刘晓彤, 许晓坤, 张友明

 , 李爱英
, 李爱英 微生物技术国家重点实验室, 山东大学微生物技术研究院, 山东大学-亥姆霍兹生物技术研究所, 山东 青岛 266237
收稿日期:2018-05-08;修回日期:2018-07-11;网络出版日期:2018-07-19
基金项目:国家自然科学基金(31670097);山东省重点研发计划(2015GSF12101);山东省重大专项(2015ZDJS04001);山东省自然科学基金(ZR2017MC031)
*通信作者:张友明, Tel:+86-532-67720918, E-mail:zhangyouming@sdu.edu.cn
李爱英, Tel:+86-532-67722928, E-mail:ayli@sdu.edu.cn
摘要:微生物来源的syrbactins属于十二元内酰胺短肽类化合物,包括丁香霉素(syringolins)、滑杆菌素(glidobactins)、cepafungins和luminmycins等。其中,syringolins、glidobactins、luminmycins等几个化合物生物合成基因簇已被克隆、测序和异源表达。研究发现它们的十二元内酰胺环骨架都是由非核糖体肽合成酶(NRPS)-聚酮合酶(PKS)复合体采用模块化组装的方式,将系列底物组装而成。这类化合物因具有优异的蛋白酶体抑制活性而受到广泛关注。本文从syrbactins的分子结构、生物合成、作用机理等几个方面进行综述,介绍了近年来syrbactins的研究进展。
关键词:Syrbactins蛋白酶体抑制剂生物合成
Advances in biosynthesis of proteasome inhibitor syrbactins
Xiaotong Liu, Xiaokun Xu, Youming Zhang
, Aiying Li State Key Laboratory of Microbial Technology, Microbial Biotechnology Institute, Shandong University-Helmholtz Institute of Biotechnology, Shandong University, Qingdao 266237, Shandong Province, China
Received 8 May 2018; Revised 11 July 2018; Published online 19 July 2018
*Corresponding author: Youming Zhang, Tel: +86-532-67720918, E-mail: zhangyouming@sdu.edu.cn
Aiying Li, Tel: +86-532-67722928, E-mail: ayli@sdu.edu.cn
Supported by the National Natural Science Foundation of China (31670097), by the Key Research and Development Program of Shandong Province (2015GSF12101), by the Major Special Project in Shandong Province (2015ZDJS04001) and by the Natural Science Foundation of Shandong Province (ZR2017MC031)
Abstract: Syrbactins are microbial natural products, mainly composed of syringolins, glidobactins, cepafunins and luminmycins. At present, several biosynthetic gene clusters of syringolins and other compounds have been cloned, sequenced and heterologously expressed. These studies revealed that, despite the structural differences between syrbactins, they are synthesized in vivo in a similar mode and share a NRPS-PKS hybrid assembly biosynthetic pathway for the formation of their core skeletons. They have drawn continuous research attentions due to the irreversible proteasome inhibition activity. This review focuses on the research advances in syrbactins and highlights their structure, biosynthesis and mode-of-action.
Keywords: syrbactinsproteasome inhibitionbiosynthesis
Syrbactins是一类微生物来源的天然产物,包含四组化合物[syringolins (丁香霉素)、glidobactins (滑杆菌素)、cepafungins和luminmycins等]。作为高效蛋白酶体抑制剂,这个家族化合物具有多种生物学活性(如诱导植物抗病性、抗菌、抗肿瘤等)。它们共有的母核结构是十二元内酰胺环,由非核糖体肽合成酶(nonribosomal-peptide-synthases,NRPS)-聚酮合酶(polyketide-synthases,PKS)复合体将底物进行模块化组装而成。本文将重点阐述syrbactins的分子结构、生物合成以及它们独特的作用机理,并介绍近年来syrbactins的研究进展。
1 Syrbactins家族成员 “Syrbactins”名称源于最先发现的两组化合物“syringolins”和“glidobactins”单词的组合。Glidobactins A-G是1988年由Oka等从Polyangium brachysporum K481-B101 (ATCC 53080)(已重新分类为伯克氏菌目)中分离到[1-2]。Syringolin A是1998年由Dudler等从丁香假单胞菌(Pseudomonas syringae) B301D-R中分离得到[3]。1999年和2012年由不同课题组分离得到syringolins B-F和syringolins G-H[4-5]。第三组cepafungins Ⅰ-Ⅲ是1990年从洋葱伯克氏菌(Burkholderia cepacia)中分离到[6](cepafungin Ⅱ实际上就是glidobactin A,所以cepafungins也常常被定义为glidobactins的不同组分)[7]。第四组luminmycins A-C是2012年由本课题组成员从发光杆菌(Photorhabdus luminescens)中直接克隆沉默基因簇并进行异源表达得到[8];luminmycin D分离自非共生发光杆菌(P. asymbiotica)[9]。
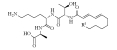
Syrbactins家族化合物的核心结构是由2个非蛋白质氨基酸组成的十二元内酰胺环,环外连一条不饱和脂肪酸或二肽短链。除了开环的luminmycins B和C,syrbactins家族的其他成员均表现出明显的抗菌和抗肿瘤活性,这也从侧面证明了十二元内酰胺环是syrbactins家族化合物的活性中心[8](表 1)。
表 1. 目前分离得到的syrbactins家族化合物 Table 1. Compounds in syrbactins family isolated so far
| Name | Structure | Substituent | Source | |
| Glidobactins A-G | 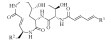 | A:R1=(CH2)6-CH3 B:R1=(CH2)2-CH=CH(Z)-(CH2)4-CH3 C:R1=(CH2)8-CH3 D:R1=(CH2)4-CH(OH)-CH2-CH3 E:R1=CH2-CH(OH)-(CH2)4-CH3 F:R1=(CH2)4-CH3 G:R1=(CH2)6-CH3 | R2=H R2=H R2=H R2=H R2=H R2=H R2=OH | Polyangium brachysporum sp. nov. No. K481-B101 (ATCC 53080) (now Burkholderia) |
| Syringolins A-H | 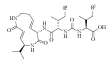 | A:R1=H C:R1=Me D:R1=H F:R1=Me | R2=H R2=H R2=Me R2=Me | Pseudomonas syringae pv. Syringae B301D-R |
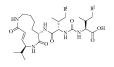 | B:R1=H E:R1=H G:R1=Me H:R1=Me | R2=H R2=Me R2=Me R2=H | ||
| Cepafungins Ⅰ-Ⅲ | 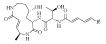 | Ⅰ:R1=(CH2)5-CH(CH3)-CH3 Ⅱ:R1=(CH2)6-CH3 Ⅲ:R1=(CH2)3-CH(CH3)-CH3 | Burkholderia cepacia | |
| Luminmycins A-D | 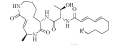 | A:R1=CH2-CH3 D:R1=CH(CH3)-CH3 | Photorhabdus luminescens Photorhabdus asymbiotica | |
 | B:R1=CH2-CH3 C:R1=CH2-CH3 | |||
 |
表选项
2 Syrbactins的生物合成 2.1 Syrbactins生物合成的基因簇和酶复合体 以syringolins为例,其基因簇含有sylA-E 5个基因,sylA基因编码LuxR类型转录激活因子,负责正向调控sylB基因和sylCDE操纵子的表达,将其突变失活后不再有syringolin A产生[10],sylB产物可能负责赖氨酸残基双键的合成,本课题组成员等通过异源表达不完整的syringolins生物合成基因簇sylCDE,分离得到2个赖氨酸饱和的化合物syringolins G-H,侧面证明该基因功能[5],sylE可能编码转运蛋白,参与syringolins的胞外分泌[10],而sylC和sylD是结构基因,sylC编码1个NRPS模块,sylD编码2个NRPS模块和1个PKS模块,负责线性合成syringolins的骨架结构[10]。每个NRPS模块有3个功能结构域:腺苷酰化结构域(adenylation domain,A domain)、缩合结构域(condensation domain,C domain)和肽酰基载体蛋白(peptide carrier protein,PCP),分别负责氨基酸底物的识别活化、肽键缩合、硫化等反应;其PKS模块也包括3个核心结构域:酮基合成酶(ketosynthase,KS)、酰基转移酶(acyhransferase,AT)和酰基载体蛋白(acylcarrier protein,ACP),分别负责二碳缩合、酰基转移和延伸底物引入。此外,syrbactins的PKS模块中还含有2个修饰酮基的功能域:酮基还原酶(ketoreductase,KR)、脱水酶(dehydratase,DH)以及负责将聚肽-酮杂合链释放和环化的硫化结构域(thioesterase,TE)[11](图 1)。
 |
| 图 1 几种syrbactins的生物合成基因簇示意图 Figure 1 Biosynthetic gene clusters of syrbactins. |
| 图选项 |
Glidobactins生物合成基因簇克隆自伯克氏菌,共含有8个基因glbA-H[12]。glbA是正调控基因[12],glbB可能负责赖氨酸的羟基化,本课题组将该基因敲除后glidobactin A消失,产生去羟基的衍生物luminmycin A (数据未发表),glbF和glbC是结构基因,分别与sylC和sylD对应,glbF编码1个NRPS模块,glbC编码2个NRPS和1个PKS模块[12],glbD编码转运蛋白,glbE编码一类MbtH-like分子伴侣,可能具有提高GlbF稳定性的作用[12],glbG编码酮异构酶,glbH功能未知[12]。本课题组将glbG和glbH敲除后,glidobactin A产量明显下降,同时还有一些类似物产生(数据未发表)(图 1)。
除了上述两种类型的基因簇,2014年,Biggins等从类鼻疽伯克氏菌(Burkholderia pseudomallei)中发现了syrbactins基因簇(SYR),包含syrA-I 9个基因,通过在基因簇前插入诱导型启动子得到glidobactin C和deoxyglidobactin C两个化合物[13](图 1)。
2.2 Syrbactins母核的生物合成过程 研究发现,虽然syrbactins结构不同,来源不同,但其母核的生物合成机制非常相似:十二元内酰胺环母核骨架都是由非核糖体肽合成酶(NRPS)-聚酮合酶(PKS)复合体采用模块化组装的方式,将系列底物组装而成。下面以syringolins和glidobactins为例阐述syrbactins母核生物合成过程(图 2)。
 |
| 图 2 Syringolin A的生物合成过程 Figure 2 The biosynthetic pathway of syringolin A. A: SylC catalyzes the ureido dipeptide formation; B: GlbF catalyzes the N-acylation of threonine; C: The NRPS/PKS assembly line of syringolin A. |
| 图选项 |
母核链的引发和起始:Syringolins生物合成的起始由SylC模块催化完成。2009年,Dudler[14]和Wuest[15]课题组通过同位素标记实验证明SylC能够活化两分子氨基酸并利用重碳酸盐/CO2形成脲键,将两分子氨基酸连接在一起,最终形成特殊的Val(Ile)-CO-Val(Ile)-S-SylC起始结构。在glidobactins的生物合成中,链的起始是由GlbF模块催化完成的:首先GlbF中的A domain选择和活化苏氨酸,在ATP的参与下形成腺苷化苏氨酸;活化的腺苷化苏氨酸连接到PCP的巯基基团上,然后C domain利用脂酰辅酶A作为供体,将活化的苏氨酸变成脂酰化苏氨酸,形成fatty acyl-Thr- S-pantetheinyl-GlbF起始结构[16]。
母核链的延伸和转移:与一般的NRPS催化机制类似,syrbactins生物合成中每一个NRPS模块负责将1个氨基酸整合到产物的骨架中(经历底物氨基酸的活化、转移及缩合构成新肽键),从而产生延长了1个氨基酸的新的肽酰-S-载体复合物和游离的载体。肽链的延伸顺序与基因簇中模块排列顺序一致。当肽链延伸到最后PKS模块的ACP上,在KS模块作用下延长1个二碳单位,β羰基经过KR、DH几个模块催化最终形成双键结构。
母核链的终止、释放与环化:在最后一个模块的C末端通常有起终止延伸和释放产物功能的TE结构域。最后在TE模块作用下中间产物被释放,经过分子内亲核攻击而合成环化产物。
除母核之外,这些化合物基因簇中还有一些推测负责母核的结构修饰(如差向异构化、甲基化、杂环化或氧化等过程)等相关基因,但研究较少。在glidobactins、cepafungins和luminmycins等化合物结构中的脂酰链的选择以及与母核的结合也鲜有研究[16]。
3 Syrbactins的生物活性和构效优化 3.1 Syrbactins作用靶标——蛋白酶体 蛋白酶体是真核生物中普遍存在的酶复合体,它是泛素-蛋白酶体通路(ubiquitin-proteasome pathway,UPP)的核心组分,是真核细胞高效并高选择性地降解错误折叠、过量蛋白的主要方式。真核细胞26S蛋白酶体包含有1个20S核心颗粒和2个19S调节颗粒。所有的20S颗粒都是由4个七元环按照顺序α1-7-β1-7-β1-7-α1-7堆积而成的筒状结构。其中β1、β2和β5三种亚基分别具有类胱天蛋白酶(caspase-like)、类胰蛋白酶(tryptic-like)和类胰凝乳蛋白酶(chymotryptic-like)活性[17]。它们的N末端苏氨酸残基是蛋白酶体的3个活性位点,用于蛋白质水解反应[17]。
原核生物,主要是放线菌细胞中,也存在类似于真核细胞的蛋白酶体系统——泛素样蛋白-蛋白酶体系统(prokaryotic ubiquitin-like protein-proteasome system,PPS)。在特定环境下,原核类泛素蛋白(prokaryotic ubiquitin-like protein,Pup)选择性标记胞内蛋白,并介导靶蛋白进入原核蛋白酶体降解。目前对于原核蛋白酶体的研究主要是在结核分枝杆菌(Mycobacterium tuberculosis)中进行的。结核分枝杆菌20S蛋白酶体与真核细胞20S蛋白酶体核心颗粒结构相似,同样是由4个堆叠的七元环组成。不同的是,结核分枝杆菌20S蛋白酶体包含有14个相同的α亚基和14个相同的β亚基,且14个β亚基均具有胰凝乳蛋白酶活性[18-19]。
3.2 Syrbactins可作为蛋白酶体抑制剂类抗肿瘤药物 2008年,Groll等首次阐明了syringolin A和glidobactin A作为不可逆蛋白酶体抑制剂与蛋白酶体的相互作用机制,并证明syringolin A十二元内酰胺环上C-4位置的双键能够与20S蛋白酶体N末端苏氨酸残基上的羟基发生Michael 1, 4-加成反应,从而对蛋白酶体产生不可逆的抑制[20] (图 3)。
 |
| 图 3 Syringolin A与20S蛋白酶体相互作用 Figure 3 Binding mode of syringolin A with 20S proteasome. |
| 图选项 |
肿瘤细胞快速增殖的特性使其基因组不稳定,更加依赖于蛋白酶体高表达来清除细胞内错误折叠或损伤的蛋白质。事实上,原发性肿瘤中蛋白酶体水平和活性远高于正常组织[21]。所以与正常细胞相比较,癌细胞对蛋白酶体抑制剂更为敏感。抑制蛋白酶体活性能够促进细胞调亡,尤其是处于快速分裂中的癌细胞的凋亡,这使得蛋白酶体成为抗肿瘤药物筛选的分子靶标[22-23]。
Syrbactins家族化合物作为一类结构新颖的蛋白酶体抑制剂具有优异的抗肿瘤活性,其对人神经母细胞瘤、人类多发性骨髓瘤和卵巢癌细胞均有显著抑制效果[24-26]。Cepafungin Ⅰ对酵母蛋白酶体的IC50仅为4 nmol/L,是syrbactins家族中抗肿瘤活性最高的化合物,被认为是最有开发潜力的抗癌药物先导化合物之一[27]。
3.3 Syrbactins可作为抗感染药物 结核病(tuberculosis TB)是由结核分枝杆菌感染引起的一种严重威胁人类健康的慢性传染病。结核分枝杆菌进入人体后,能够通过蛋白酶体及时清除由于受到宿主防御系统抵制而损伤的蛋白质,从而使自身能够在宿主细胞内长期存在。据统计,全球至少有20亿人潜伏感染结核分枝杆菌,最终患结核病的感染者约占5%-10%。因此,如何抑制这些潜伏的结核分枝杆菌成为结核病防控的重要研究内容。
同哺乳动物蛋白酶体具有7类β-亚基的结构相比,结核分枝杆菌蛋白酶体的内核仅含有一类β-亚基,活性位点更加广泛,是一种很有潜力但非传统的药物靶标。Syrbactins家族化合物是结核分枝杆菌20S蛋白酶体的选择性抑制剂。2016年,Totaro等通过理性设计合成的syringolin同系物1,减弱了对人20S蛋白酶体的抑制,但却将对结核分枝杆菌20S蛋白酶体的抑制活性提高了70多倍,成为结核病药物开发的新起点[28]。
3.4 对植物抗病性的诱导作用 丁香假单胞菌(P. syringae)分泌的syringolin A是一种植物抗病诱导剂,它能够被水稻、小麦等植物识别,进而激活宿主防御基因,诱导植物产生获得抗病性(acquired resistance,AR)[3]。
3.5 Syrbactins的构效优化 目前几个课题组通过化学合成的方式合成了一系列syrbactins的同系物,希望通过构效优化提高syrbactins的抗菌和抗肿瘤活性。
Glidobactin A衍生物:1988年,Oka等将glidobactin A酶解消化得到核心结构——十二元内酰胺环,然后通过DCC/HOBt耦合反应为其加上不同的侧链合成了30多种glidobactins衍生物。生物活性表明脂肪酸侧链的长度和饱和程度被证明是影响这类化合物生物活性的一个重要因素[25]。
Syringolins衍生物:2009年至2011年间,Clerc等先后合成了脂链syringolin A (sylA-Lip)(2)、聚乙二醇syringolin A (sylA-PEG)(3)以及syringolin A和glidobactin A的杂合化合物sylA-glbA (4)[29-31]。2014年,Chiba等基于syringolin A合成了系列化合物[32]。令人振奋的是,这些化合物与天然产物相比均有更好的蛋白酶体抑制活性,尤其是19a-e这一组化合物对蛋白酶体胰凝乳蛋白酶活性位点的表观抑制常数Ki'仅为0.14-1.10 nmol/L (抑制效力是syringolin A的800倍,是glidobactin A的50倍),具有良好的蛋白酶体抑制剂开发潜力,具有比天然syringolin A更好的抗肿瘤活性,能够抑制人神经母细胞瘤、多发性骨髓瘤、卵巢癌细胞、表皮癌细胞等多种癌细胞的增殖,其中19a(5)对人骨髓瘤细胞RPMI8226的IC50仅为2.2 nmol/L,可与已经上市的蛋白酶体抑制剂类抗癌药物硼替佐米相媲美。此外,研究发现syringolins侧链的氨基酸构型,尤其是紧连赖氨酸残基的第一个氨基酸的构型对其活性也有影响,侧链为D型氨基酸的syringolin A比L型syringolin A的活性低几十倍[30, 33](图 4)。
 |
| 图 4 几种化学合成的syrbactins同系物 Figure 4 Structural diversity of syrbactins analogs. |
| 图选项 |
2016年,Bachmann等化学合成的syrbactins类似物TIR-199(6)具有选择性作用于肾癌细胞的优异活性,是天然化合物syringolin A的250多倍,曾被美国国家癌症研究所列入优先研发药物之列,最近该课题组合成了其一系列硫代化合物,提高了其水溶性,而且发现化合物7和8具有选择性作用于免疫蛋白酶体β2亚基的抑制活性[34-35](图 4)。
2018年,Yoshida等通过一系列实验证明syringolin类似物syringolog-1(5)是20S蛋白酶体β5和β2两个活性位点的双重抑制剂,它不仅抑制正常多发性骨髓瘤细胞,而且对耐硼替佐米骨髓瘤细胞株表现出显著的抗肿瘤活性[36]。这些研究为新一代蛋白酶体抑制剂类抗癌药物的开发提供了理论基础。
4 合成生物学技术用于syrbactins生物合成基因挖掘和合成途径改造 4.1 GenBank中syrbactins生物合成基因簇的挖掘和直接克隆技术(Red/ET大片段DNA直接克隆技术) 随着越来越多的微生物基因组被测序,GenBank数据库中大量的细菌基因组及宏基因组信息为发现潜在的syrbactins化合物生物合成基因提供了重要的数据资源。除了上述这些已经成功表达的syrbactins生物合成基因簇以外,本课题组已经在其他菌种如类鼻疽伯克氏菌(B. pseudomallei)、荚壳伯克氏菌PG1 (B. glumae PG1)中发现与syrbactins类似的生物合成基因簇,并尝试对PG1中的同源基因簇进行原位激活实验。
Syrbactins产生菌多数难以直接进行遗传操作,且syrbactins生物合成基因数量多,基因簇一般都较长,很难直接扩增或合成。而本课题组建立的基于大肠杆菌原噬菌体RecE/RecT DNA重组酶的“Red/ET大片段DNA直接克隆技术”可以克服这个技术瓶颈[37-39]。
“Red/ET大片段DNA直接克隆技术”是指将线性dsDNA载体[包括质粒的复制起点(ori)、选择性标记基因(sm)和相应同源臂与线性化的基因组] DNA混合,共转化到含有RecE/RecT重组酶的E. coli细胞内。RecE/RecT重组酶可以介导线-线DNA同源重组(即线性载体通过末端的同源臂与基因组片段上的靶标DNA发生重组,形成能在E. coli细胞内复制的质粒)(图 5)。利用该技术,本课题组从发光杆菌(P. luminescens)染色体上克隆了包括luminmides和luminmycins/glidobactins的沉默基因簇(10-52 kb)[8, 40],从假单胞菌(P. syringae)基因组中克隆了syringolins的生物合成基因簇(22 kb)[5, 41],并成功地在异源宿主中进行了表达,表明该技术在天然产物基因簇直接克隆上具有很大优势。
 |
| 图 5 Red/ET大片段DNA直接克隆示意图 Figure 5 Diagram of Red/ET DNA direct cloning. |
| 图选项 |
4.2 Syrbactins生物合成基因簇异源表达底盘生物的筛选和优化 创新药物的人工生物合成还需要有适配的底盘生物提供一个利于生产的内环境。微生物会通过复杂的系统精细地控制syrbactins生物合成相关酶系的表达,从而控制最后活性产物的产量;在细胞工厂里,只有通过精细地微调各部分之间的比例才能最大化地释放细胞的合成潜能,高效地生产目的产物。
已发现从土壤中分离到的伯克氏菌DSM7029能够产生聚酮合酶与非核糖体多肽合成酶(PKS/NRPS)杂合的化合物glidobactin,表明该菌具有合成聚酮类和非核糖体多肽类化合物所必需的基本元件和PKS/NRPS激活所需要的因子磷酸泛酰巯基乙胺转移酶。本课题组在该菌株中建立了一套简单高效的遗传操作系统,并利用该系统实现了DSM7029作为天然产物通用底盘菌的改造和优化[42]。
此外,以E. coli Nissle1917 (欧洲市场的一株常用益生菌)作为底盘细胞,具有生长速度快、遗传操作方便、可表达天然产物合成基因等优势;而且Red/ET DNA同源重组技术可以在其细胞内高效工作,在其中直接完成药物基因簇的修饰和表达[8]。利用E. coli Nissle1917,本课题组实现了luminmides和luminmycins/glidobactins等基因簇的异源表达,且在E. coli Nissle1917导入粘细菌来源的磷酸泛酰基巯基乙胺转移酶基因mtaA后,插入组成型启动子显著提高glidobactin的产量(10倍)[40]。这都为优化syrbactins人工合成途径和高效适配的底盘宿主打下了良好的基础。
5 总结和展望 在中国,癌症已成为疾病死因之首。抗癌药物研发刻不容缓。用于癌症治疗的蛋白酶体抑制剂品种不多,主要分为肽类和非肽类(图 6)。肽类中,最早上市的硼替佐米(Bonezomib)作为一线药物已被用于多发性骨髓瘤、套细胞淋巴瘤治疗,对胰腺癌、非霍奇金氏淋巴瘤都有抑制效果,但其副作用明显,会导致神经退行病症[43-45]。卡非佐米(Carfilzomib)对复发难治多发性骨髓瘤具有持久的抗瘤活性,该药物在临床Ⅲ期试验中与地塞米松(Dexamethasone)联合使用能显著延长患者生存期,但它在使用中会出现中性粒细胞减少症和血小板减少的情况[46]。Ixazomib是FDA批准的首个可口服的用于治疗多发性骨髓瘤的蛋白酶体抑制剂[22]。Marizomib是一种具有稳定的β-内酯结构的非肽类蛋白酶体抑制剂,更有利于促进肿瘤细胞凋亡,2014年被美国FDA授予“孤儿药”资格,用于治疗复发性恶性胶质瘤[47]。还有一些二代肽类蛋白酶抑制剂正在临床前研究中。
 |
| 图 6 已经批准上市的几种蛋白酶体抑制剂抗癌药物 Figure 6 Structures of several proteasome inhibitors that have already been approved to go on the market. |
| 图选项 |
在国内,蛋白酶体抑制剂研发多是集中在药理方面,生物合成研究鲜见报道,已经有几家研究机构在进行化合物的化学合成和修饰。目前我国尚无自主知识产权的蛋白酶体抑制剂类抗肿瘤药物。
不同于上述已经用于临床的蛋白酶体抑制剂,syrbactins家族化合物的十二元内酰胺环活性中心可与蛋白酶体β亚基N末端的酶活中心的苏氨酸羟基发生共价加成反应,这种作用是不可逆的。更重要的是这类化合物独特的化学结构和作用机理使其对蛋白酶体β亚基有更强的选择性,所以syrbactins被认为具有成为新一代抗癌药物先导化合物的潜力。
近年来,通过化学合成改造syrbactins结构来提高其生物活性已经初见成效,比如2016年合成的syrbactins类似物TIR-199具有选择性作用于肾癌细胞的优异活性,但其水溶性低的缺点阻碍了其应用,没有达到治疗肿瘤的理想效果。
除了化学合成,通过生物技术来对syrbactins进行构效优化似乎更加符合临床重大需求。鉴于syrbactins的骨架结构是由合成酶的催化模块和功能结构域的排列顺序来决定,这种模块化的装配催化机制可以允许研究人员对其相关结构域和模块的功能进行重排,可以人为控制其聚酮或聚肽骨架结构的合成。因此通过合成生物学的方法构建人工的syrbactins生物合成途径、在适配底盘细胞中高效表达、筛选高活性低毒性的蛋白酶体抑制剂将是一个很有前景的研究方向,有助于推进蛋白酶体抑制剂类抗肿瘤药物的新药研发,但目前尚未有这方面的报道。
伴随着大片段直接克隆、多片段有序拼接等生物工程技术的成熟,包括syringolins、glidobactins、luminmycins在内的几个syrbactins家族化合物的生物合成基因簇已经被克隆和异源表达。可以从GenBank等数据库或者相应菌株中挖掘更多的syrbactins类似基因簇,以进一步深入研究其生物合成机制,为合理设计和改造syrbactins化合物积累资源,提供理论支持。
过去的30年中,人们在syrbactins的化学结构、生物活性、作用机理和化学合成等方面的研究取得了一系列重要进展[48]。尽管如此,对于这类化合物的认识仍然存在盲点。继续开展对这类化合物的研究,对抗肿瘤新药的开发具有重要意义。
References
| [1] | Oka M, Yaginuma K, Numata K, Konishi M, Oki T, Kawaguchi H. Glidobactins A, B and C, new antitumor antibiotics. Ⅱ. Structure elucidation. The Journal of Antibiotics, 1988, 41(10): 1338-1350. DOI:10.7164/antibiotics.41.1338 |
| [2] | Oka M, Ohkuma H, Kamei H, Konishi M, Oki T, Kawaguchi H. Glidobactins D, E, F, G and H; minor components of the antitumor antibiotic glidobactin. The Journal of Antibiotics, 1988, 41(12): 1906-1909. DOI:10.7164/antibiotics.41.1906 |
| [3] | W?spi U, Blanc D, Winkler T, Rüedi P, Dudler R. Syringolin, a novel peptide elicitor from Pseudomonas syringae pv. syringae that induces resistance to Pyricularia oryzae in rice.. Molecular Plant-Microbe Interactions, 1998, 11(8): 727-733. DOI:10.1094/MPMI.1998.11.8.727 |
| [4] | W?spi U, Hassa P, Staempfli AA, Molleyres LP, Winkler T, Dudler R. Identification and structure of a family of syringolin variants: Unusual cyclic peptides from Pseudomonas syringae pv. syringae that elicit defense responses in rice. Microbiological Research, 1999, 154(1): 89-93. DOI:10.1016/S0944-5013(99)80040-8 |
| [5] | Bian XY, Huang F, Stewart FA, Xia LQ, Zhang YM, Müller R. Direct cloning, genetic engineering, and heterologous expression of the syringolin biosynthetic gene cluster in E. coli through Red/ET recombineering. ChemBioChem, 2012, 13(13): 1946-1952. DOI:10.1002/cbic.201200310 |
| [6] | Shoji J, Hinoo H, Kato T, Hattori T, Hirooka K, Tawara K, Shiratori O, Terui Y. Isolation of cepafungins Ⅰ, Ⅱ and Ⅲ from Pseudomonas species. The Journal of Antibiotics, 1990, 43(7): 783-787. DOI:10.7164/antibiotics.43.783 |
| [7] | Terui Y, Nishikawa J, Hinoo H, Kato T, Shoji J. Structures of cepafungins Ⅰ, Ⅱ and Ⅲ. The Journal of Antibiotics, 1990, 43(7): 788-795. DOI:10.7164/antibiotics.43.788 |
| [8] | Bian XY, Plaza A, Zhang YM, Müller R. Luminmycins A-C, cryptic natural products from Photorhabdus luminescens identified by heterologous expression in Escherichia coli. Journal of Natural Products, 2012, 75(9): 1652-1655. DOI:10.1021/np300444e |
| [9] | Theodore CM, King JB, You JL, Cichewicz RH. Production of cytotoxic glidobactins/luminmycins by Photorhabdus asymbiotica in liquid media and live crickets. Journal of Natural Products, 2012, 75(11): 2007-2011. DOI:10.1021/np300623x |
| [10] | Amrein H, Makart S, Granado J, Shakya R, Schneider-Pokorny J, Dudler R. Functional analysis of genes involved in the synthesis of Syringolin A by Pseudomonas syringae pv. syringae B301D-R. Molecular Plant-Microbe Interactions, 2004, 17(1): 90-97. DOI:10.1094/MPMI.2004.17.1.90 |
| [11] | Walsh CT. Polyketide and nonribosomal peptide antibiotics: modularity and versatility. Science, 2004, 303(5665): 1805-1810. DOI:10.1126/science.1094318 |
| [12] | Schellenberg B, Bigler L, Dudler R. Identification of genes involved in the biosynthesis of the cytotoxic compound glidobactin from a soil bacterium. Environmental Microbiology, 2007, 9(7): 1640-1650. DOI:10.1111/j.1462-2920.2007.01278.x |
| [13] | Biggins JB, Kang HS, Ternei MA, DeShazer D, Brady SF. The chemical arsenal of Burkholderia pseudomallei is essential for pathogenicity. Journal of the American Chemical Society, 2014, 136(26): 9484-9490. DOI:10.1021/ja504617n |
| [14] | Ramel C, Tobler M, Meyer M, Bigler L, Ebert MO, Schellenberg B, Dudler R. Biosynthesis of the proteasome inhibitor Syringolin A: the ureido group joining two amino acids originates from bicarbonate. BMC Biochemistry, 2009, 10: 26. DOI:10.1186/1471-2091-10-26 |
| [15] | Imker HJ, Walsh CT, Wuest WM. SylC catalyzes ureido-bond formation during biosynthesis of the proteasome inhibitor Syringolin A. Journal of the American Chemical Society, 2009, 131(51): 18263-18265. DOI:10.1021/ja909170u |
| [16] | Imker HJ, Krahn D, Clerc J, Kaiser M, Walsh CT. N-Acylation during glidobactin biosynthesis by the tridomain nonribosomal peptide synthetase module GlbF. Chemistry & Biology, 2010, 17(10): 1077-1083. |
| [17] | Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chemical Reviews, 2007, 107(3): 687-717. DOI:10.1021/cr0502504 |
| [18] | Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science, 2008, 322(5904): 1104-1107. DOI:10.1126/science.1163885 |
| [19] | Hu GQ, Lin G, Wang M, Dick L, Xu RM, Nathan C, Li HL. Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Molecular Microbiology, 2006, 59(5): 1417-1428. DOI:10.1111/j.1365-2958.2005.05036.x |
| [20] | Groll M, Schellenberg B, Bachmann AS, Archer CR, Huber R, Powell TK, Lindow S, Kaiser M, Dudler R. A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature, 2008, 452(7188): 755-758. DOI:10.1038/nature06782 |
| [21] | Chen L, Madura K. Increased proteasome activity, ubiquitin-conjugating enzymes, and eEF1A translation factor detected in breast cancer tissue. Cancer Research, 2005, 65(13): 5599-5606. DOI:10.1158/0008-5472.CAN-05-0201 |
| [22] | ?led? P, Baumeister W. Structure-driven developments of 26S proteasome inhibitors. Annual Review of Pharmacology and Toxicology, 2016, 56(1): 191-209. DOI:10.1146/annurev-pharmtox-010814-124727 |
| [23] | Rentsch A, Landsberg D, Brodmann T, Bülow Leila, Girbig AK, Kalesse M. Synthesis and pharmacology of proteasome inhibitors. Angewandte Chemie International Edition, 2013, 52(21): 5450-5488. DOI:10.1002/anie.201207900 |
| [24] | Coleman CS, Rocetes JP, Park DJ, Wallick CJ, Warn-Cramer BJ, Michel K, Dudler R, Bachmann AS. Syringolin A, a new plant elicitor from the phytopathogenic bacterium Pseudomonas syringae pv. syringae, inhibits the proliferation of neuroblastoma and ovarian cancer cells and induces apoptosis. Cell Proliferation, 2006, 39(6): 599-609. DOI:10.1111/cpr.2006.39.issue-6 |
| [25] | Oka M, Numata K, Nishiyama Y, Kamei H, Konishi M, Oki T, Kawaguchi H. Chemical modification of the antitumor antibiotic glidobactin. The Journal of Antibiotics, 1988, 41(12): 1812-1822. DOI:10.7164/antibiotics.41.1812 |
| [26] | Archer CR, Koomoa DLT, Mitsunaga EM, Clerc J, Shimizu M, Kaiser M, Schellenberg B, Dudler R, Bachmann AS. Syrbactin class proteasome inhibitor-induced apoptosis and autophagy occurs in association with p53 accumulation and Akt/PKB activation in neuroblastoma. Biochemical Pharmacology, 2010, 80(2): 170-178. DOI:10.1016/j.bcp.2010.03.031 |
| [27] | Stein ML, Beck P, Kaiser M, Dudler R, Becker CFW, Groll M. One-shot NMR analysis of microbial secretions identifies highly potent proteasome inhibitor. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(45): 18367-18371. DOI:10.1073/pnas.1211423109 |
| [28] | Totaro KA, Barthelme D, Simpson PT, Jiang XJ, Lin G, Nathan CF, Sauer RT, Sello JK. Rational design of selective and bioactive inhibitors of the Mycobacterium tuberculosis proteasome. ACS Infectious Diseases, 2017, 3(2): 176-181. DOI:10.1021/acsinfecdis.6b00172 |
| [29] | Clerc J, Groll M, Illich DJ, Bachmann AS, Huber R, Schellenberg B, Dudler R, Kaiser M. Synthetic and structural studies on Syringolin A and B reveal critical determinants of selectivity and potency of proteasome inhibition. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106(16): 6507-6512. DOI:10.1073/pnas.0901982106 |
| [30] | Clerc J, Schellenberg B, Groll M, Bachmann AS, Huber R, Dudler R, Kaiser M. Convergent synthesis and biological evaluation of Syringolin A and derivatives as eukaryotic 20S proteasome inhibitors. European Journal of Organic Chemistry, 2010, 2010(21): 3991-4003. DOI:10.1002/ejoc.201000317 |
| [31] | Clerc J, Li N, Krahn D, Groll M, Bachmann AS, Florea BI, Overkleeft HS, Kaiser M. The natural product hybrid of Syringolin A and Glidobactin A synergizes proteasome inhibition potency with subsite selectivity. Chemical Communications, 2011, 47(1): 385-387. DOI:10.1039/C0CC02238A |
| [32] | Chiba T, Hosono H, Nakagawa K, Asaka M, Takeda H, Matsuda A, Ichikawa S. Total synthesis of Syringolin A and improvement of its biological activity. Angewandte Chemie International Edition, 2014, 53(19): 4836-4839. DOI:10.1002/anie.201402428 |
| [33] | Kolodziejek I, Misas-Villamil JC, Kaschani F, Clerc J, Gu C, Krahn D, Niessen S, Verdoes M, Willems LI, Overkleeft HS, Kaiser M, van der Hoorn RAL. Proteasome activity imaging and profiling characterizes bacterial effector Syringolin A. Plant Physiology, 2011, 155(1): 477-489. DOI:10.1104/pp.110.163733 |
| [34] | Bachmann AS, Opoku-Ansah J, Ibarra-Rivera TR, Yco LP, Ambadi S, Roberts CC, Chang CA, Pirrung MC. Syrbactin structural analog TIR-199 blocks proteasome activity and induces tumor cell death. Journal of Biological Chemistry, 2016, 291(16): 8350-8362. DOI:10.1074/jbc.M115.710053 |
| [35] | Bakas NA, Schultz CR, Yco LP, Roberts CC, Chang CA, Bachmann AS, Pirrung MC. Immunoproteasome inhibition and bioactivity of thiasyrbactins. Bioorganic & Medicinal Chemistry, 2018, 26(2): 401-412. |
| [36] | Yoshida T, Ri M, Kanamori T, Aoki S, Ashour R, Kinoshita S, Narita T, Totani H, Masaki A, Ito A, Kusumoto S, Ishida T, Komatsu H, Kitahata S, Chiba T, Ichikawa S, Iida S. Potent anti-tumor activity of a syringolin analog in multiple myeloma: a dual inhibitor of proteasome activity targeting β2 and β5 subunits. Oncotarget, 2018, 9(11): 9975-9991. |
| [37] | Fu J, Bian XY, Hu SB, Wang HL, Huang F, Seibert PM, Plaza A, Xia LQ, Müller R, Stewart AF, Zhang YM. Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nature Biotechnology, 2012, 30(5): 440-446. DOI:10.1038/nbt.2183 |
| [38] | Wang HL, Li Z, Jia RN, Yin J, Li AY, Xia LQ, Yin YL, Müller R, Fu J, Stewart AF, Zhang YM. ExoCET: exonuclease in vitro assembly combined with RecET recombination for highly efficient direct DNA cloning from complex genomes. Nucleic Acids Research, 2018, 46(5): e28. DOI:10.1093/nar/gkx1249 |
| [39] | Wang HL, Li Z, Jia RN, Hou Y, Yin J, Bian XY, Li AY, Müller R, Stewart AF, Fu J, Zhang YM. RecET direct cloning and Redαβ recombineering of biosynthetic gene clusters, large operons or single genes for heterologous expression. Nature Protocols, 2016, 11(7): 1175-1190. DOI:10.1038/nprot.2016.054 |
| [40] | Bian XY, Huang F, Wang HL, Klefisch T, Müller R, Zhang YM. Heterologous Production of glidobactins/luminmycins in Escherichia coli Nissle containing the glidobactin biosynthetic gene cluster from Burkholderia DSM7029. ChemBioChem, 2014, 15(15): 2221-2224. DOI:10.1002/cbic.v15.15 |
| [41] | Huang F, Tang JL, He L, Ding XZ, Huang SY, Zhang YM, Sun YJ, Xia LQ. Heterologous expression and antitumor activity analysis of syringolin from Pseudomonas syringae pv. syringae B728a. Microbial Cell Factories, 2018, 17: 31. DOI:10.1186/s12934-018-0859-1 |
| [42] | Wang X, Zhou HB, Chen HN, Jing XS, Zheng WT, Li RJ, Sun T, Liu JQ, Fu J, Huo LJ, Li YZ, Shen YM, Ding XM, Müller R, Bian XY, Zhang YM. Discovery of recombinases enables genome mining of cryptic biosynthetic gene clusters in Burkholderiales species. Proceedings of the National Academy of Sciences of the United States of America, 2018, 115(18): E4255-E4263. |
| [43] | Price CTD, Al-Quadan T, Santic M, Rosenshine I, Kwaik YA. Host proteasomal degradation generates amino acids essential for intracellular bacterial growth. Science, 2011, 334(6062): 1553-1557. DOI:10.1126/science.1212868 |
| [44] | Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade?: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist, 2003, 8(6): 508-513. DOI:10.1634/theoncologist.8-6-508 |
| [45] | Murray MY, Auger MJ, Bowles KM. Overcoming bortezomib resistance in multiple myeloma. Biochemical Society Transactions, 2014, 42(4): 804-808. DOI:10.1042/BST20140126 |
| [46] | Dimopoulos MA, Goldschmidt H, Niesvizky R, Joshua D, Chng WJ, Oriol A, Orlowski RZ, Ludwig H, Facon T, Hajek R, Weisel K, Hungria V, Minuk L, Feng SB, Zahlten-Kumeli A, Kimball AS, Moreau P. Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open-label, randomised, phase 3 trial. The Lancet Oncology, 2017, 18(10): 1327-1337. DOI:10.1016/S1470-2045(17)30578-8 |
| [47] | Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angewandte Chemie International Edition, 2003, 42(3): 355-357. DOI:10.1002/anie.200390115 |
| [48] | Krahn D, Ottmann C, Kaiser M. The chemistry and biology of syringolins, glidobactins and cepafungins (syrbactins). Natural Product Reports, 2011, 28(11): 1854-1867. DOI:10.1039/c1np00048a |
