromosome segment substitution line, SL15. The plant height of the two lines was obviously different, and application of exogenous gibberellinA3 removed this difference. QTL mapping placed qPH3.1 within a 4.0cM interval, explaining 32.3% of the phenotypic variance. Furthermore, eight homozygous segmental isolines (SILs) developed from two larger F2 populations further narrowed down qPH3.1 to within a 12.6kb interval. ZmGA3ox2, an ortholog of OsGA3ox2, which encodes a GA3 beta-hydroxylase, was positionally cloned. Association mapping identified two polymorphisms in ZmGA3ox2 that were significantly associated with plant height across two experiments. Quantitative RT-PCR showed that SL15 had higher ZmGA3ox2 expression relative to Zong3. The resultant higher GA1 accumulation led to longer internodes in SL15 because of increased cell lengths. Moreover, a large deletion in the coding region of ZmGA3ox2 is responsible for the dwarf mutant d1-6016. The successfully isolated qPH3.1 enriches our knowledge on the genetic basis of plant height in maize, and provides an opportunity for improvement of plant architecture in maize breeding.
DOI:10.1105/tpc.7.1.75URLPMID:7696880 [本文引用: 1]
Abstract The Anther ear1 (An1) gene product is involved in the synthesis of ent-kaurene, the first tetracyclic intermediate in the gibberellin (GA) biosynthetic pathway. Mutations causing the loss of An1 function result in a GA-responsive phenotype that includes reduced plant height, delayed maturity, and development of perfect flowers on normally pistillate ears. The an1::Mu2-891339 allele was recovered from a Mutator (Mu) F2 family. Using Mu elements as molecular probes, an An1-containing restriction fragment was identified and cloned. The identity of the cloned gene as An1 was confirmed by using a reverse genetics screen for maize families that contain a Mu element inserted into the cloned gene and then by demonstrating that the insertion causes an an1 phenotype. The predicted amino acid sequence of the An1 cDNA shares homology with plant cyclases and contains a basic N-terminal sequence that may target the An1 gene product to the chloroplast. The sequence is consistent with the predicted subcellular localization of AN1 in the chloroplast and with its biochemical role in the cyclization of geranylgeranyl pyrophosphate, a 20-carbon isoprenoid, to ent-kaurene. The semidwarfed stature of an1 mutants is in contrast with the more severely dwarfed stature of GA-responsive mutants at other loci in maize and may be caused by redundancy in this step of the GA biosynthetic pathway. DNA gel blot analysis indicated that An1 is a single-copy gene that lies entirely within the deletion of the an1-bz2-6923 mutant. However, homozygous deletion mutants accumulated ent-kaurene to 20% of the wild-type level, suggesting that the function of An1 is supplemented by an additional activity.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
DOI:10.1073/pnas.42.4.185URLPMID:16589846 [本文引用: 1]
Not Available
[本文引用: 1]
DOI:10.1093/pcp/pcq153URLPMID:20937610 [本文引用: 1]
DELLA proteins are nuclear-localized negative regulators of gibberellin signaling found ubiquitously throughout higher plants. Dominant dwarfing mutations of DELLA proteins have been primarily responsible for the dramatic increases in harvest index of the 'green revolution'. Maize contains two genetic loci encoding DELLA proteins, dwarf plant8 (d8) and dwarf plant 9 (d9). The d8 gene and three of its dominant dwarfing alleles have been previously characterized at the molecular level. Almost 20 years after the initial description of the mutant, this investigation represents the first molecular characterization of d9 and its gibberellin-insensitive mutant, D9-1. We have molecularly, subcellularly and phenotypically characterized the gene products of five maize DELLA alleles in transgenic Arabidopsis. In dissecting the molecular differences in D9-1, a critical residue for normal DELLA function has been uncovered, corresponding to E600 of the D9 protein. The gibberellin-insensitive D9-1 was found to produce dwarfing and, notably, earlier flowering in Arabidopsis. Conversely, overexpression of the D9-1 allele delayed flowering in transgenic maize, while overexpression of the d9 allele led to earlier flowering. These results corroborate findings that DELLA proteins are at the crux of many plant developmental pathways and suggest differing mechanisms of flowering time control by DELLAs in maize and Arabidopsis.
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]
[本文引用: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}