
* 通讯作者(Corresponding author): 张洪博, E-mail:hbzhang@swu.edu.cn 收稿日期:2014-09-16 基金:
摘要
关键词:酵母表面展示; Smart cDNA文库; DNA结合蛋白; 酵母单杂交;
Screening of Promoter-Binding Factors of Tobacco
CHEN Hong, NIU Hai-Xia, WANG Wen-Jing, MA Hao-Ran, LI Jia-Na, CHAI You-Rong, ZHANG Hong-Bo
Abstract
Keyword:Yeast surface display; Smart cDNA Library; DNA-binding protein; Yeast one-hybrid;
Show Figures
Show Figures


引言在DNA结合蛋白的筛选研究中, 酵母单杂交系统是一个常用工具, 但该系统在应用中易受酵母内源干扰而影响DNA结合蛋白的筛选效率[ 1, 2]。酵母表面展示系统(yeast surface display system)将外源基因与酵母表面展示载体的酵母细胞壁甘露糖蛋白基因融合, 从而使导入酵母的外源基因蛋白分子在信号肽引导下分泌到酵母细胞表面[ 3, 4, 5], 可通过接近体外实验的方法研究蛋白的分子间相互作用[ 6, 7, 8, 9], 有利于克服酵母内源干扰。同用于蛋白表面展示研究的大肠杆菌和噬菌体相比, 酵母具有完整的蛋白修饰系统, 更适合真核生物的蛋白功能研究[ 10, 11, 12, 13]。虽然已有不少应用酵母表面展示技术筛选蛋白互作分子的研究[ 5, 9, 14], 但利用该系统筛选DNA结合蛋白的研究还很有限。基于pYD1表达载体和EBY100菌株的酵母表面展示系统是一个常用体系。该系统在应用研究中, 需先构建质粒文库, 再将质粒文库导入酵母获得表面展示文库, 这一过程费时费力, 对酵母表面展示文库的构建效率影响较大。Clontech公司的Smart cDNA文库构建系统是一个基于酵母的简易高效率的文库构建系统。将pYD1载体改造成一个可与Clontech公司的Smart cDNA文库构建系统相匹配的酵母表达载体, 可极大地简化酵母表面展示文库的构建工作。
烟草腐胺甲基转移酶基因 PMT(putrescine N-methyltransferase)是尼古丁合成的关键基因[ 15, 16], 在烟草中有5个成员( PMT1a、 PMT1b、 PMT2、 PMT3和 PMT4)[ 17], 控制着尼古丁分子的吡咯烷环合成代谢[ 15, 16]。茉莉素是调控尼古丁合成代谢的重要植物激素, 包括 PMT在内的大部分尼古丁合成关键基因都受茉莉素诱导表达[ 18, 19, 20, 21, 22]。删除突变实验证明 PMT1a启动子中含有一段由G-box、AT-rich和GCC- box-like元件构成的茉莉素应答必要片段[ 22, 23], 该DNA片段也存在于其他 PMT基因启动子中, 在后来的研究中被称为GAG片段[ 23, 24]。虽然近期的研究已鉴定了若干尼古丁合成调控因子, 如ERF转录因子家族成员ORC1和JAP1[ 25], bHLH转录因子NbbHLH1/2[ 26]和MYC2a/b等[ 27], 但是, 茉莉素途径调控 PMT基因表达的分子机制仍不清楚, 还无法从调控机制上解释GAG片段的3个元件在茉莉素信号应答中的必要性及其相互关系。因此, 进一步筛选 PMT基因启动子GAG片段的DNA结合蛋白对揭示尼古丁合成代谢调控机理有重要意义。然而, 我们近期利用酵母单杂交系统对GAG片段结合蛋白进行的多次筛选, 结果均不理想, 而且已报道的GAG片段结合蛋白大多无法在酵母单杂交系统中激活GAG片段操纵的报告基因表达[ 23, 24], 推测是由于酵母内源因子干扰或GAG片段的结构特殊性。在以酵母筛选 PMT基因启动子GAG片段结合蛋白的研究中, 对酵母内源干扰的克服是影响筛选效率的一个重要因素。
本研究对基于pYD1表达载体和EBY100菌株的酵母表面展示系统的pYD1载体进行了改造, 使其可与Clontech公司的Smart cDNA文库构建系统相匹配, 简化了酵母表面展示文库的构建工作; 并以烟草 PMT基因启动子GAG片段的结合蛋白MYC2a[ 24]作为阳性对照, 以GAG片段作为DNA探针, 建立了利用酵母表面展示系统筛选DNA结合蛋白的试验体系。然后, 利用改进的酵母表面展示系统对GAG片段的结合蛋白进行了筛选, 并对筛选到的DNA结合蛋白与GAG片段的结合特性进行了分析。
1 材料与方法1.1 研究材料大肠杆菌( Escherichia coli)菌株DH5α由本实验室提供。酵母( Saccharomyces cerevisiae)菌株EBY100、pYD1载体、链亲和素磁珠及TRIzol试剂购自Invitrogen公司。pGADT7-Rec2载体及酵母培养所需试剂购自Clontech公司。其他生化试剂购自生工生物工程公司。
1.2 pYD1-Rec载体构建将pYD1和pGADT7-Rec2质粒分别用 EcoR I和 Xho I双酶切, 电泳分离酶切产物后, 回收pYD1载体骨架和pGADT7-Rec2载体酶切产物的114 bp长度片段, 经连接反应后转化大肠杆菌DH5α, 筛选阳性克隆并测序鉴定, 所获得的重组质粒命名为pYD1-Rec。
1.3 pYD1-MYC2a阳性对照载体的构建依据前期研究报道的方法克隆烟草 MYC2a基因[ 24], 然后用特异引物5'-AAAGGTACCGGGTC AGTCCCATTTTGGG-3'和5'-GCGCCTTGAGTCTTT ACTAAC-3'扩增 MYC2a基因的DNA结合域片段, 经 Kpn I酶切后, 克隆至由 Kpn I和 Eco I CRI双酶切的pYD1-Rec载体, 获得pYD1-MYC2a阳性对照载体。
1.4 酵母转化在YPD固体培养基上画线活化酵母菌株EBY100, 并按照Clontech公司的酵母培养手册(Yeast Protocols Handbook)提供的方法挑取酵母菌落制备热激转化感受态。在进行常规酵母质粒转化时, 将0.3 μg质粒与30 μg鱼精DNA加入50 µL酵母感受态中, 混匀后加入6 µL DMSO和300 µL含40% PEG-4000的LiOAc/TE溶液(按Clontech公司酵母培养手册方法配制), 轻轻混匀, 于30℃温箱孵育30 min, 42℃水浴热激15 min, 离心收集酵母菌体, 涂布在营养缺陷型培养基SD/-Ura/-Trp平板上, 于30℃培养。酵母文库按照Clontech公司BD Matchmaker Library Construction & Screening Kits操作手册提供的方法制备。
1.5 DNA探针制备依据前期报道的方法[ 24]克隆烟草 PMT1a基因启动子的GAG片段, 然后用生物素标记的正向引物5'-Biotin-ACTAGTCTAACCCTGCACG-3'和反向引物5'-TCTAGAAGTGGAGGGCGC-3'进行PCR扩增, 获得生物素标记的DNA探针, 通过电泳分离和切胶回收纯化DNA探针, 将探针终浓度稀释至250~300 ng µL-1。
1.6 酵母表面展示系统筛选DNA结合蛋白的试验体系将分别导入pYD1-Rec (阴性对照)和pYD1-MYC2a (阳性对照)载体的EBY100细胞在葡萄糖为碳源的SD/-Ura/-Trp培养基平板上画线培养后, 接种至相同碳源的SD/-Ura/-Trp液体培养基, 30℃过夜振荡培养。然后, 离心收集培养的酵母细胞, 重悬于半乳糖为碳源的SD/-Ura/-Trp液体培养基, 并稀释至OD600=0.5, 于20℃继续振荡培养24 h诱导表面展示蛋白表达。同时, 用葡萄糖为碳源的SD/-Ura液体培养基培养EBY100细胞用于封闭试验体系的非特异结合。
配制新鲜DNA结合酵母分离缓冲液1×buffer [25 mmol L-1 HEPES-KOH, pH 7.4, 30 mmol L-1 KCl, 5 mmol L-1 MgCl2, 1 mmol L-1 EDTA, 0.2 mg mL-1 poly(dI-dC)和0.1% BSA]。离心收集完成了表面展示蛋白诱导表达的酵母细胞, 并依据其OD600吸收值, 用1×buffer稀释至每毫升含1×107个细胞备用。同时, 离心收集用作封闭菌液的EBY100细胞, 以1×buffer稀释至每毫升含1×107菌液备用。
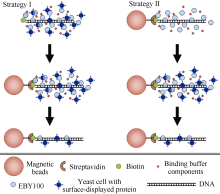
DNA结合酵母筛选方案如 图1所示。
图1
Fig. 1
| Figure OptionViewDownloadNew Window | |
 | 图1 酵母表面展示技术分离DNA结合蛋白实验方案示意图Fig. 1 Schematic diagram of DNA-binding protein isolation using yeast surface display |
方案一: (1)将带链亲和素的磁珠10 µL (约100 µg磁珠)加入1 mL 1×buffer, 混匀, 置磁力架上, 待磁珠被吸附后去上清液, 如此重复3次平衡磁珠; (2)分别将上述制备的阴性对照和阳性对照菌液1 µL, 加入1 mL封闭菌液(即以1∶1000的比例将对照菌液用EBY100封闭细胞稀释), 混匀后加入生物素标记的DNA探针(1.5 µg, 约20 pmol), 于25℃自旋式混匀仪上, 以20转 min-1孵育30 min, 使生物素标记的DNA探针与酵母细胞结合; (3)加入20 µL平衡处理的链亲和素磁珠, 继续在25℃自旋式混匀仪上, 以20转 min-1孵育30 min, 完成磁珠与DNA探针复合物的结合; (4)在磁力架的帮助下, 移除未结合菌液, 并用1×buffer洗涤3次, 将结合了酵母细胞的磁珠悬于100 µL 1×TE溶液, 分别涂布于YPD及SD/-Ura/-Trp培养基平板, 于30℃恒温箱培养2 d, 观察统计菌落数。
方案二: (1)用方案一之(1)的方法平衡磁珠; (2)将平衡过的磁珠和1.5 µg (约20 pmol)生物素标记的DNA探针加入1 mL 1×buffer (DNA探针的终浓度为20 pmol 100 µg-1磁珠), 混匀后于25℃自旋式混匀仪上以20转 min-1孵育30 min, 然后在磁力架的帮助下除去未结合的DNA探针, 用1×buffer洗涤3次后重悬于100 µL 1×buffer备用; (3)在1 mL封闭菌液中, 加入20 µL结合了DNA探针的磁珠, 于25℃自旋式混匀仪上以20转 min-1孵育30 min, 在磁力架的帮助下移除封闭菌液, 用1×buffer洗涤3次备用; (4)在封闭过的磁珠探针复合体中分别加入上述制备的阴性对照和阳性对照菌液1 µL, 加入1 mL封闭菌液, 混匀后于25℃自旋式混匀仪上以20转 min-1孵育30 min, 然后, 在磁力架的帮助下移除未结合的菌液, 用1×buffer洗涤3次, 将结合了酵母细胞的磁珠悬于100 µL 1×TE溶液, 分别涂布于YPD和SD/-Ura/-Trp培养基平板, 于30℃恒温箱培养2 d, 观察统计菌落数。
1.7 烟草cDNA酵母表面展示文库构建以TRIzol试剂提取茉莉素处理6 h的烟草根总RNA, 将总RNA在无RNase活性的DNaseI作用下, 去除DNA污染, 进行纯化, 获得构建cDNA文库所需RNA。然后, 按照Clontech公司BD Matchmaker Library Construction & Screening Kits的操作手册进行烟草根RNA的cDNA合成、扩增和纯化。
用 Sma I限制性内切酶对pYD1-Rec载体酶切后, 电泳切胶回收载体DNA, 使载体DNA浓度达到1 µg µL-1; 将纯化后的双链cDNA与pYD1-Rec载体, 按照Clontech公司BD Matchmaker Library Construction & Screening Kits操作手册提供的酵母转化方案共转化酵母EBY100, 涂布于SD/-Ura/-Trp培养基平板上, 于30℃培养24~48 h, 待酵母菌落生长至直径约0.5~1.0 mm时, 将酵母菌落通过刮板的方式悬浮于葡萄糖为碳源的SD/-Ura/-Trp液体培养基, 并于30℃振荡培养5 h, 获得烟草cDNA的酵母表面展示文库。然后, 离心收集酵母文库, 并重悬于半乳糖为碳源的SD/-Ura/-Trp液体培养基, 诱导表达酵母表面展示蛋白。
1.8 酵母表面展示文库筛选根据1.6中的筛选方案二, 以生物素标记的GAG片段作DNA探针, 对完成表面展示蛋白诱导表达的1×107酵母表面展示文库细胞进行筛选, 将筛选到的酵母单菌落分别培养后, 提取酵母质粒转化大肠杆菌DH5α, 然后从阳性菌株分离质粒并测序。
1.9 蛋白与DNA探针的体外结合分析按照前期报道的方法[ 24]进行阳性对照MYC2a的DNA结合域蛋白表达载体构建、蛋白表达及纯化, 其PCR扩增引物为5°-AAAACCATGGAGAATAAG AAGAAGAAAAGGTCAC-3°和5°-AAAACTCGAG GCGTGTTTCAGCAACTCT-3°。筛选出的2个ERF转录因子ERF71和ERF72, 分别用特异引物扩增(ERF71: 5°-AAACCATGGAAGATCATCAACAAAG-3°和5°-AAACTCGAGAAAATCCCAAATAGATTCATG ATG-3°; ERF72: 5°-ATGGACTCCAACGAAGATC-3°和5°-AAACTCGAGAAAATACTCAAATTCAATAAC CTTCTCTTC-3°; 下画线标记序列为引入的酶切位点), 并通过 Nco I和 Xho I酶切位点构建至pET28b原核蛋白表达载体(Novagen公司); 克隆ERF72时, pET28b载体的 Nco I切点酶切后需要补平。构建完成的原核表达载体被导入大肠杆菌菌株BL21(DE3), 在含1 mmol L-1 IPTG的LB培养基中于37℃培养3 h诱导蛋白表达。按Invitrogen公司Ni+亲和柱的说明书纯化带组氨酸标签的重组蛋白。
在Gel-shift (凝胶迁移率滞阻试验)试验中, 将4 μg纯化蛋白与5 ng生物素标记的GAG片段DNA探针在30 μL结合缓冲液[25 mmol L-1 Hepes-KOH, pH 7.4, 50 mmol L-1 KCl, 0.1 mmol L-1 EDTA, 5%甘油, 0.5 mmol L-1 DTT和200 ng poly(dI-dC)]中混匀, 于室温孵育15 min, 用0.5×TBE缓冲液配制的4% (w/v)聚丙烯酰胺凝胶分离, 然后通过半干电转移方法将分离的反应物转移至尼龙膜上, 并用紫外交联后, 以Roche公司的生物素检测试剂盒检测试验结果。
1.10 蛋白与DNA探针在酵母中的结合分析通过酵母单杂交试验检验筛选出的DNA结合蛋白与GAG片段在酵母中的相互作用, 所用试验系统为Clontech公司的MATCHMAKER酵母单杂交系统, 所用激活域表达载体为pGADT7-Rec2。GAG片段的4次重复序列被分别克隆至pHISi-1和pLacZi载体的报告基因启动子中, 随后这2个载体被共同导入酵母菌株YM4271, 获得酵母单杂交所需的报告菌株。MYC2a、ERF71和ERF72的DNA结合域片段用1.9的克隆方法导入酵母单杂交激活域表达载体pGADT7-Rec2, 然后分别转化酵母报告菌株, 在营养缺陷型培养基SD/-Leu/-Ura/-His上于30℃培养, 在用于检测蛋白与DNA互作的培养基中加入15 mmol L-1 3-AT (3-氨基-1, 2, 4-三唑)。酵母培养所需营养缺陷型培养基的配制及蛋白与GAG片段在酵母中的相互作用分析按照Clontech公司Match-Maker酵母单杂交系统提供的方法操作。
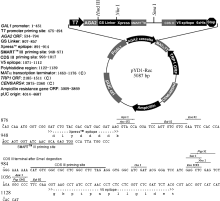
2 结果与分析2.1 酵母表面展示系统的改造通过 EcoR I和 Xho I双酶切将酵母单杂交系统载体pGADT7-Rec2中的Smart重组片段转移至酵母表面展示载体pYD1, 获得了重组载体pYD1-Rec, 其结构如 图2所示。pYD1-Rec载体具有Clontech公司Smart文库构建系统必须的Smart III和CDS III引物序列, 并有线性化pYD1-Rec载体的 Sma I酶切位点, 可与Smart文库构建试剂盒匹配用于酵母表面展示文库构建, 简化了酵母表面展示文库的构建过程。
图2
Fig. 2
| Figure OptionViewDownloadNew Window | |
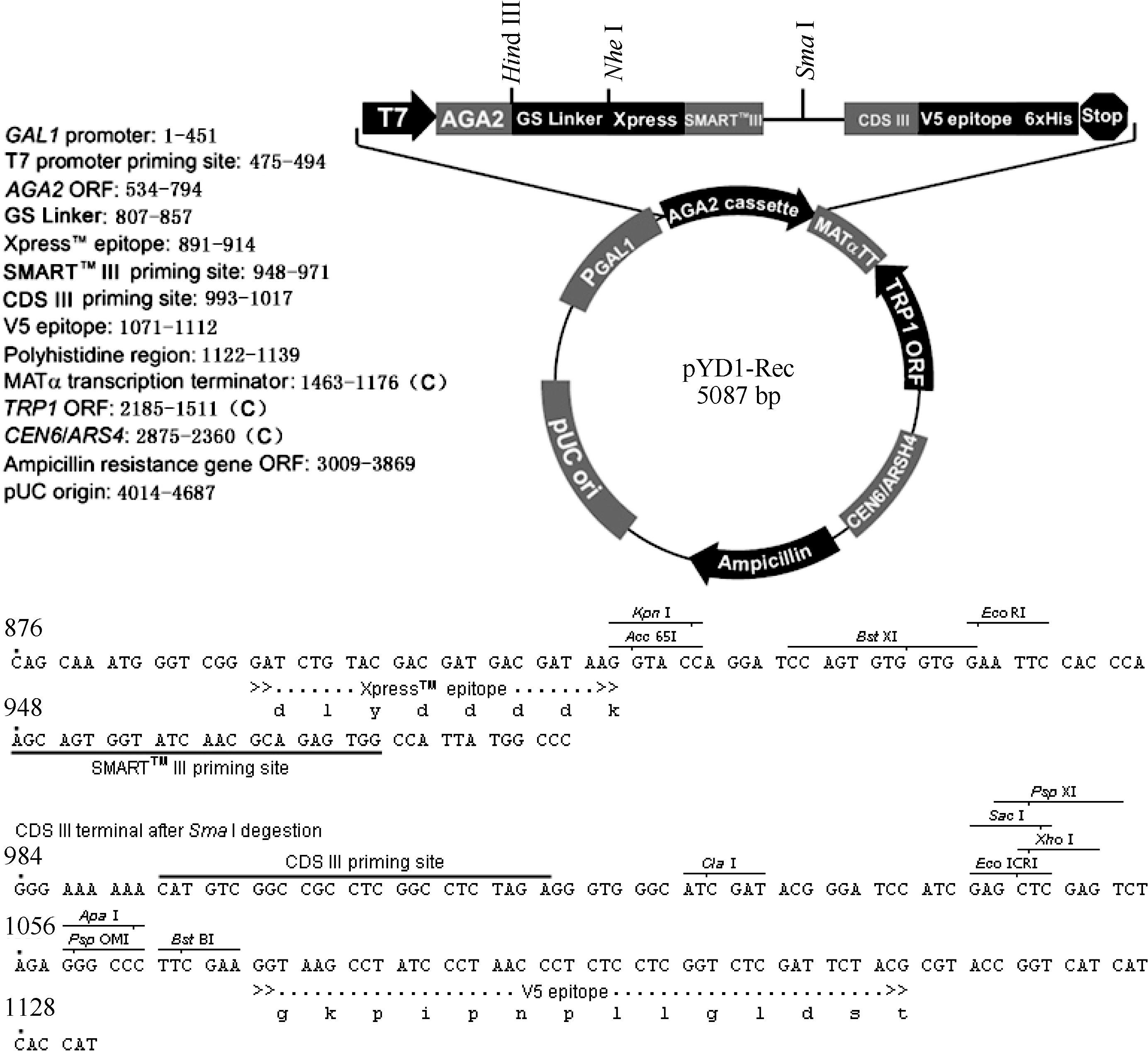 | 图2 pYD1-Rec 载体结构Fig. 2 Structure of pYD1-Rec vector |
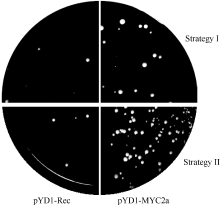
2.2 以酵母表面展示系统筛选DNA结合蛋白试验体系的建立采用2种试验方案进行了比较研究。如1.6所述, 方案一先让DNA探针和酵母细胞结合, 然后用链亲和素磁珠分离结合了DNA探针的酵母细胞; 方案二则用预先结合了DNA探针的磁珠分离可结合DNA探针的酵母细胞。 表1所示为按照1.6所述试验方案通过3次重复试验得到的筛选效率对比结果。 图3所示, 为筛选结果的酵母生长情况。从对阳性对照的筛选结果中可以看出, 用方案二的方法筛选DNA探针结合酵母的效率较高, 更有利于分离含有DNA探针结合蛋白的酵母细胞。同时, 对阴性对照的筛选结果显示2种实验方案都会有极少数非特异结合酵母得到分离。
表1
Table 1
表1(Table 1)
| 表1 DNA结合蛋白筛选对照试验结果 Table 1 Result of DNA-binding protein isolation | ||||||||||||||||||||||
图3
Fig. 3
| Figure OptionViewDownloadNew Window | |
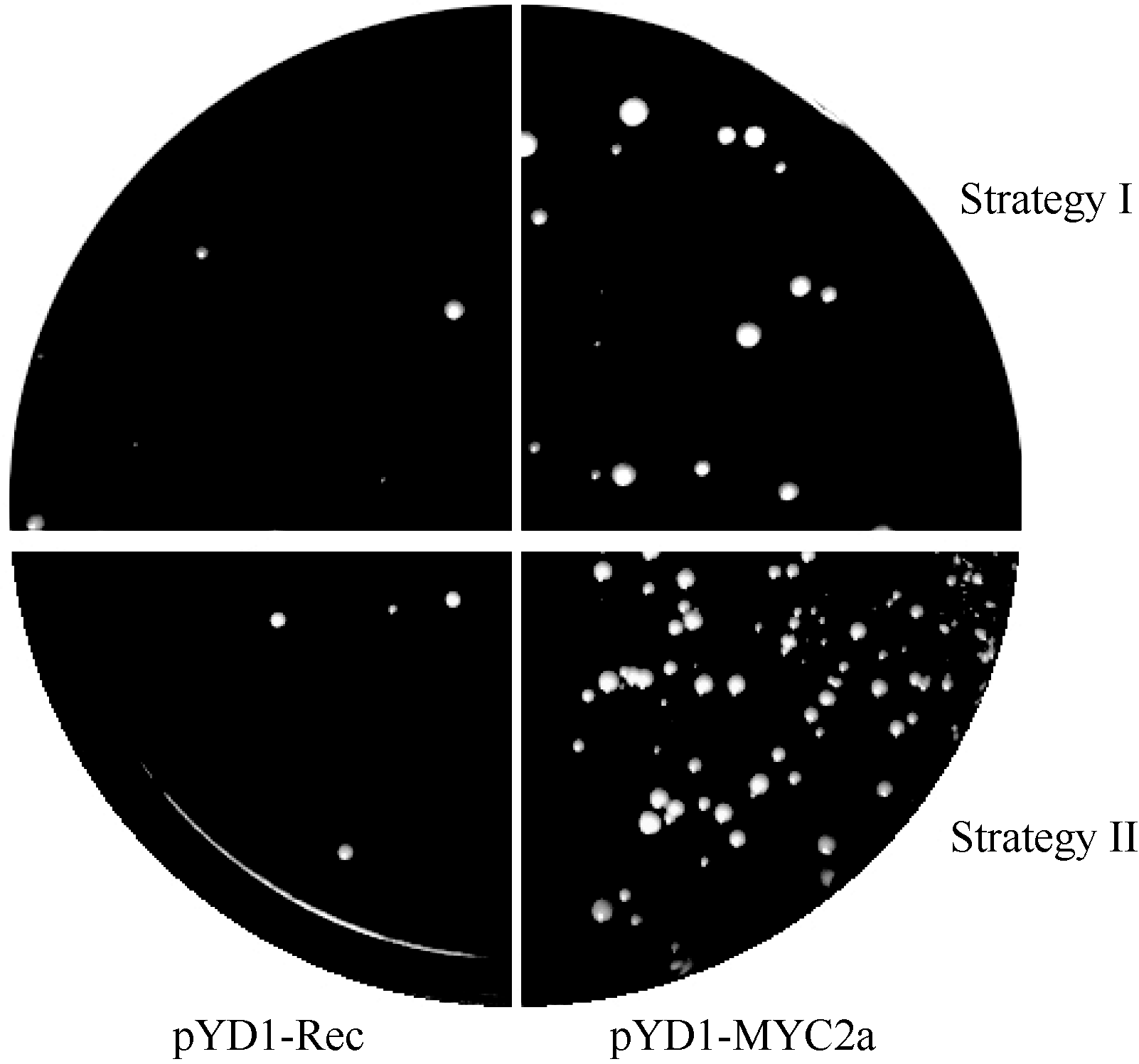 | 图3 酵母生长情况Fig. 3 Growth of yeast cells |
上述结果说明本研究设计的试验方案在利用酵母表面展示系统筛选DNA结合蛋白的研究中具有可行性。由于应用方案二筛选DNA探针结合酵母细胞的效率较高, 因此, 在利用酵母表面展示系统筛选烟草 PMT基因启动子GAG片段结合蛋白的研究中采用了方案二的试验体系。
2.3 PMT基因启动子GAG片段结合蛋白的酵母表面展示系统筛选 利用启动子元件分析比较详细的烟草尼古丁合成基因 PMT启动子的茉莉素应答核心元件GAG片段作为DNA探针, 对烟草cDNA酵母表面展示文库进行了筛选。由于烟草尼古丁在根部合成, 且 PMT基因受茉莉素诱导表达[ 15, 16, 18, 19, 20, 22, 27], 因此, cDNA酵母表面展示文库的构建选用从茉莉素处理的烟草根中提取的总RNA以增加 PMT基因调控因子在酵母表面展示文库中的比例。按照方案二的试验体系对烟草根cDNA酵母表面展示文库进行了2次筛选, 每次筛选约1×107酵母细胞。为更好地去除非特异结合细胞, 在筛选中用1×buffer洗涤了4次。通过2次筛选共获得约180个阳性克隆, 对阳性克隆载体中插入的cDNA片段长度分析后对其中80个克隆进行了测序分析, 其中有bHLH转录因子基因1个, ERF转录因子基因2个, 这2类转录因子可能与GAG片段中的G-box和GCC-box-like元件互作; 在其余克隆中还有1个MYB转录因子基因及一些非转录因子基因。
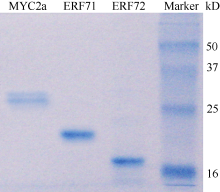
2.4 筛选出的ERF转录因子的原核蛋白表达及纯化为验证筛选出的DNA结合蛋白与所用DNA探针的特异结合, 通过蛋白与DNA的体外结合试验检测了筛选的ERF转录因子与GAG片段的相互作用。将筛选到的ERF转录因子的DNA结合域分别克隆到pET28b载体中与载体的6×His标签融合, 并导入大肠杆菌BL21中, 进行原核蛋白诱导表达。同时, 将前期报道的GAG片段结合蛋白MYC2a[ 24]的DNA结合域按照文献所述方法进行原核蛋白诱导表达, 作为检测转录因子蛋白与GAG片段互作的阳性对照。经过IPTG诱导原核蛋白表达后, 利用Ni2+离子琼脂糖树脂对含有上述蛋白表达载体的大肠杆菌BL21细胞裂解物进行纯化, 获得相应纯化蛋白。 图4所示为克隆到的2个ERF转录因子的序列结构。 图5所示为纯化的ERF转录因子及MYC2a转录因子蛋白。
图4
Fig. 4
| Figure OptionViewDownloadNew Window | |
 | 图4 ERF转录因子的序列结构 灰色背景部分为ERF结构域。Sequences in gray block are the ERF domains.Fig. 4 Sequences of ERF transcription factors |
图5
Fig. 5
| Figure OptionViewDownloadNew Window | |
 | 图5 ERF转录因子的蛋白表达纯化Marker为蛋白分子量标准。 Marker indicates protein molecular weight marker.Fig. 5 Purified proteins for ERF transcription factors |
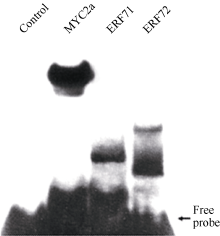
2.5 筛选出的ERF转录因子与 PMT基因启动子GAG探针的体外结合 Gel-shift (凝胶迁移率滞阻实验)是体外检测蛋白与DNA互作的主要方法。在获得筛选到的ERF转录因子及阳性对照MYC2a转录因子DNA结合域的原核表达蛋白后, 利用生物素标记的GAG片段作为DNA探针, 通过Gel-shift试验检测这些蛋白与GAG片段的相互作用。如 图6所示, 无蛋白的泳道没有滞阻条带, 有MYC2a转录因子DNA结合域蛋白的阳性对照泳道及有ERF转录因子DNA结合域蛋白的泳道都有滞阻条带。这些结果说明筛选到的ERF转录因子可以与GAG片段互作, 也表明本研究建立的酵母表面展示系统试验体系可以成功应用于DNA结合蛋白的筛选。
图6
Fig. 6
| Figure OptionViewDownloadNew Window | |
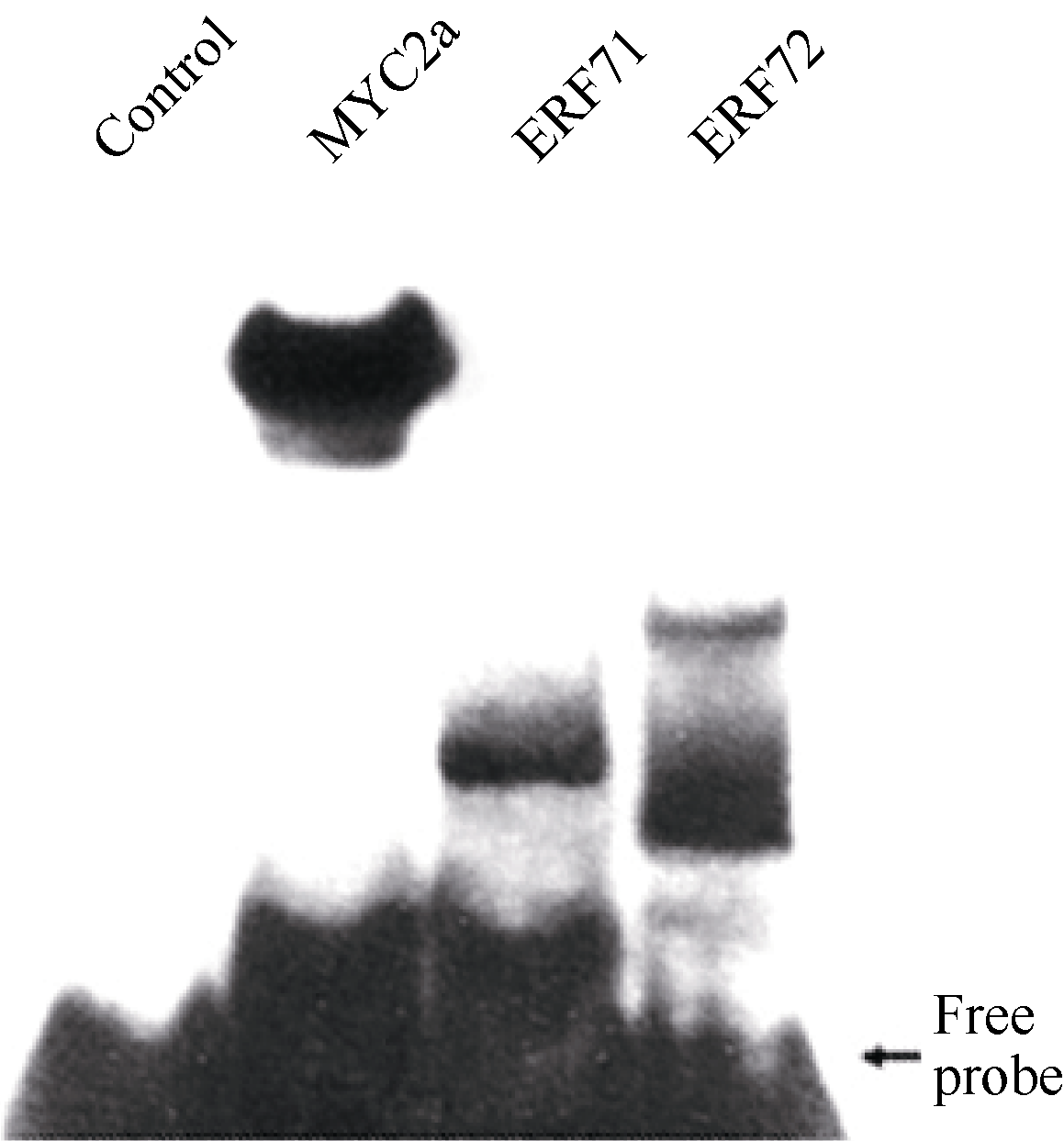 | 图6 ERF转录因子与GAG片段的体外互作分析Control: 未加蛋白阴性对照; MYC2a: 阳性对照。Fig. 6 Interaction in vitro between ERF transcription factors and GAG fragmentControl: negative control without protein; MYC2a: positive control. |
2.6 筛选出的ERF转录因子与 PMT基因启动子GAG片段在酵母中的互作分析 将MYC2a、ERF71和ERF72分别导入GAG片段的酵母单杂交报告菌株, 检测其与GAG片段的相互作用。 图7所示, 分别导入MYC2a、ERF71和ERF72的酵母均无法在含15 mmol L-1 3-AT的SD/- Leu/-Ura/-His/3-AT营养缺陷型培养基上生长( 图7-B), 而且在无3-AT的SD/-Leu/-Ura/-His营养缺陷型培养基上生长的菌落也无法激活β-半乳糖苷酶报告基因的表达( 图7-C)。表明ERF71和ERF72同MYC2a一样虽然可在体外试验中与GAG片段结合, 但都不能在酵母单杂交试验中与GAG片段互作。
图7
Fig. 7
| Figure OptionViewDownloadNew Window | |
 | 图7 ERF转录因子与GAG片段在酵母中的互作分析A: 酵母在SD/-Leu/-Ura/-His培养基上的生长; B: 酵母在SD/-Leu/-Ura/-His/3-AT培养基上的生长; C: β-半乳糖苷酶活性分析; D: 平板上的酵母菌株分布图; MYC2a为GAG结合蛋白对照。Fig. 7 Interaction in vivo between ERF transcription factors and GAG fragmentA: growth of yeast on SD/-Leu/-Ura/-His plate; B: growth of yeast on SD/-Leu/-Ura/-His/3-AT plate; C: β-galactopyranoside activity assay; D: labeling of yeast strains; MYC2a is used as a control that binds GAG fragment. |
3 讨论酵母表面展示系统将外源蛋白展示在细胞表面, 在克服酵母内源因子干扰方面具有独特优势。将酵母表面展示系统应用于DNA结合蛋白筛选, 可以克服酵母单杂交系统的不足, 提高对易受酵母内源干扰DNA结合蛋白的筛选效率。然而, 酵母表面展示系统的应用目前还主要局限于蛋白间相互作用研究及蛋白结合小分子的筛选[ 5, 9, 14], 并未在DNA结合蛋白筛选研究中得到广泛应用, 这在一定程度上受到现有酵母表面展示系统的文库构建困难及筛选试验体系不成熟的影响。本研究对酵母表面展示系统常用载体pYD1进行了改造, 如 图2所示, 改造后获得的pYD1-Rec载体具有Clontech公司的Smart cDNA文库构建系统所需的全部载体元件, 可以与之相匹配, 完成cDNA酵母表面展示文库的构建, 从而简化了酵母表面展示文库的构建步骤, 降低了其构建难度, 并提高了构建效率。同时, 本研究利用结构分析比较清楚的烟草 PMT基因启动子茉莉素应答核心元件GAG片段作为DNA探针, 以前期证明的GAG片段结合蛋白MYC2a作为阳性对照, 通过对2种试验方案的DNA结合蛋白分离效率比较, 建立了一个比较高效的利用酵母表面展示系统分离DNA结合蛋白的试验体系。在该试验体系中, 先将带链亲和素的磁珠与生物素标记DNA探针结合, 并以EBY100酵母菌液封闭磁珠和DNA探针复合物的非特异结合位点, 然后, 用该复合物从表达表面展示蛋白的酵母细胞中分离结合DNA探针的酵母细胞。与先让DNA探针和酵母细胞结合, 然后用链亲和素磁珠分离结合DNA探针的酵母细胞的试验方案相比, 该方案的优点可能在于DNA与磁珠的预先结合降低了试验体系对DNA探针和磁珠之间相互作用的干扰, 从而提高了磁珠对结合酵母DNA探针的分离效率。上述两方面的研究工作为酵母表面展示系统在DNA结合蛋白筛选研究中的应用奠定了技术基础。
在利用改进的酵母表面展示系统进行的DNA结合蛋白筛选应用研究中, 我们对烟草 PMT基因启动子GAG片段的DNA结合蛋白进行了筛选。 PMT基因启动子GAG片段的结构分析比较清楚, 但是其DNA结合蛋白的酵母单杂交筛选效率极低, 即使一些已经证明的GAG片段结合蛋白也无法在酵母单杂交体系中激活报告基因表达( 图6)[ 23, 24]。一个可以克服酵母内源干扰的DNA结合蛋白筛选系统, 对 PMT基因启动子结合蛋白的分离及分子调控机制研究具有重要推动作用。本研究以GAG片段为DNA探针对烟草根cDNA酵母表面展示文库进行筛选, 并获得若干转录因子蛋白, 其中包括2个可能结合GAG片段GCC-like-box的ERF转录因子。通过Gel-shift试验证明这2个ERF转录因子可与GAG片段互作。然而, 在酵母单杂交试验中, 这2个ERF转录因子与MYC2a一样, 无法在酵母中结合GAG片段并激活由GAG片段操纵的报告基因表达。这一研究结果充分证明酵母表面展示系统可以有效克服酵母内源干扰, 弥补酵母单杂交技术的不足, 用于分离以酵母单杂交系统无法筛选的DNA结合蛋白, 因此, 该研究为易受酵母内源因子干扰DNA结合蛋白的筛选提供了一个新路径。
4 结论在改造蛋白表达载体pYD1基础上, 建立了以酵母表面展示系统筛选DNA结合蛋白的试验体系, 并成功应用这一系统对烟草 PMT基因启动子茉莉素应答核心元件GAG片段的DNA结合蛋白进行了筛选, 获得若干在体外结合GAG片段但不能在酵母中结合GAG片段的DNA结合蛋白。研究结果表明酵母表面展示系统可以克服内源干扰, 弥补酵母单杂交技术在DNA结合蛋白筛选方面的不足。
The authors have declared that no competing interests exist.
作者已声明无竞争性利益关系。
参考文献View Option
原文顺序
文献年度倒序
文中引用次数倒序
被引期刊影响因子
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}