唐仕伟, 李辉, 崔时媛, 张正坦, 谢志平


上海交通大学生命科学技术学院, 上海 200240
收稿日期:2018-09-15;修回日期:2018-11-14;网络出版日期:2018-12-25
基金项目:上海市教育委员会科研创新计划(2017-01-07-00-02-E00035)
*通信作者:谢志平, Tel: +86-21-34204090;E-mail:zxie@sjtu.edu.cn.
摘要:[目的] 构建一套用于酿酒酵母基因功能研究的质粒。该套质粒结合pUG系列和pFA6a系列的优点,同时采用同尾酶实现蛋白表位标签的串联插入。[方法] 利用PCR技术分别克隆pUG系列质粒的loxP位点、pFA6a质粒多酶切位点和ADH1终止子模块;通过重组连接各片段,构建pCLHN-TRP和pCLHN-URA质粒。在此基础上利用同尾酶实现多种蛋白表位标签的单个或串联重复插入,获得一系列蛋白表位标记质粒。最后,以ATG1、COX4和NHX1为例验证本质粒系列的性能。[结果] 在本项工作中,我们共构建2种基因敲除用质粒和17种表位标记用质粒(涵盖1-8 FLAG、1-12 V5、3-9 HA、2-8 MYC、GFP和mCherry)。在几个靶基因上的应用证实了本套质粒的实用性。尤其值得指出的是,通过组合采用不同重复度的串联表位标签,在同一张膜上同时检测表达差异极大的不同蛋白而不使高表达蛋白信号饱和成为可能。[结论] 本文所构建的pCLHN质粒系列是对现有酵母质粒工具的有益补充。
关键词:酵母细胞基因敲除蛋白标签同尾酶表达量
Construction of a novel plasmid system for epitope tagging and gene deletion in Saccharomyces cerevisiae
Shiwei Tang, Hui Li, Siwon Choi, Zhengtan Zhang, Zhiping Xie
School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China
*Corresponding author: Xie Zhiping, Tel: +86-21-34204090; E-mail:zxie@sjtu.edu.cn.
Foundation item: Supported by the Shanghai Municipal Education Commission (2017-01-07-00-02-E00035)
Abstract: [Objective] For yeast gene functional studies, we constructed a plasmid set combining the advantages of pUG and pFA6a plasmid series, and allowing convenient insertion of tandem epitope tags using isocaudomers. [Methods] We cloned the loxP locus of pUG plasmids, the multiple restriction site of pFA6a plasmids and ADH1 terminator cassette by PCR. Through homologous recombination of the DNA fragments, we obtained plasmid pCLHN-TRP and pCLHN-URA. Using introduced isocaudomer sites, we then constructed additional tagging plasmids containing either single or multiple tandem copies of various epitope tags. Finally, we tested the performance of our plasmids using ATG1, COX4 and NHX1 as target genes. [Results] We constructed two plasmids for gene deletion purposes, and seventeen plasmids for epitope tagging purposes (covering 1-8 FLAG, 1-12 V5, 3-9 HA, 2-8 MYC, GFP and mCherry). Testing on selected target genes demonstrated the usability of these plasmids. In particular, by combining different copy numbers of tandem epitopes, it was feasible to properly detect proteins with drastically different expression levels on the same blot without saturating the signal of high expression targets. [Conclusion] The pCLHN plasmids constitute a beneficial complement to existing yeast plasmid tools.
Keywords: yeast cellsgene deletionepitope taggingisocaudomerexpression level
酿酒酵母作为重要的真核模式生物,在基因功能研究中起到了非常重要作用[1-3]。1996年酿酒酵母完整基因组序列发表,标志着其基因功能分析进入到一个新的阶段[4]。在酵母系统中,基因敲除(gene deletion)[5-7]和表位标记(epitope tagging)[8-9]是基因功能研究的两个非常重要的基本操作工具。酵母高效的同源重组机制使人们可以方便地将DNA片段特异性地整合到基因组序列上,达到基因敲除和蛋白表位标记的目的。这种操作一个顺带的好处是,表位标记标签在基因组原位插入,可以在基本不改变蛋白原有表达水平的情况下实现目的蛋白的检测[10]。
在酵母中广泛应用的表位标记质粒主要是pFA6a系列为基础衍生的C端表位标记质粒系统[11-13]。目前在酵母系统中应用的多串联蛋白标签融合质粒有13MYC、5FLAG、3HA和12V5等[11, 13-14]。在蛋白检测与纯化实验中,不同表达水平的目的蛋白需要不同重复度的蛋白标签来满足实验要求。然而,在酵母中表达的融合蛋白标签质粒大部分只涵盖一部分长度的蛋白标签,同时包含短标签和长标签(多重复度)的系列化标签质粒较少。
酵母基因敲除常用的质粒系统包括pUG系列衍生质粒系统[15-16]。该系列质粒采用Cre-loxP系统实现筛选标记的重复利用。Cre是一种序列特异的DNA重组酶;loxP位点则是Cre识别的位点。pUG系列质粒在URA3、TRP1等常用的筛选标记两端安插loxP序列。利用这段两端带有loxP序列的DNA替换被敲除的基因后,再通过另一个质粒表达Cre酶,从而重组去除loxP中间的筛选基因。
在本文中,我们构建了新型的蛋白表位标记和基因敲除质粒。该套质粒不仅具有pUG系列loxP系统较好的筛选标记回复能力和pFA6a质粒表位标记的优点,而且通过引入同尾酶,可以方便地构建多重复标签质粒。在该套质粒系统上,我们分别在蛋白标签上游和筛选标记下游设计了T1(D1)/LRS位点,从而满足基因敲除和表位标记引物通用性设计。
1 材料和方法 1.1 材料 本文所用的限制性内切酶包括Nhe I (#R3131S,NEB公司)、Avr II (#R0174L,NEB公司)和Afl II (#R0520S,NEB公司);Q5?高保真DNA聚合酶(#M0493L)和T4连接酶(#B0202S)均购自NEB公司;重组酶购自诺维赞公司(C112-01/02);质粒提取试剂盒购自康为世纪生物科技有限公司(CW0500S);所用引物合成于GENEWIZ公司;所用V5 (T40006M)、FLAG (M22001M)、HA (M2003M)和MYC (M2002M)一抗均购自AbMART公司;山羊抗小鼠二抗(ZB-5305)购自北京中杉金桥公司;DH5α购自生工有限公司(B528413);DNA marker购自康为世纪生物科技有限公司(DM2000、DM15000)和北京擎科新业生物技术有限公司(1 kb DNA Ladder)。
1.2 质粒载体构建 为了构建pCLHN-URA质粒,以质粒pUG72、pFA6a-MX6和酵母基因组为模板,利用Q5高保真DNA聚合酶,分别用引物UG-F (10 μmol/L)和UG-R (10 μmol/L) (表 1)扩增pUG72质粒的loxP- URA3-loxP等区域;用引物FA6A-F (10 μmol/L)和FA6A-R (10 μmol/L) (表 1)扩增质粒pFA6a的多酶切位点区域并在引物上添加Nhe I和Avr II酶切位点;用引物ADH1-F (10 μmol/L)和ADH1-R (10 μmol/L)扩增ADH1终止序列。同理,为了构建pCLHN-TRP质粒,以质粒pUG76、pFA6a-MX6和酵母基因组为模板,分别用引物UG-F (10 μmol/L)和UG-R (10 μmol/L)扩增pUG76的loxP-TRP1-loxP等区域(图 1)。引物FA6A-F (10 μmol/L)和FA6A-R (10 μmol/L) (表 1)扩增质粒pFA6a-MX6的多酶切位点区域并在引物上添加Nhe I和Avr II酶切位点。用引物ADH1-F (10 μmol/L)和ADH1-R (10 μmol/L)扩增ADH1终止序列。将DNA片段重组后转化至DH5α感受态细胞,筛选出阳性克隆菌,进行菌落PCR验证,最后测序。
 |
| 图 1 构建pCLHN-URA/TRP质粒的示意图 Figure 1 Schematic representation of the construction of pCLHN-URA/TRP plasmids. The multiple restriction enzyme site from pFA6a-MX6 plasmid, the loxP system cassette from the pUG plasmid and the terminator of ADH1 were amplified by PCR using primers FA6A-F/R, UG-F/R and ADH1-F/R, respectively (see Table 1). The target fragments were then recombined to construct pCLHN-URA/TRP plasmids |
| 图选项 |
表 1. 本文所用到的质粒 Table 1. Plasmids used in this study
| Plasmid | Description | Selection marker | Reference |
| pUG72 | Gene deletion | URA3 | [16] |
| pUG76 | Gene deletion | TRP1 | [16] |
| pFA6a-MX6 | Gene tagging | KAN | [13] |
| pCLHN-TRP | Gene deletion | TRP1 | This study |
| pCLHN-URA | Gene deletion | URA3 | This study |
| pCLHN-FLAG | Gene tagging | URA3/TRP1 | This study |
| pCLHN-2FLAG | Gene tagging | URA3/TRP1 | This study |
| pCLHN-4FLAG | Gene tagging | URA3/TRP1 | This study |
| pCLHN-8FLAG | Gene tagging | URA3/TRP1 | This study |
| pCLHN-V5 | Gene tagging | URA3/TRP1 | This study |
| pCLHN-2V5 | Gene tagging | URA3/TRP1 | This study |
| pCLHN-4V5 | Gene tagging | URA3/TRP1 | This study |
| pCLHN-8V5 | Gene tagging | URA3/TRP1 | This study |
| pCLHN-12V5 | Gene tagging | URA3/TRP1 | This study |
| pCLHN-3HA | Gene tagging | URA3/TRP1 | This study |
| pCLHN-6HA | Gene tagging | URA3/TRP1 | This study |
| pCLHN-9HA | Gene tagging | URA3/TRP1 | This study |
| pCLHN-2MYC | Gene tagging | URA3/TRP1 | This study |
| pCLHN-4MYC | Gene tagging | URA3/TRP1 | This study |
| pCLHN-8MYC | Gene tagging | URA3/TRP1 | This study |
| pCLHN-GFP | Gene tagging | URA3/TRP1 | This study |
| pCLHN-mCherry | Gene tagging | URA3/TRP1 | This study |
表选项
1.3 利用同尾酶技术构建pCLHN多重复标签质粒 在成功构建pCLHN-URA和pCLHN-TRP质粒后,通过PCR技术分别扩增MYC、HA、V5、FLAG、GFP和mCherry片段,并整合到pCLHN-URA和pCLHN-TRP质粒上。由此构建了单蛋白标签质粒pCLHN-(MYC/HA/V5/FLAG/GFP/mCherry)- URA3/TRP1。在单蛋白标签质粒的基础之上,通过(Nhe I+Afl II)和(Avr II+Afl II)酶切的方式(其中Afl II酶切位点也可以用其他位于Avr II下游的单酶切位点替换)获得目的片段。用T4连接酶连接纯化后的目的片段,构建多标签重复质粒(图 2)。最后将连接产物转化至DH5α感受态细胞,并对阳性克隆进行菌落PCR验证,鉴定后送去测序。
 |
| 图 2 构建pCLHN系列多重复标签质粒 Figure 2 Construction of pCLHN plasmids with tandem tags. Using the characteristics of isocaudomers Nhe I and Avr II, plasmids containing tandem tags were constructed by the ligation of two fragments that were produced by the digestion of single-tagged plasmids with (Nhe I+Afl II) and (Avr II+Afl II) enzyme pairs |
| 图选项 |
以构建pCLHN-8FLAG为例,在构建pCLHN- 4FLAG质粒之后,将该质粒分别用(Nhe I+Afl II)和(Avr II+Afl II)酶切,分别获得酶切小片段和大片段。最后用T4连接酶将目的片段连接成完整的pCLHN-8FLAG质粒。
1.4 菌株的构建 本文使用的是TN124背景的酿酒酵母(Saccharomyces cerevisiae)[17]。作者挑选了ATG1、NHX1和COX4作为目标基因,其中蛋白Atg1的表达量约为1070 molecules/cell,蛋白大小为101 kDa;Nhx1表达量为521 molecules/cell,蛋白大小70.1 kDa;Cox4表达量为9410 molecules/cell,蛋白大小17.1 kDa。上述数据来源于SGD数据库(https://www.yeastgenome.org/)。以pCLHN-(XHA、XV5、XMYC、XFLAG、GFP和mCherry)-URA系列标签质粒为模板(表 1),用引物ATG1-T1/LRS、COX4-T1/LRS、NHX1-T1/LRS和VPS8-T1/LRS (表 2)来扩增目的片段,用于蛋白表位标记。PCR产物含有两端约40 bp目标基因同源序列以及表达筛选标记基因URA3/TRP1的基因片段,用于重组插入目的基因的编码框C端。目的基因的敲除所需DNA片段采用ATG1-D1和ATG1-LRS (表 2)引物获得。将PCR产物用PEG/LiAc方法转化至酵母中[18],获得酵母单克隆菌落。
表 2. 用于质粒构建以及表位标记的引物 Table 2. Primers used in this study
| Name | Sequence (5′?3′) |
| UG-F | AGGGATAACAGGGTAATATCTGCAGGTCGACAACCCTTAAT |
| UG-R | GCCGCGTTCTAACGACAATATGTCCATATGGTGCACTCTC |
| FA6A-F | TATTGTCGTTAGAACGCGGCTACAATTAATACATAACCTT |
| FA6A-R | GCTAGCGATGTTAATTAACCCAGGAATCCCACGACCTGCAGCGTACGAAG |
| ADH1-F | ATCGCTAGCCCTAGGTGAGGCGCGCCACTTCTAAATAAGCGAATTTCTTAT |
| ADH1-R | GCAGATATTACCCTGTTATCCCTAGCGGATCTGCCGGTAGAG |
| CLHN-F | TTCCTGGGTTAATTAACATCGCTAGCAGAATTGGCAAGATGCGTC |
| CLHN-R | TTCTTCAGCAACTTCAAGAGAAAGTGCAACGAAGGAGTCACTTTCC |
| ATG1-T1 | CAGGTTGAAAATATTGAGGCAGAAGATGAACCACCAAAATGCTTCGTACGCTGCAGGTCG |
| ATG1-D1 | ATGGGAGACATTAAAAATAA AGATCACACA ACCTCTGTGAGCTTCGTACGCTGCAGGTCG |
| ATG1-LRS | GGTCATTTGTACTTAATAAGAAAACCATATTATGCATCACGCATAGGCCACTAGTGGATC |
| ATG1(YZ)-F | TTTTACAACA CCAGACGAGAAATTAAGAAA |
| ATG1(YZ)-R | ACTTGAAAATATAGCAGGTCATTTGTACTT |
| COX4-T1 | CTAAACCCTGTTGGTGTTCCAAATGATGACCACCATCACGCTTCGTACGCTGCAGGTCG |
| COX4-LRS | AAGTAAAAGAGAAACAGAAGGGCAACTTGAATGATAAGAGCATAGGCCACTAGTGGATC |
| VPS8-T1 | TGTTTAATTTGCCAGACGGAATCTAACCCAAAAATAGTAGCTTCGTACGCTGCAGGTCG |
| VPS8-LRS | TTTTATGTAACCAAAGTTGTATTAAATATTTAGAAATGGCATAGGCCACTAGTGGATC |
| NHX1-T1 | GCTACGCAATCACCTGCAGATTTCTCTTCCCAAAACCACGCTTCGTACGCTGCAGGTCG |
| NHX1-LRS | ATTTATATTAGAAACAAGGAAACCATACACTTTAAAGTGCATAGGCCACTAGTGGATC |
表选项
1.5 敲除菌株的验证 利用验证引物ATG1(YZ)-F和ATG1(YZ)-R (表 2,引物设计为位置分别在ATG1启动子5′端上游和下游终止子),通过酵母菌落PCR的方式,验证敲除菌株YST45正确性。
1.6 蛋白表位标记的检测 用引物CLHN-F和CLHN-R (表 2)进行菌落PCR验证。将验证正确的酵母细胞培养到对数期(OD600=1.0),取3OD[OD=OD600×体积(mL)]酵母细胞,用TCA方法提取酵母蛋白,并进行BCA蛋白检测。用Western blotting检测MYC、FLAG、HA和V5标签:5%脱脂奶粉孵育1 h,之后用MYC、FLAG、HA和V5一抗(AbMART公司)(1:5000稀释)孵育2 h后洗膜5次,1%脱脂奶粉+1:10000的Anti-mouse二抗(W402B,Promega公司)孵育2 h后洗膜5次,加入底物显色曝光。内参抗体为GAPDH (M20028,AbMART公司)。
1.7 ALP (alkaline phosphatase)酶活检测 挑取目的菌株于营养培养基中,将细胞在YPD培养基中培养至对数期,然后分别取3OD细胞转至氮饥饿培养基(SD-N)中饥饿4 h,诱导自噬。本文所用到的pho8Δ60 pho13Δ菌株中,自噬的诱导水平可以通过检测碱性磷酸酶的活性来定量分析。取3OD细胞(OD600值为0.6–0.8),加入裂解液(20 mmol/L PIPES pH 6.8,50 mmol/L KCl,100 mmol/L KOAc,10 mmol/L MgSO4,10 μmol/L ZnSO4,1 mmol/L PMSF,0.5% Triton X-100) 150 μL和玻璃粉100 μL振荡破碎,离心。取上清4 μL,加入裂解液和含有ρ-对硝基苯磷酸二钠盐(ρ-nitrophenylphosphate,1.3 mmol/L)底物的反应液(250 mmol/L Tris-HCl pH 8.5,10 mmol/L MgSO4,10 μmol/L ZnSO4,0.4% Triton X-100) 80 μL来检测酶的活性,30 ℃反应20–40 min后加入终止液(1 mol/L glycine,pH 11.0) 100 μL来终止反应,测量400 nm光吸收。
自噬水平按公式(1)计算。
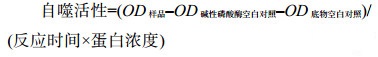 | 公式(1) |
相对自噬水平按照公式(2)计算。
 | 公式(2) |
1.8 显微镜观察 取OD600值为0.6–0.9的对数生长期酵母细胞1 OD置于EP管中。取200 μL细胞液滴于涂有Concanavalin A (刀豆蛋白A)的载玻片上,静置2 min,盖上盖玻片,置于IX83荧光倒置显微镜(奥林巴斯)下,观察细胞中荧光蛋白的分布情况。最后通过ImageJ软件分析目的蛋白在细胞当中的定位。
2 结果和分析 2.1 CLHN-(V5、FLAG、HA、MYC、GFP和mCherry)单标签质粒构建 为了构建在酵母系统中应用的多重复标签质粒,我们首先构建了单标签质粒。pCLHN-URA和pCLHN-TRP质粒(图 1)是本套质粒的骨架质粒。其设计初衷主要在3个方面:(1)方便筛选标记在酵母系统当中的重复使用;(2)表位标记与敲除引物设计的通用性(图 3);(3)利用同尾酶快速和简便地构建多重复标签质粒。
 |
| 图 3 pCLHN系列质粒中的通用引物设计 Figure 3 Universal primer design in pCLHN plasmids. The illustration shows the T1(D1)/LRS sites on the plasmid, which allows universal design of gene knockout and epitope tagging primers |
| 图选项 |
在酵母系统当中,pFA6a系列具有良好的C端表位标记能力,而pUG系列拥有loxP系统的筛选标记回复能力,故分别挑选了pFA6a的多酶切位点序列(加入了Nhe I和Avr II酶切位点)、pUG系列的loxP系统序列和ADH1终止子序列,构建了pCLHN-URA和pCLHN-TRP质粒(图 1)。
在获得CLHN-URA和CLHN-TRP质粒基础之上,通过以PCR扩增不同蛋白标签(MYC、FLAG、HA、V5、GFP和mCherry),通过重组方式,构建了一系列的单标签蛋白质粒(表 1)。
2.2 多标签质粒的构建 在成功构建的单标签质粒基础之上,利用pCLHN系列质粒具有Nhe I和Avr II同尾酶特点,将单标签质粒分别利用(Nhe I+Afl II)和(Avr II+ Afl II)酶切获得各自带有一段标签序列的载体和插入片段,最后通过T4连接酶连接方式构建标签加倍的质粒。以构建pCLHN-8FLAG-URA质粒为例(图 2),我们将构建好的pCLHN-4FLAG-URA质粒分别用(Nhe I+Afl II)和(Avr II+Afl II)酶切,分别得到各自含有4FLAG的小片段和载体,最后利用T4连接酶将目的片段连接成完整的pCLHN-8FLAG质粒(图 4)。其他多重复标签质粒的构建也是按照此方式。
 |
| 图 4 8FLAG多标签质粒的构建 Figure 4 Construction of the plasmid containing 8 FLAG tags. A: M1, DNA marker (DM15000); M2, DNA marker (DM2000); lane 1 is the CLHN-4FLAG-URA plasmid; lane 2 is the CLHN-4FLAG-URA plasmid digested with Avr II and Afl II; lane 3 is the same plasmid digested with Nhe I and Afl II. B: M, DNA marker (DM15000); lane 1 is the resulting CLHN-8FLAG-URA plasmid, constructed using T4 ligase |
| 图选项 |
2.3 敲除菌株的验证 作为应用实例,我们利用本文所构建的系列质粒对ATG1[19-20]进行了基因敲除和C端表位标记。在基因敲除实验中,我们采用引物ATG1(YZ)-F和ATG1(YZ)-R (表 2)验证转化株YST45 (图 5)。结果显示YST45菌落经PCR成功扩增出目的DNA条带,相应的对照组(ATG1基因没有敲除的YST40) PCR条带大小与预期一致,说明YST45中ATG1基因被有效地敲除。
 |
| 图 5 ATG1敲除菌株的PCR验证 Figure 5 Validation of ATG1 knockout strain by PCR. M: DNA marker (1 kb DNA Ladder); lane 1: YST45 strain (atg1△); lane 2: TN124 strain (control) |
| 图选项 |
2.4 经不同重复度表位标记后的目标蛋白表达检测 对于表位标记实验,在PCR验证多标签序列成功整合到ATG1的C端基础上,我们采用蛋白免疫印记检测表位标记效果。结果显示,信号水平随标签重复度增加而增强(图 6)。
 |
| 图 6 Atg1表位标记融合蛋白的免疫检测 Figure 6 Detection of Atg1 protein with different tandem tags. Yeast cells expressing Atg1 with different tandem tags were grown to mid-log phase in YPD, and then collected for protein extraction. For each sample, 5 μg of total protein was loaded and analyzed by Western blotting with antibodies against HA, V5, MYC, FLAG and GAPDH |
| 图选项 |
2.5 经不同重复度表位标记后的目标蛋白功能检测 为了验证不同长度表位标记对于目的蛋白的功能影响,我们通过pho8△60实验来检测融合不同长度标签Atg1蛋白的自噬水平。该方法通过碱性磷酸酶(ALP)活性指示自噬水平。结果显示,同一种类表位标记,数量越多,其对目的基因功能影响越大,而不同种类表位标记,其对于目的蛋白的功能影响水平是不同的(图 7)。
 |
| 图 7 Atg1蛋白表位标记菌株的自噬水平 Figure 7 Levels of autophagy in strains expressing tagged Atg1 proteins. The indicated strains were grown to mid-log phase in YPD, and then shifted to nitrogen starvation medium (SD-N) for 4 hours. The levels of autophagy were measured by the pho8Δ60 assay. Error bar=standard deviation, n=3 |
| 图选项 |
2.6 不同表达量目标蛋白的同时检测 此外,我们以低表达的NHX1和高表达的COX4基因为例尝试同时检测表达差异较大的靶标。Nhx1表达量约为521 molecules/cell,分子量70.1 kDa;Cox4表达量约为9410 mol/cell,分子量17.1 kDa。二者表达量差异接近一个数量级。结果显示,通过融合不同重复数量的V5标签,可以做到让低水平表达的Nhx1-4V5蛋白能够被有效检测,而高水平表达的Cox4-1V5蛋白信号又不至于过饱和(图 8)。
 |
| 图 8 Nhx1和Cox4表位标记融合蛋白的免疫检测 Figure 8 Detection of Nhx1 and Cox4 protein with V5 tags. Yeast cells expressing Atg1 with different tandem tags were grown to mid-log phase in YPD, and then collected for protein extraction. For each sample, 5 μg of total protein was loaded and analyzed by Western blotting with antibodies against V5 and GAPDH |
| 图选项 |
2.7 目标蛋白荧光蛋白标记的显微镜观察 我们挑选了Cox4、Nhx1和Vps8作为目标蛋白,以质粒pCLHN-GFP和pCLHN-mCherry为模板,通过PCR的方式扩增荧光蛋白GFP和mCherry片段,并整合到目的蛋白的C端。显微镜结果显示,目的蛋白能够被有效标记,荧光信号指示了被标记的蛋白在细胞中的定位情况(图 9)。
 |
| 图 9 荧光蛋白标记的Cox4、Nhx1和Vps8 Figure 9 Localization of Cox4, Nhx1 and Vps8 tagged with different fluorescent proteins in yeast cells. Yeast cells expressing the indicated fusion proteins were grown to mid-log phase in YPD, and then collected for fluorescent microscope. Scale bar=5 μm |
| 图选项 |
3 讨论 通过连接HA、FLAG等表位标记标签,人们无需为每个蛋白制备专用的抗体即可方便地采用免疫共沉淀、免疫电镜、ELISA等手段研究蛋白表达和功能。由于细胞中不同蛋白的表达水平跨度很大,为了在同一个实验中更好地检测不同表达水平的目的蛋白,需要使用不同重复度的标签质粒调整每个蛋白的信号水平:表达水平低的蛋白,需要多重复度的蛋白标签使其易于检测;而表达水平高的蛋白,需要低重复度的蛋白标签避免信号过饱和。本套质粒中的标签重复个数跨度大,同时基本设计便于继续扩展,可以满足检测不同表达水平蛋白的需求。
此外,本套质粒还可作为其他系列化功能标签质粒构建的基础,例如单个或串联荧光蛋白标签。荧光蛋白不仅可以用于蛋白表达检测,而且可以用来研究目的蛋白在活细胞中的定位和蛋白相互作用[21-23]。
References
| [1] | Tong AHY, Evangelista M, Parsons AB, Xu H, Bader GD, Pagé N, Robinson M, Raghibizadeh S, Hogue CWV, Bussey H, Andrews B, Tyers M, Boone C. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science, 2001, 294(5550): 2364-2368. DOI:10.1126/science.1065810 |
| [2] | Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M'Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Véronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science, 1999, 285(5429): 901-906. |
| [3] | Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends in Biotechnology, 1998, 16(9): 373-378. DOI:10.1016/S0167-7799(98)01214-1 |
| [4] | Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG. Life with 6000 genes. Science, 1996, 274(5287): 546-567. DOI:10.1126/science.274.5287.546 |
| [5] | Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Research, 1993, 21(14): 3329-3330. DOI:10.1093/nar/21.14.3329 |
| [6] | Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast, 1998, 14(2): 115-132. DOI:10.1002/(ISSN)1097-0061 |
| [7] | Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast, 1999, 15(14): 1541-1553. DOI:10.1002/(ISSN)1097-0061 |
| [8] | Schneider BL, Seufert W, Steiner B, Yang QH, Futcher AB. Use of polymerase chain reaction epitope tagging for protein tagging in Saccharomyces cerevisiae. Yeast, 1995, 11(13): 1265-1274. DOI:10.1002/(ISSN)1097-0061 |
| [9] | Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature, 2003, 425(6959): 737-741. DOI:10.1038/nature02046 |
| [10] | Lichty JJ, Malecki JL, Agnew HD, Michelson-Horowitz DJ, Tan S. Comparison of affinity tags for protein purification. Protein Expression and Purification, 2005, 41(1): 98-105. DOI:10.1016/j.pep.2005.01.019 |
| [11] | Noguchi C, Garabedian MV, Malik M, Noguchi E. A vector system for genomic FLAG epitope-tagging in Schizosaccharomyces pombe. Biotechnology Journal, 2008, 3(9/10): 1280-1285. |
| [12] | Sato M, Dhut S, Toda T. New drug-resistant cassettes for gene disruption and epitope tagging in Schizosaccharomyces pombe. Yeast, 2005, 22(7): 583-591. DOI:10.1002/(ISSN)1097-0061 |
| [13] | Gadaleta MC, Iwasaki O, Noguchi C, Noma KI, Noguchi E. New vectors for epitope tagging and gene disruption in Schizosaccharomyces pombe. Biotechniques, 2013, 55(5): 257-263. |
| [14] | B?hler J, Wu JQ, Longtine M S, Shah NG, Mckenzie III A, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast, 1998, 14(10): 943-951. DOI:10.1002/(ISSN)1097-0061 |
| [15] | Güldener U, Heck S, Fiedler T, Beinhauer J, Hegemann JH. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Research, 1996, 24(13): 2519-2524. DOI:10.1093/nar/24.13.2519 |
| [16] | Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Research, 2002, 30(6): e23. DOI:10.1093/nar/30.6.e23 |
| [17] | Noda T, Matsuura A, Wada Y, Ohsumi Y. Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochemical and Biophysical Research Communications, 1995, 210(1): 126-132. DOI:10.1006/bbrc.1995.1636 |
| [18] | Hu YJ, Gao F, Zhu CB, Zhu BQ. A modified method for yeast transformation with PEG/LiAc. Biotechnology, 1998, 8(5): 22-26. (in Chinese) 胡又佳, 高枫, 朱春宝, 朱宝泉. PEG/LiAc转化酵母细胞方法的改进. 生物技术, 1998, 8(5): 22-26. |
| [19] | Chan EY, Tooze SA. Evolution of Atg1 function and regulation. Autophagy, 2009, 5(6): 758-765. DOI:10.4161/auto.8709 |
| [20] | Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Molecular Biology of the Cell, 2009, 20(7): 2004-2014. DOI:10.1091/mbc.e08-12-1250 |
| [21] | Laird DW, Jordan K, Thomas T, Qin H, Fistouris P, Shao Q. Comparative analysis and application of fluorescent protein-tagged connexins. Microscopy Research & Technique, 2001, 52(3): 263-272. |
| [22] | Müller-Taubenberger A. Application of fluorescent protein tags as reporters in live-cell imaging studies. Methods in Molecular Biology, 2006, 346: 229-246. |
| [23] | Dash AK, Yende AS, Tyagi R K. Novel application of red fluorescent protein (DsRed-Express) for the study of functional dynamics of nuclear receptors. Journal of Fluorescence, 2017, 27(4): 1225-1231. DOI:10.1007/s10895-017-2109-z |
