陈南1, 朱璟1, 叶梅霞1,2, 金一1, 何晓青1

 , 邬荣领1,2
, 邬荣领1,2 1.北京林业大学生物科学与技术学院, 北京 100083;
2.北京林业大学计算生物学中心, 北京 100083
收稿日期:2016-07-30;修回日期:2016-11-03;网络出版日期:2016-11-03
基金项目:国家自然科学基金青年科学基金(51108029);中央高校基本科研业务费专项(TD2012-03);水利部公益性行业科研专项(201201032)
*通信作者:何晓青, E-mail: lenahe@bjfu.edu.cn
摘要: [目的]通过实验室培养模拟自然环境微生物相互作用,进而找到影响细菌基因型和表型的基因。[方法]将大肠杆菌和金黄色葡萄球菌在实验室条件下进行单独培养和两两混合培养并连续转接,通过得到的数量表型与最大生长速率表型做全基因组关联分析(GWAS),对得到的与表型相关的SNP进行注释与分析。[结果]162个SNP位点影响到大肠杆菌原始菌株与共培养菌株的生长,36个SNP位点影响大肠杆菌菌株在单独培养和共同培养的生长。总共有85个SNP位点影响金黄色葡萄球菌的原始菌株与单独培养。其中5个基因在之前文献中已有报道。对影响不同时间点细菌数量变化形状的SNP位点进行功能注释,大肠杆菌中有706个与生长性能相关。金黄色葡萄球菌中,129个和不同的生长性能相关。大肠杆菌SNP位点的13个基因在之前的研究中已有报道。[结论]混合培养和单独培养都检测到与生长相关的显著基因,本研究表明了GWAS在研究细菌互作进化机制方面的潜力。
关键词: 微生物竞争 全基因组重测序 全基因组关联分析
Interactions between Escherichia coli and Staphylococcusaureus determined by genome-wide association analysis
Nan Chen1, Jing Zhu1, Meixia Ye1,2, Yi Jin1, Xiaoqing He1
, Rongling Wu1,2 1.College of Biological Sciences and Technology, Beijing Forestry University, Beijing 100083, China;
2.Center for Computational Biology, Beijing Forestry University, Beijing 100083, China
Received 30 July 2016; Revised 03 November 2016; Published online 03 November 2016
*Corresponding author: Xiaoqing He, E-mail: lenahe@bjfu.edu.cn
Supported by the National Natural Science Foundation of China (51108029), by the Fundamental Research Funds for the Central University (TD2012-03) and by the Non-Profit Industry Financial Program of Ministry of Water Resources (201201032)
Abstract: [Objective]We studied the interactions in a co-culture of two bacteria.[Methods]By pairwise co-culturing of 36 Escherichia coli and 36 Staphylococcus aureus strains, we monitored the growth of each species in an interaction environment. We identified numerous Single Nucleotide Polymorphisms (SNPs) by whole-genome sequencing used as genetic markers to predict variations in phenotypic traits. Genome-wide association study (GWAS) was applied to identify loci that controlled competition between the two species.[Results]In E. coli, 162 significant SNPs affected the change of maximum growth rate by comparing initials strains with those grown in co-culture, and 36 significant SNPs affected the change of maximum growth rate comparing monoculture and co-culture strains. Five of the significant E. coli genes we identified after annotation this time were also reported in other evolutionary studies. We also identified 85 significant SNPs in S. aureus that affected the change of maximum growth rate by comparing initial strains with those grown in monoculture. About the change of bacterial numbers, we found that 706 significant SNPs were associated in E. coli and 129 in S. aureus. Thirteen of the E. coli significant genes in this study were also verified in previous evolutionary reports.[Conclusion]We found several significant genes both in monoculture and co-culture affecting the interaction of E. coli and S. aureus. GWAS has the potential to study interspecific interactions of bacteria.
Key words: microbial competition whole-genome sequencing genome-wide association studies
在自然环境中发生着众多微生物间的竞争,这些相互作用对于地球生化多样性是非常重要的[1]。理解物种适应新环境对阐明生物系统动力学和预测物种对环境变化的反应至关重要[2]。当面对环境变化时,物种相互作用不仅引起生态物种丰度和分布的变化,也影响物种的进化,同时也会影响生态系统功能[3]。大量例子证明,共同进化有利于加速物种的进化[4],且物种的共同进化区别于物种单独生长时的进化[5]。尽管提出了多种生态机制解释为什么物种相互作用促进适应环境变化[6],但其遗传基础仍然未知。直到近几年才有了少量关于表型变化对应基因型变化的研究[7]。
为了获得更多的信息,基因组进化研究需要结合微生物实验的全基因组测序 (whole-genome sequencing,WGS)[8]。目前,WGS被用在识别自然群体中以表型变异、适应性为基础的基因变异[9],全基因组测序提供了比此前更全面的微生物基因组结构和内容的分析[10-11]。与传统的测序方法相比,通过识别单核苷酸的差异,WGS提供了表型性状的遗传基础信息[12]。微生物的全基因组测序能发现自适应进化细菌的基本参数,包括突变的数量,突变基因的功能等。在细菌方面,适应性进化的一些研究已经包括基因组重测序[13]。
全基因组关联研究 (Genome-wide association study,GWAS) 是对多个个体在全基因组范围的遗传变异多态性进行检测,获得基因型,进而将基因型与可观测的性状 (即表型) 进行群体水平的统计学分析,根据统计量或P值筛选出最有可能影响该性状的遗传变异。GWAS与以往的候选基因研究策略不同,众多功能不明的基因及大量基因间区域的SNP都能为复杂性状的研究提供线索,不必再受预先设定的候选基因的限制[14]。GWAS在微生物方面的应用是快速识别调节表型变异遗传因素的一个潜在而有效的方法,更容易鉴别出导致某种表型变化的遗传基础[15],识别重要表型潜在的遗传因素,但很少被应用于细菌[16]。大肠杆菌和金黄色葡萄球菌在自然界中广泛存在,研究这两种菌的不同菌株之间的相互作用具有代表性。而且,大肠杆菌和金黄色葡萄球菌具有相对较小的基因组,也易于获得不同基因型的菌株,因而是全基因组研究非常好的模式生物。
细菌的相互作用类型主要为竞争作用[17]。本研究中我们在同一培养基中培养大肠杆菌和金黄色葡萄球菌不同菌株的配对组合,观察各菌株在不同时间点的浓度以及培养前后的最大生长速率。之后用GWAS的方法来识别在人为生态环境中控制特定生态过程的基因。本文使用的是传统分析方法——基于高密度单核苷酸多态性 (Single nucleotide polymorphism,SNP) 标记的全基因组关联分析[18],用基因组中数量巨大的SNP为遗传标记,在全基因组水平上进行对照分析或相关性分析,通过比较发现影响复杂疾病或性状的功能基因的方法[19]。该法最初应用于人类全基因组范围内筛选出与复杂疾病相关的SNP,进而确定致病基因。随着各物种全基因组信息的陆续公布,GWAS已经成功应用在很多领域的复杂数量性状候选基因的检测中[20]。
1 材料和方法 1.1 材料
1.1.1 菌株来源: 大肠杆菌共36株,其中24株购自中国普通微生物菌种保藏中心 (CGMCC),12株购自中国工业微生物菌种保藏中心 (CICC);金黄色葡萄球菌共36株,其中7株购自中国普通微生物菌种保藏中心 (CGMCC),4株购自中国工业微生物菌种保藏中心 (CICC),6株购自中国农业菌种保藏中心 (ACCC),1株购自中国林业微生物菌种保藏管理中心 (CFCC),5株购自中国典型培养物保藏中心 (CCTCC),5株购自中国药学微生物菌种保藏管理中心 (CPCC),2株购自中国医学微生物菌种保藏管理中心 (CMCC),5株由中国农业大学动物医学院赠与,1株诱变菌株。所有菌株均保存于–80 ℃。:
1.1.2 试剂: 溶菌酶、溶葡萄球菌素均为美国Sigma公司产品;营养肉汤培养基 (Nutrient Broth),胰蛋白胨大豆琼脂培养基 (TSA) 为Oxiod公司产品;细菌基因组DNA提取试剂盒,荧光定量试剂盒购自天根生化科技有限公司;引物由北京博迈德生物技术有限公司合成。: 1.2 接种与培养 将大肠杆菌和金黄色葡萄球菌各36株菌株冻存液在TSA平板上划线后37 ℃培养24 h,挑取单菌落于20 mL NB培养基中37 ℃、130 r/min过夜培养后,利用流式细胞术 (FCM),对样本进行染色后35 ℃避光孵育10 min,上机检测,数据分析[21]后计算出各菌液样本浓度。取适量稀释后的菌液接种于稀释10倍的NB培养液中,使其终浓度在5×103 cells/mL左右;混合培养的实验组分别接入等同于单独培养的菌液量,使得各菌种的终浓度均在5×103 cells/mL左右。将菌液置于恒温摇床中30 ℃、130 r/min培养。培养4 d后记为第1个时间点。从各瓶中取出1 mL菌液分别转接入新鲜的10倍稀释NB培养液中,同时取出5 mL菌液用于提取DNA。将新转接的培养基继续振荡培养3 d后进行同样的操作,记为第2个时间点,以此类推每周转接2次,每次间隔为4 d或3 d,持续8周,共16个时间点。分别取36对菌的原始菌株、第16个时间点的36对单独分离株和36对混合分离株各3个单菌落于NB原液培养基中过夜活化后,取100 μL接入新鲜的NB原液培养基中,每小时用酶标仪 (Infinite M200 PRO,TECAN,瑞士) 测定菌液的OD600值,绘制生长曲线并计算各菌株的最大生长速率。
1.3 qPCR测定菌液浓度 根据大肠杆菌β-D-葡萄糖苷酶编码基因uidA和金黄色葡萄球菌的耐热核酸酶nuc基因的序列,设计大肠杆菌uidA基因的引物uidA-F (5′-CGCTG AAGAGATGCTCGACT-3′) 和uidA-R (5′-CACGC TTGGGTGGTTTTTGT-3′),扩增片段的长度为217 bp。金黄色葡萄球菌nuc基因的引物nuc-F (5′-AGATAACGGCGTA AATAGA-3′) 和nuc-R (5′-ACTTGCTTCAGGACC ATA-3′),扩增片段长度为226 bp。将提取的DNA通过qPCR的方法测得每个时间点菌液浓度。
1.4 全基因组重测序 取1 mL过夜培养菌液用细菌DNA提取试剂盒提取基因组,经检测质量合格后由测序公司利用Hiseq 2500测序平台进行细菌全基因组重测序 (诺禾致源,北京)。去除所含低质量碱基 (质量值≤38) 超过一定比例 (默认40 bp)、N碱基达到一定比例 (默认10 bp)、去除与接头之间重叠超过一定阈值 (默认15 bp) 的reads,并去除重复的reads。有效测序数据通过BWA软件比对到参考基因组上,参考基因组选择Escherichia coli str. K-12 substr.MG1655和Staphylococcus aureus subsp. aureus NCTC 8325。利用SAMtools软件过滤掉质量值低于20、测序深度低于4×的SNP的结果,即保证至少有4条以上且质量值均高于20的reads支持,进一步保证分析结果的准确性。
1.5 GWAS分析 通过重测序技术在全基因组范围内筛选出了高密度的分子标记SNP位点。本研究采用最大似然方法来分析每个SNP位点与单独培养细菌各个时间点所获得的生长数据及最大生长速率值之间的关系,找到影响对应表型的显著SNP。通过基因注释,定位显著SNP位于哪些基因内或基因间隔区,进一步找到影响表型的差异基因。考虑到细菌每个SNP位点有2种不同单倍体基因型Q (编号为1) 和q (编号为2),用yi表示一种细菌菌株i的数量值,在指定SNP位点表型评估的似然用公式 (1) 表示。
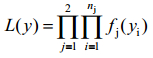 | 公式(1) |
利用最大似然估计对模型参数进行估计。通过把这些估计值插入到零和备假设下的似然,计算每个标记的对数似然比 (LR)。LR值用来和临界阈值比较,临界阈值是排列检验通过重排所有菌株的表型1000次得到的[22]。
2 结果和分析 2.1 全基因组测序结果 大肠杆菌和金黄色葡萄球菌基因组的重测序信息统计在表 1和表 2中进行了概述。对所有特定的区域进行评估来确定具体的SNP。从36株大肠杆菌菌株中共确定了65533个SNP位点,密度大约为每60个/bp,从36株金黄色葡萄球菌菌株中确定了86516个SNP位点,密度大约为40个/bp。
表 1. 大肠杆菌重测序数据统计 Table 1. Resequencing statistics for E. coli
| Number | Sample ID | Insert size/bp | Reads length/bp | Raw data/Mb | Filtered reads/% | Clean data/Mb | Clean data Q20/% | Clean data Q30/% |
| 1 | E1 | 500 | (100:100) | 992 | 30.18 | 693 | 91.75 | 77.57 |
| 2 | E2 | 500 | (100:100) | 1421 | 27.95 | 1024 | 95.07 | 86.00 |
| 3 | E3 | 500 | (100:100) | 1015 | 31.37 | 696 | 91.18 | 78.30 |
| 4 | E4 | 500 | (100:100) | 1332 | 30.15 | 932 | 90.34 | 78.48 |
| 5 | E5 | 500 | (100:100) | 1710 | 28.20 | 1228 | 95.17 | 86.92 |
| 6 | E6 | 500 | (100:100) | 682 | 27.17 | 497 | 94.28 | 82.75 |
| 7 | E7 | 500 | (100:100) | 720 | 33.01 | 483 | 91.42 | 76.84 |
| 8 | E8 | 500 | (100:100) | 930 | 25.82 | 690 | 92.02 | 79.76 |
| 9 | E9 | 500 | (100:100) | 1060 | 24.68 | 798 | 92.36 | 80.12 |
| 10 | E10 | 500 | (100:100) | 853 | 30.27 | 595 | 91.82 | 77.81 |
| 11 | E11 | 500 | (125:125) | 935 | 18.47 | 762 | 94.80 | 88.80 |
| 12 | E12 | 500 | (100:100) | 1344 | 26.64 | 986 | 95.21 | 87.08 |
| 13 | E14 | 500 | (100:100) | 945 | 22.37 | 733 | 92.95 | 81.26 |
| 14 | E17 | 500 | (100:100) | 1070 | 28.60 | 764 | 95.04 | 85.96 |
| 15 | E18 | 500 | (100:100) | 1316 | 26.83 | 963 | 95.17 | 86.36 |
| 16 | E19 | 500 | (100:100) | 895 | 28.24 | 643 | 91.44 | 78.50 |
| 17 | E20 | 500 | (100:100) | 831 | 23.76 | 633 | 93.47 | 81.72 |
| 18 | E21 | 500 | (100:100) | 1418 | 27.25 | 1032 | 95.28 | 87.16 |
| 19 | E22 | 500 | (100:100) | 1183 | 26.51 | 869 | 95.12 | 86.12 |
| 20 | E23 | 450 | (125:125) | 1086 | 12.41 | 952 | 94.63 | 88.78 |
| 21 | E25 | 500 | (125:125) | 680 | 20.09 | 544 | 94.82 | 88.88 |
| 22 | E26 | 500 | (100:100) | 1084 | 30.29 | 756 | 95.36 | 86.96 |
| 23 | E27 | 500 | (100:100) | 1472 | 26.93 | 1076 | 95.15 | 86.85 |
| 24 | E28 | 500 | (100:100) | 1116 | 18.07 | 915 | 95.87 | 86.03 |
| 25 | E29 | 500 | (100:100) | 1120 | 28.80 | 798 | 95.58 | 87.39 |
| 26 | E30 | 500 | (100:100) | 1016 | 17.9 | 834 | 97.28 | 90.04 |
| 27 | E31 | 500 | (100:100) | 940 | 30.85 | 650 | 95.69 | 88.71 |
| 28 | E32 | 500 | (100:100) | 964 | 32.68 | 649 | 94.83 | 86.15 |
| 29 | E34 | 500 | (100:100) | 882 | 23.07 | 679 | 92.66 | 80.95 |
| 30 | E35 | 500 | (100:100) | 954 | 36.55 | 605 | 94.28 | 85.02 |
| 31 | E40 | 500 | (125:125) | 1123 | 22.03 | 876 | 94.82 | 88.70 |
| 32 | E41 | 500 | (100:100) | 995 | 31.08 | 686 | 91.07 | 78.07 |
| 33 | E42 | 500 | (125:125) | 900 | 18.34 | 735 | 94.90 | 88.81 |
| 34 | E43 | 500 | (100:100) | 1151 | 28.82 | 820 | 95.97 | 89.31 |
| 35 | E44 | 500 | (125:125) | 814 | 14.72 | 694 | 96.01 | 91.24 |
| 36 | E45 | 500 | (100:100) | 1096 | 31.37 | 752 | 94.94 | 86.43 |
表选项
表 2. 金黄色葡萄球菌重测序数据统计 Table 2. Resequencing statistics of S. aureus
| Number | Sample ID | Insert size/bp | Reads length/bp | Raw data/Mb | Filtered reads/% | Clean data/Mb | Clean data Q20/% | Clean data Q30/% |
| 1 | S1 | 500 | (100:100) | 1012 | 18.86 | 821 | 97.01 | 90.85 |
| 2 | S2 | 500 | (100:100) | 1215 | 22.99 | 935 | 93.32 | 82.34 |
| 3 | S3 | 500 | (100:100) | 1012 | 9.88 | 912 | 98.54 | 94.28 |
| 4 | S4 | 500 | (100:100) | 1247 | 9.54 | 1128 | 97.61 | 91.41 |
| 5 | S5 | 500 | (100:100) | 1220 | 10.68 | 1090 | 96.51 | 88.97 |
| 6 | S6 | 500 | (100:100) | 1166 | 8.78 | 1064 | 97.46 | 90.96 |
| 7 | S7 | 500 | (100:100) | 1094 | 8.99 | 996 | 97.37 | 90.67 |
| 8 | S8 | 500 | (100:100) | 1272 | 9.31 | 1153 | 97.74 | 92.00 |
| 9 | S9 | 500 | (100:100) | 1099 | 15.29 | 931 | 97.53 | 92.29 |
| 10 | S10 | 500 | (100:100) | 1159 | 13.04 | 1008 | 97.67 | 92.30 |
| 11 | S11 | 500 | (100:100) | 1468 | 14.44 | 1256 | 97.48 | 91.83 |
| 12 | S12 | 500 | (100:100) | 1235 | 8.89 | 1125 | 96.58 | 88.98 |
| 13 | S14 | 500 | (100:100) | 1237 | 9.75 | 1116 | 96.28 | 88.32 |
| 14 | S17 | 500 | (100:100) | 1134 | 10.24 | 1018 | 96.63 | 89.24 |
| 15 | S18 | 500 | (100:100) | 1173 | 9.30 | 1064 | 97.76 | 92.11 |
| 16 | S19 | 500 | (100:100) | 1088 | 11.45 | 964 | 97.01 | 89.67 |
| 17 | S20 | 500 | (100:100) | 1220 | 9.94 | 1099 | 96.56 | 89.03 |
| 18 | S21 | 500 | (100:100) | 1015 | 16.51 | 848 | 96.07 | 87.04 |
| 19 | S22 | 500 | (100:100) | 1194 | 13.93 | 1028 | 98.18 | 94.17 |
| 20 | S23 | 500 | (100:100) | 1056 | 18.80 | 858 | 97.52 | 92.65 |
| 21 | S25 | 500 | (125:125) | 1668 | 6.69 | 1556 | 96.30 | 92.37 |
| 22 | S26 | 500 | (100:100) | 1120 | 28.52 | 800 | 95.58 | 87.75 |
| 23 | S27 | 500 | (125:125) | 1263 | 8.71 | 1153 | 95.90 | 91.63 |
| 24 | S28 | 500 | (125:125) | 1536 | 5.95 | 1444 | 96.39 | 92.50 |
| 25 | S29 | 500 | (100:100) | 1212 | 9.15 | 1101 | 96.39 | 88.50 |
| 26 | S30 | 500 | (100:100) | 1215 | 14.90 | 1034 | 94.95 | 85.49 |
| 27 | S31 | 500 | (100:100) | 1821 | 15.68 | 1535 | 97.37 | 91.55 |
| 28 | S32 | 500 | (125:125) | 1417 | 5.86 | 1334 | 96.80 | 93.42 |
| 29 | S34 | 500 | (100:100) | 1043 | 9.18 | 947 | 97.66 | 91.64 |
| 30 | S35 | 500 | (100:100) | 1525 | 10.52 | 1364 | 97.24 | 90.42 |
| 31 | S40 | 500 | (125:125) | 1105 | 7.63 | 1021 | 96.32 | 92.49 |
| 32 | S41 | 500 | (125:125) | 1116 | 4.74 | 1063 | 96.39 | 92.50 |
| 33 | S42 | 500 | (125:125) | 1191 | 5.19 | 1129 | 96.42 | 92.58 |
| 34 | S43 | 500 | (125:125) | 943 | 4.25 | 903 | 96.35 | 92.41 |
| 35 | S44 | 500 | (125:125) | 1309 | 7.53 | 1211 | 96.37 | 92.56 |
| 36 | S45 | 500 | (125:125) | 1075 | 4.41 | 1027 | 96.31 | 92.33 |
表选项
2.2 最大生长速率GWAS分析结果 如图 1和图 2所示,分别测定原始菌株、单独培养分离株和混合培养分离株的最大生长速率,11株大肠杆菌 (E30,E10,E41,E12,E11,E3,E5,E32,E23,E44,E22) 和8株金黄色葡萄球菌 (S2,S30,S23,S18,S19,S45,S25,S26) 单独培养分离株的生长速率显著高于原始菌株。这些数据与实验室条件能提高生长速率的假设一致。6株大肠杆菌 (E17,E18,E21,E45,E12,E44) 和16株金黄色葡萄球菌 (S42,S5,S7,S8,S6,S20,S14,S11,S22,S21,S12,S23,S10,S29,S25,S26) 混合培养分离株的生长速率比原始菌株和单独培养分离株显著要快,这可能是混合培养过程中生长适应的结果。3株金黄色葡萄球菌 (S32,S34,S35) 混合培养分离株比单独培养分离株和原始株慢很多。
 |
| 图 1. 大肠杆菌培养前后最大生长速率比较 Figure 1. Maximum growth rates for E. coli strains after co-culture and monoculture. A: original strains; B: monoculture; C: co-culture; means followed by same letters (a, b, c, ab) were not significantly different at p≤0.05 in statistical analysis of each strain (Duncan's multiple range test) |
| 图选项 |
 |
| 图 2. 金黄色葡萄球菌培养前后最大生长速率比较 Figure 2. Maximum growth rates for S. aureus strains after co-culture and monoculture. A: original strains; B: monoculture; C: co-culture; means followed by same letters (a, b, c, ab) were not significantly different at p≤0.05 in statistical analysis of each strain (Duncan's multiple range test) |
| 图选项 |
利用最大生长速率数据进行GWAS分析,数据显示162个SNP影响到大肠杆菌原始株与共培养菌株的生长,36个SNP影响大肠杆菌菌株在单独培养和共培养的生长。总共有85个SNP影响金黄色葡萄球菌的原始菌株与单独培养分离株。
对影响原始株与共培养分离大肠杆菌,单独培养与共培养分离株大肠杆菌,和原始株与单独培养金黄色葡萄球菌最大生长速率的基因进行功能注释,其中5个基因在之前文献中已有报道,分别是rpoB[13, 23-24],rpoC[23]在之前的进化实验中确定为影响RNA转录的调控;另外3个分别是负责监管RNA合成的relA[13],影响甲硫氨酸生物合成的metL[24],和编码苹果酸脱氢酶、参与碳水化合物的分解代谢的mdh[25]。
2.3 不同时间点菌体数量GWAS分析结果 我们使用36对大肠杆菌和金黄色葡萄球菌菌株在培养过程中不同时间点的数量来确定不同的表型与基因多态性之间的关系。曼哈顿图显示了在GWAS分析结果中重要的SNP位点。大肠杆菌中65533个SNP位点中,有706个与生长性能相关 (P < 0.05)。金黄色葡萄球菌的86516个SNP位点中,129个与生长性能相关 (P < 0.05)。在混合培养中,609个SNP位点与大肠杆菌的生长有关 (图 3),17个SNP位点和金黄色葡萄球菌的生长有关 (图 4)。在单独培养中,97个SNP位点与大肠杆菌的生长有关 (图 5),112个SNP位点与金黄色葡萄球菌的生长有关 (图 6)。在大肠杆菌混合培养的所有时间点中,第1个时间点得到的SNP数量在所有时间点中最多,共有541个。
 |
| 图 3. 混合培养大肠杆菌显著SNP曼哈顿图 Figure 3. Manhattan plots of GWAS results for E. coli grown in co-culture. Genomic location was plotted against ?log10(P), with 609 SNPs identified at multiple time points; each dot corresponds to a single SNP. A: time point 1; B: time point 2; C: time point 3; D: time point 15; E: time point 16. |
| 图选项 |
 |
| 图 4. 混合培养金黄色葡萄球菌显著SNP曼哈顿图 Figure 4. Manhattan plots of GWAS results for S. aureus in co-culture. Genomic location is plotted against ?log10(P), with 112 SNPs identified at multiple time points; each dot corresponds to a single SNP. A: time point 4; B: time point 7; C: time point 10. |
| 图选项 |
 |
| 图 5. 单独培养大肠杆菌显著SNP曼哈顿图 Figure 5. Manhattan plots of GWAS results for E.coli grown in monoculture. Genomic location was plotted against ?log10(P), with 97 SNPs identified at multiple time points; each dot corresponds to a single SNP. |
| 图选项 |
 |
| 图 6. 单独培养金黄色葡萄球菌显著SNP曼哈顿图 Figure 6. Manhattan plots of GWAS results for S. aureus in monoculture. Genomic location is plotted against ?log10(P), with 17 SNPs identified at multiple time points; each dot corresponds to a single SNP. A: time point 1; B: time point 4; C: time point 6; D: time point 8; E: time point 16. |
| 图选项 |
SNP分布在整个基因组的基因上或者在假设的基因和基因间区域参与代谢调控。在大肠杆菌中,混合培养确定了60个显著SNP (第1个时间点56个,第2个时间点2个,第3个时间点2个)。在金黄色葡萄球菌的混合培养中,第1个时间点只确定了24个,第7个时间点之后确定了6个显著SNP,在单独培养中观察到39个显著SNP (第1个时间点2个,第4个时间点17个,第6个时间点1个,第16个时间点19个)。
对影响不同时间点细菌数量变化的显著SNP进行功能注释,大肠杆菌SNP的13个基因在之前的研究中已有报道。其中2个,murE[23]和phoQ[25]分别在适应以丙三醇为基础的培养环境和异丁醇压力下影响肽聚糖代谢。topA[8]基因编码DNA拓扑异构酶,被用在进化实验的评估上。ydcI[13],rpoS[13, 26]和rpoB[13, 23-24] 3个基因被证实影响RNA的转录调控。在本次研究中,识别的7个基因也与蛋白质的合成有关,这也证明了本实验方法的有效性。
3 讨论 新一代测序技术使微生物基因组综合分析成为可能[27]。而且,WGS促进了大肠杆菌中各种表型进化的多个代谢途径的发现[24]。有研究通过基因组重测序发现,大肠杆菌在进化实验过程中的多个进化提高了对异丁醇的耐受性[25]。S. aureus NCTC 8325是21世纪初期首批进行基因组测序的菌株之一。这项研究覆盖了836个片段序列,3206852 bp在公共数据库中可用。
GWAS在动植物的研究尤其是人类医学方面已经被广泛应用[14, 28],但是很少被应用于细菌方面[29]。虽然这样的研究还在起步阶段,但早期研究表明这种方法在影响表型多样性位点的识别方面是可靠的。因此,GWAS可以作为一个发现新细菌临床相关特征的强大工具[30]。当菌株生活环境中选择压力等附加条件改变时,应用本研究中的进化方法或许可以更好地理解其中的生态相互作用。尽管全基因组关联研究中存在多重检验等问题,但本实验采用相应的校正方法对检验结果的P值进行校正,使P值小于0.05,降低了假阳性率[14, 31]。
在本研究中,我们用短期实验室进化实验和基因组测序的方法研究大肠杆菌和金黄色葡萄球菌适应性进化机制,将多对大肠杆菌和金黄色葡萄球菌菌株在同一培养基中共同培养,观察两种细菌增长的动态变化,力求找到调节种群间相互作用的目的基因。据了解,这是首次将GWAS应用在大肠杆菌和金黄色葡萄球菌共培养的研究上。
Lawrence等的研究[2]表明,很多物种会改变其他物种的生存环境,产生大量不可预知的进化结果。该研究假设自然界中普遍存在一些物种进化到可以消耗其他物种产生的废弃物的程度[6]。LaCroix等的研究[24]表明实验室自适应进化实验可以用来检测导致大肠杆菌表型优化的突变。
物种在变化环境中的适应性进化机制,有助于我们合理预测细菌对环境的适应性变化,提出有效的抗菌治疗方案等[6]。简单的实验设计能够明确这项研究中的因果关系。利用未知相互作用的物种构建的人工微生物群落为解决关于自然交互起源的许多重要问题提供平台[32]。这项研究的发现对于详细了解传染病的传播动力学、微生物群落的功能组建等方面都将是至关重要的。同时该发现也会促进复杂微生物系统间相互作用关系及关键参数的发现[33]。由于倾向于使用相同资源的物种共同生长时也可能会产生竞争关系,因此,细菌种间关系到底是竞争还是合作的关键可能在于它们长期互相适应的潜力和生态位的重叠程度[34]。
在本次实验中,我们用WGS的方法来分析这36株大肠杆菌和36株金黄色葡萄球菌,得到了基因组中大量的SNP位点 (大肠杆菌高质量数据28.45 G,金黄色葡萄球菌40.84 G),其中多数是新发现的。另外,在基因注释过程中确定了大量可以作为遗传标记预测表型性状变化的SNP位点。GWAS被应用于分析大肠杆菌和金黄色葡萄球菌的基因功能,包括信号导致菌株之间的遗传分化。本研究中混合培养和单独培养都检测到与生长相关的显著基因,表明了GWAS在研究细菌互作进化机制方面的潜力。
参考文献
| [1] | Bell TH, Callender KL, Whyte LG, Greer CW. Microbial competition in polar soils: a review of an understudied but potentially important control on productivity.Biology, 2013, 2(2): 533–554DOI:10.3390/biology2020533. |
| [2] | Lawrence D, Fiegna F, Behrends V, Bundy JG, Phillimore AB, Bell T, Barraclough TG. Species interactions alter evolutionary responses to a novel environment.PLoS Biology, 2012, 10(5): 2794–2804. |
| [3] | Brown JH, Whitham TG, Morgan Ernest SK, Gehring CA. Complex species interactions and the dynamics of ecological systems: long-term experiments.Science, 2001, 293(5530): 643–650DOI:10.1126/science.293.5530.643. |
| [4] | Brockhurst MA, Morgan AD, Rainey PB, Buckling A. Population mixing accelerates coevolution.Ecology Letters, 2003, 6(11): 975–979DOI:10.1046/j.1461-0248.2003.00531.x. |
| [5] | Schluter D, Price TD, Grant PR. Ecological character displacement in darwin's finches.Science, 1985, 227(4690): 1056–1059DOI:10.1126/science.227.4690.1056. |
| [6] | Turcotte MM, Corrin MSC, Johnson MTJ. Adaptive evolution in ecological communities.PLoS Biology, 2012, 10(5): e1001332DOI:10.1371/journal.pbio.1001332. |
| [7] | Plata G, Henry CS, Vitkup D. Long-term phenotypic evolution of bacteria.Nature, 2015, 517(7534): 369–372. |
| [8] | Barrick JE, Lenski RE. Genome dynamics during experimental evolution.Nature Reviews Genetics, 2013, 14(12): 827–839DOI:10.1038/nrg3564. |
| [9] | Gouin A, Legeai F, Nouhaud P, Whibley A, Simon JC, Lemaitre C. Whole-genome re-sequencing of non-model organisms: lessons from unmapped reads.Heredity, 2015, 114(5): 494–501DOI:10.1038/hdy.2014.85. |
| [10] | Berscheid A, Sass P, Weber-Lassalle K, Cheung AL, Bierbaum G. Revisiting the genomes of the Staphylococcus aureus strains NCTC 8325 and RN4220.International Journal of Medical Microbiology, 2012, 302(2): 84–87DOI:10.1016/j.ijmm.2012.01.002. |
| [11] | Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, Mohaideen N, Ioerger TR, Sacchettini JC, Lipsitch M, Flynn JL, Fortune SM. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection.Nature Genetics, 2011, 43(5): 482–486DOI:10.1038/ng.811. |
| [12] | Price JR, Didelot X, Crook DW, Llewelyn MJ, Paul J. Whole genome sequencing in the prevention and control of Staphylococcus aureus infection.Journal of Hospital Infection, 2013, 83(1): 14–21DOI:10.1016/j.jhin.2012.10.003. |
| [13] | Conrad TM, Joyce AR, Applebee MK, Barrett CL, Xie B, Gao Y, Palsson B?. Whole-genome resequencing of Escherichia coli K-12 MG1655 undergoing short-term laboratory evolution in lactate minimal media reveals flexible selection of adaptive mutations.Genome Biology, 2009, 10(10): R118DOI:10.1186/gb-2009-10-10-r118. |
| [14] | Wang JY, Wang HX, Chi RB, Guo JF, Wu Y. Progresses in research of genome-wide association studies in livestock and poultry.Scientia Agricultura Sinica, 2013, 46(4): 819–829(in Chinese).王继英, 王海霞, 迟瑞宾, 郭建凤, 武英. 全基因组关联分析在畜禽中的研究进展.中国农业科学, 2013, 46(4): 819–829. |
| [15] | Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis.Bioinformatics, 2007, 23(10): 1294–1296DOI:10.1093/bioinformatics/btm108. |
| [16] | Sheppard SK, Didelot X, Meric G, Torralbo A, Jolley KA, Kelly DJ, Bentley SD, Maiden MCJ, Parkhill J, Falush D. Genome-wide association study identifies vitamin B5 biosynthesis as a host specificity factor in Campylobacter.Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(29): 11923–11927DOI:10.1073/pnas.1305559110. |
| [17] | Foster KR, Bell T. Competition, not cooperation, dominates interactions among culturable microbial species.Current Biology, 2012, 22(19): 1845–1850DOI:10.1016/j.cub.2012.08.005. |
| [18] | Abasht B, Lamont SJ. Genome-wide association analysis reveals cryptic alleles as an important factor in heterosis for fatness in chicken F2 population.Animal Genetics, 2007, 38(5): 491–498DOI:10.1111/j.1365-2052.2007.01642.x. |
| [19] | Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, Meng DZ, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JDG, Michael T, Nemri A, Roux F, Salt DE, Tang CL, Todesco M, Traw MB, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines.Nature, 2010, 465(7298): 627–631DOI:10.1038/nature08800. |
| [20] | 张莉. 绵羊肉用性状全基因组关联分析. 中国农业科学院博士学位论文, 2013. |
| [21] | Prest EI, Hammes F, K?tzsch S, van Loosdrecht MCM, Vrouwenvelder JS. Monitoring microbiological changes in drinking water systems using a fast and reproducible flow cytometric method.Water Research, 2013, 47(19): 7131–7142DOI:10.1016/j.watres.2013.07.051. |
| [22] | Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping.Genetics, 1994, 138(3): 963–971. |
| [23] | Herring CD, Raghunathan A, Honisch C, Patel T, Applebee MK, Joyce AR, Albert TJ, Blattner FR, van den Boom D, Cantor CR, Palsson B?. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale.Nature Genetics, 2006, 38(12): 1406–1412DOI:10.1038/ng1906. |
| [24] | LaCroix RA, Sandberg TE, O'Brien EJ, Utrilla J, Ebrahim A, Guzman GI, Szubin R, Palsson BO, Feist AM. Use of adaptive laboratory evolution to discover key mutations enabling rapid growth of Escherichia coli K-12 MG1655 on glucose minimal medium.Applied and Environmental Microbiology, 2015, 81(1): 17–30DOI:10.1128/AEM.02246-14. |
| [25] | Minty JJ, Lesnefsky AA, Lin FM, Chen Y, Zaroff TA, Veloso AB, Xie B, McConnell CA, Ward RJ, Schwartz DR, Rouillard JM, Gao Y, Gulari E, Lin XN. Evolution combined with genomic study elucidates genetic bases of isobutanol tolerance in Escherichia coli.Microbial Cell Factories, 2011, 10(1): 18DOI:10.1186/1475-2859-10-18. |
| [26] | Honisch C, Raghunathan A, Cantor CR, Palsson B?, van den Boom D. High-throughput mutation detection underlying adaptive evolution of Escherichia coli-K12.Genome Research, 2004, 14(12): 2495–2502DOI:10.1101/gr.2977704. |
| [27] | Forde BM, O'Toole PW. Next-generation sequencing technologies and their impact on microbial genomics.Briefings in Functional Genomics, 2013, 12(5): 440–453DOI:10.1093/bfgp/els062. |
| [28] | Zhou JP, Pei ZY, Chen YB, Chen RS. Strategies of genome-wide association study based on high-throughput sequencing.Hereditas (Beijing), 2014, 36(11): 1099–1111(in Chinese).周家蓬, 裴智勇, 陈禹保, 陈润生. 基于高通量测序的全基因组关联研究策略.遗传, 2014, 36(11): 1099–1111. |
| [29] | Alam MT, Petit RA Ⅲ, Crispell EK, Thornton TA, Conneely KN, Jiang YX, Satola SW, Read TD. Dissecting vancomycin-intermediate resistance in Staphylococcus aureus using genome-wide association.Genome Biology and Evolution, 2014, 6(5): 1174–1185DOI:10.1093/gbe/evu092. |
| [30] | Read TD, Massey RC. Characterizing the genetic basis of bacterial phenotypes using genome-wide association studies: a new direction for bacteriology.Genome Medicine, 2014, 6(11): 109DOI:10.1186/s13073-014-0109-z. |
| [31] | Pan DD, Li ZB, Zhang W, Li QZ. The review of genome-wide association studies.Chinese Journal of Applied Probability and Statistics, 2014, 30(1): 84–103(in Chinese).潘东东, 李正帮, 张维, 李启寨. 全基因组关联研究综述.应用概率统计, 2014, 30(1): 84–103. |
| [32] | Andrade-Domínguez A, Salazar E, Vargas-Lagunas MDC, Kolter R, Encarnación S. Eco-evolutionary feedbacks drive species interactions.The ISME Journal, 2014, 8(5): 1041–1054DOI:10.1038/ismej.2013.208. |
| [33] | Hibbing ME, Fuqua C, Parsek MR, Peterson SB. Bacterial competition: surviving and thriving in the microbial jungle.Nature Reviews Microbiology, 2010, 8(1): 15–25DOI:10.1038/nrmicro2259. |
| [34] | Ren D, Madsen JS, S?rensen SJ, Burm?lle M. High prevalence of biofilm synergy among bacterial soil isolates in cocultures indicates bacterial interspecific cooperation.The ISME Journal, 2015, 9(1): 81–89DOI:10.1038/ismej.2014.96. |
