 , 王逸1
, 王逸1 , 周丽华2, 江峰3, 袁勇1
, 周丽华2, 江峰3, 袁勇11. 广东工业大学环境科学与工程学院, 广州 510006;
2. 广东工业大学生物医药学院, 广州 510006;
3. 陆军研究院工程设计研究所, 南京 210000
收稿日期: 2020-12-30; 修回日期: 2021-03-01; 录用日期: 2021-03-01
基金项目: 国家自然科学基金(No.41907122,51678162,41877045,21876032)
作者简介: 刘创创(1994-), 男, E-mail: sasedliu@qq.com
通讯作者(责任作者): 王逸, E-mail: wangyiii@gdut.edu.cn
摘要:锆基纳米材料具有很好的物理化学稳定性及较高的比表面积,适用于解决传统过渡金属基催化剂活化过硫酸盐时金属浸出等问题.本研究将氯化锆-尿素络合物通过热解法合成了一种氮掺杂碳/氧化锆复合材料(N-C/ZrO2),用于活化过硫酸盐降解水中苯酚,并采用SEM、XRD、XPS和FTIR技术对N-C/ZrO2进行表征.结果表明,N-C/ZrO2催化材料是直径小于50 nm的纳米颗粒,且氮掺杂量为3.6%~4.9%;在pH=6~7的硼酸缓冲体系中,N-C/ZrO2催化过硫酸盐可在30 min内使水中苯酚(20 mg·L-1)的降解率达99%,且催化剂具有良好的化学稳定性和可重复利用性.淬灭和电子顺磁共振(EPR)结果表明,在N-C/ZrO2/PMS体系中,苯酚的降解是以1O2为主要氧化物质的非自由基氧化过程.本研究为Zr基纳米材料在活化过硫酸盐降解污染物领域的应用提供了理论基础.
关键词:过一硫酸盐二氧化锆苯酚单线态氧
The mechanism on activation of peroxymonosulfate using nano N-C/ZrO2 for phenol degradation
LIU Chuangchuang1
, WANG Yi1, ZHOU Lihua2, JIANG Feng3, YUAN Yong11. School of Environmental Science and Engineering, Guangdong University of Technology, Guangzhou 510006;
2. School of Biomedical and Pharmaceutical Sciences, Guangdong University of Technology, Guangzhou 510006;
3. Army Institute of Engineering Design, Nanjing 210000
Received 30 December 2020; received in revised from 1 March 2021; accepted 1 March 2021
Abstract: Zirconium-based nanomaterials with high physical and chemical stability and surface area, have emerged as catalyst for persulfate activation with less extraction than that of traditional transition metal catalyst. A nano nitride-doped carbon-zirconia (N-C/ZrO2) was fabricated via a facile thermal pyrolysis method using zirconium chloride (ZrCl4) and urea as a zirconia precursor and nitride-carbon source, respectively. The N-C/ZrO2 was used to catalyze persulfate activation for phenol degradation and its physicochemical properties were exanimated by Scanning electron microscope (SEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FTIR). The result showed that the as-fabricated N-C/ZrO2 catalyst with 3.6%~4.9% N-doping and a particle size of < 50 nm revealed an excellent catalytic capability with almost removal (99%) of 20 mg·L-1 phenol in 30 min in borate buffer solution (pH=6~7). Moreover, the N-C/ZrO2 catalyst exhibited good chemical stability after five successive degradation cycles. The singlet oxygen (1O2) as the dominant reactive species in phenol degradation was confirmed by electron paramagnetic resonance spectroscopy (EPR) and scavenger experiments. This study demonstrated that the N-doped Zr-based nanomaterials in activating persulfate can be considered as a promising technology for the removal of aqueous organic contaminant.
Keywords: peroxymonosulfatezirconiaphenolsinglet oxygen
1 引言(Introduction)高级氧化技术(AOPs)对有机污染物有很好的降解效果, 在AOPs中有高活性氧, 如羟基自由基(·OH)、硫酸根自由基(SO4·-)和超氧自由基(O2·-)等, 能将污染化合物降解为无害产物(Lei et al., 2015).过一硫酸盐(PMS)因产生的·OH和SO4·-具有高氧化还原电位而广受关注(Zou et al., 2013;许芬等, 2018).非均相催化剂是活化PMS的重要方式, 其中, Co3O4(Anipsitakis et al., 2005)、CuFe2O4(Zhang et al., 2013)、ZnO@Fe3O4(赵浩迪等, 2018)等过渡金属氧化物是研究比较多的非均相催化剂.但过渡金属氧化物的使用面临金属离子浸出问题(Zhu et al., 2013), 从而会影响其使用效果, 因此, 金属离子应以稳定形态与支持材料结合.
二氧化锆(ZrO2)是一种重要的多功能宽带隙p型半导体, 其表面上具有大量的氧空位结构.纳米ZrO2具有良好的耐化学性、硬度和离子导电性(Manna et al., 2010).由于离子传导性和表面富氧缺陷, ZrO2可用于催化剂或作为催化剂载体(Ma, 2011).研究表明, 纳米ZrO2对水体中的砷(Shao et al., 2019)、氟(Yu et al., 2019)、磷(Liu et al., 2008)、铬和钒离子(Wu et al., 2019)具有良好吸附性, 而少有****将其用于活化PMS.将纳米ZrO2用于非均相催化剂合成, 虽然富氧缺陷结构有助于催化剂形成单线态氧(Wang et al., 2016), 但较差的电子导电性将限制其用于非自由基途径的污染物降解(Sun et al., 2015;Huang et al., 2019).碳材料因具有优异性能, 如超高的空隙体积及比表面积、特有的电学性能、较高的化学热稳定性及丰富的表面基团而常被用作催化剂掺杂材料.此外, 边缘氮(edge N)在PMS活化反应中具有出色的催化性能(Guo et al., 2016;Wang et al., 2019), 这是由于N原子能有效促进相邻碳原子的转移, 使碳原子具有较高的电荷密度(Ding et al., 2020). 因此, C和N掺杂催化剂被广泛用于PMS活化(周雪君等, 2015).
基于此, 本文以ZrCl4为前驱体, 尿素作为C和N源, 利用高温热解法合成复合纳米材料.同时, 采用场发扫描电镜(SEM)、X射线衍射仪(XRD)、X射线光电子能谱(XPS)和傅立叶变换红外光谱仪(FTIR)探究复合材料的结构特征.由于水中内分泌干扰物(如雌酮、17β-雌二醇、17α-乙炔雌二醇等)通常具有酚类结构, 而酚类结构被认为是其内分泌干扰作用的主要活性官能团, 因此, 本文选取具有最简单酚类结构的苯酚作为目标污染物.通过改变尿素掺杂比、反应体系pH方式考察催化剂性能, 通过酸洗及重复实验考察材料的稳定性, 并通过自由基淬灭实验和电子自旋共振仪(EPR)等方法探究参与反应的主要活性物种和降解机理.
2 材料与方法(Materials and methods)2.1 材料与试剂D-无水葡萄糖(GR)、氯化锆(Ⅳ)(99.5%)、四氯化铪(99.99%)、二氧化锆(Ⅳ)、氮化锆(99%)、四氯化钛(AR, 99.0%)、苯甲酸、对羟基苯甲酸和2, 2-联氨-双(3-乙基苯并噻唑啉-6-磺酸)二胺盐(ABTS)购自上海麦克林生化科技有限公司, 尿素(AR, 99%)、甲醇(MeOH)、叔丁醇(TBA)、糠醇(FFA)、过硫酸氢钾(KHSO5·0.5KHSO4·0.5K2SO4 PMS)、苯酚(AR)、硼酸(99.5%)、氢氧化钾(AR, 90%)、双酚A(BPA)、5, 5-二甲基-1-吡咯啉-N-氧化物(DMPO)、2, 2, 6, 6-四甲基哌啶(TEMP)和对苯醌(p-BQ)购自阿拉丁试剂有限公司.实验中所用的去离子水由Unique-R20仪器制备, 其中, 超纯水的电阻率为18.2 MΩ·cm-1.
2.2 纳米N-C/ZrO2催化剂制备将3 g氯化锆(Ⅳ)加入到6 mL无水乙醇中, 待溶解完全后, 向溶液中加入3 g尿素, 搅拌加超声30 min溶解至形成无色均相液体, 并放置过夜.将液体置于管式炉中, 出口用洗气瓶液封, 防止空气回流.通氩气(99.999%高纯度氩气)1 h直至完全排出管中空气后停止通气, 以10 ℃·min-1的速度加热管式炉, 至800 ℃后维持3 h, 自然降温.待管式炉降至室温再次通氩气30 min, 得到纳米N-C/ZrO2.
2.3 材料测试表征制备所得的N-C/ZrO2催化剂通过场发扫描电镜(FESEM, 日本, JEOL JSM-6700F)观察形貌, 通过X-射线粉末衍射仪(XRD, 德国, Bruker D8 Advanced)和X-射线光电子能谱(XPS, 德国, Thermo Fisher Escalab 250Xi)表征材料的结构. XPS刻蚀实验采用氩离子刻蚀, 刻蚀电压为2000 eV, 刻蚀区域为1.5 mm.通过傅立叶变换红外光谱仪(FTIR, 美国, Thermofisher Nicolet IS50)探究催化剂表面官能团和化学键.
2.4 苯酚降解批实验降解实验在100 mL烧杯中进行, 该烧杯装有40 mL指定浓度(10、20、30或40 mg·L-1)的苯酚溶液, 置于(25±2) ℃室温下进行实验.首先配置指定浓度(10、20、30或40 mg·L-1)的苯酚溶液, 为防止pH改变影响实验结果, 反应体系根据不同pH缓冲需要, 利用20 mmol·L-1醋酸盐或硼酸盐调节溶液到一定pH(4.0、5.0、6.0、6.5、7.0、8.0或8.5), 随后加入催化剂, 超声5 min后加入200 mg·L-1的PMS以开始降解过程.烧杯中放置搅拌子, 转速为550 r·min-1.定点取样1.0 mL, 加入10 μL硫代硫酸钠(0.05 mol·L-1)中止反应, 之后用0.22 μm有机滤膜过滤, 装入色谱进样瓶, 最后用高效液相色谱仪测定剩余苯酚浓度.所有实验均做平行样, 图中数据均为平均值, 误差棒代表平均值的标准偏差.
2.5 检测分析方法2.5.1 苯酚检测苯酚在紫外检测波长270 nm处有较强特征峰, 因此, 采用高效液相色谱法(HPLC)检测水中酚浓度.其中, 高效液相色谱仪型号为岛津LC-16, 色谱柱为C18柱(4.6 μm×100 mm×275 mm), 色谱条件:流动相为水和乙腈(50%∶50%, 体积比), 进样体积为10 μL, 柱箱温度为40 ℃, 流速为0.8 mL·min-1.
2.5.2 PMS浓度检测采用ABTS法检测溶液中PMS浓度, 具体操作为:用去离子水配置2 mmol·L-1 ABTS溶液备用;将14.5 mL乙酸、34 g乙酸钠加入到超纯水中配成500 mL pH=4的缓冲溶液备用;在10 mL比色管中加入1 mL PMS样品、1 mL ABTS溶液、1 mL pH缓冲液和20 μL碘溶液(1.0 mmol·L-1), 用去离子水稀释到10 mL;当采用2 μmol·L-1碘离子作催化剂时, ABTS·+在415 nm处有最大吸收值(34000 L·mol-1·cm-1).如式(1)所示, 关键计量因子为2, 1 mol PMS和2 mol ABTS生成2 mol ABTS·+.
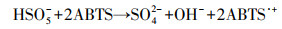 | (1) |
2.5.4 其他污染物浓度检测对羟基苯甲酸、苯甲酸和双酚A均用HPLC进行检测, 根据所检测物质的性质选择适合的紫外灯波长和流动相.其中, 检测羟基苯甲酸时选用波长为246 nm, 流动相为甲醇和0.01 mol·L-1磷酸(20%∶80%, 体积比);检测苯甲酸时选用检测器波长为224 nm, 流动相为甲醇和0.02 mol·L-1乙酸铵(10%∶90%, 体积比);检测双酚A时检测波长为276 nm, 流动相为甲醇和超纯水(65%∶35%, 体积比).
2.5.5 Zr4+的浓度检测水中Zr4+浓度使用电感耦合等离子体质谱(ICP-MS, 德国, Thermo Fisher ICAP RQ)进行检测.具体操作为:待苯酚降解反应结束后, 从反应溶液中取10 mL溶液过0.22 μm有机滤膜至进样瓶中, 随即用ICP-MS检测其Zr4+浓度.
2.6 自由基抑制实验在淬灭实验中, 将甲醇(MeOH)和叔丁醇(TBA)作为硫酸根自由基(SO4·-)和羟基自由基(·OH)的淬灭剂, 对苯醌(p-BQ)作为超氧自由基(O2·-)淬灭剂, 糠醇(FFA)和叠氮化钠(NaN3)作为单线态氧自由基捕获剂.实验条件为在2.4节实验基础上加入淬灭剂, 间隔一定反应时间取样1 mL过0.22 μm滤膜后加入硫代硫酸钠中止反应, 混匀后用HPLC检测苯酚浓度.
3 结果与讨论(Results and discussion)3.1 N-C/ZrO2催化剂的表征3.1.1 SEM分析利用SEM观察纳米N-C/ZrO2表面形貌结构, 发现催化剂为纳米颗粒且直径小于50 nm(图 1a和1b), 说明所合成的材料具有较大的比表面积, 在水中具有很好的分散性.如图 1b、1c所示, 材料受高温影响, 部分颗粒不同程度地烧结在一起.从SEM mapping图(1d~1g)可以看出, 元素Zr和O具有很高的匹配度, 且与图 1c中展示的材料形态完全一致;其次, C和N两种元素分布均匀.综合而言, 合成的材料中4种元素分布均匀, C和N元素较好地掺杂到材料中.
图 1(Fig. 1)
 |
| 图 1 纳米N-C/ZrO2复合物的SEM图(a~c)及对应的元素图(d~g) Fig. 1SEM images of nano N-C/ZrO2 composite(a~c) and element mapping(d~g) |
3.1.2 XRD谱图分析如图 2a所示, 以尿素为C源的材料与商用ZrO2及以葡萄糖为C源合成的掺杂材料不同. N-C/ZrO2和以葡萄糖为C源合成的材料在(-111)、(111)、(002)、(200)、(220)、(302)、(311)晶面与m-ZrO2匹配较好, 在(101)和(211)等晶面与t-ZrO2(四方氧化锆)匹配较好.说明采用葡萄糖合成的催化剂也具备ZrO2为基底的复合结构, 此外还说明N-C/ZrO2中含m-ZrO2和t-ZrO2两种形态的氧化锆.
图 2(Fig. 2)
 |
| 图 2 纳米N-C/ZrO2的XRD光谱图(a)及Zr 3d(b)、C 1s(c)、O 1s(d)和N 1s(e)的高分辨率XPS Fig. 2XRD spectrum(a) and high resolution XPS narrow scans for Zr 3d(b), C 1s (c), O 1s(d) and N 1 s(e) |
3.1.3 XPS谱图分析为了深入了解N和C原子在催化剂中的掺杂形态, 采用XPS对N-C/ZrO2进行分析, 图 2b~2e分别为Zr 3d、C 1s、O 1s和N 1s核心能级谱的解卷积分析, 图 3a的宽扫描谱说明材料的主要构成元素为Zr、O、C和N 4种元素.在图 2b中, Zr 3d精细峰有Zr 3d5/2(184.8 eV)和Zr 3d3/2(182.4 eV)强自旋轨道双峰, Zr 3d5/2-Zr 3d3/2自旋轨道分离为2.4 eV, 说明ZrO2存在Zr4+氧化状态(Poungchan et al., 2016). C 1s高分辨率谱图如图 2c所示, 在284.4~284.8 eV之间有一个主峰, 对应于sp2-杂化的石墨C原子, 较高结合能的微弱信号表明退火后材料中残留有C—N和C—O类官能团(Jeong et al., 2008). C 1s谱中284.6 eV处对应于sp2碳原子的一个主峰, 表明大多数碳原子构成了共轭蜂窝格结构(Sheng et al., 2011).由于N原子的引入导致类石墨结构的无序性, N-C/ZrO2的C 1s谱中仍被分配给石墨sp2碳原子尖峰, 向284.4~284.8 eV的更高结合能转移.表 1中N-C/ZrO2-3的碳原子比为40.28%, 而图 2a中N-C/ZrO2的XRD谱图没有明显的石墨峰, 说明合成材料中的碳大部分以无定形碳存在. N 1s窄峰如图 2e所示, 可以被拟合成4个峰, 分别位于398.3、400.0、401.0和403.0 eV处.其中, 位于较低结合能398.3和400.0 eV处的峰分别表示吡啶氮(pyridine N)和吡咯氮(pyrrole N), 有助于形成石墨烯层中带有一堆p电子的π-共轭体系;结合能为401.0 eV处的峰表示石墨氮(graphite N);在402.3~403.0 eV处的微弱峰通常归因于氧化氮(Xu et al., 2008).表 1中N-C/ZrO2-3的氮原子比为4.9%, 表明复合材料中氮掺杂量较高.N-C/ZrO2刻蚀10 nm和100 nm的XPS总谱与未刻蚀测得的XPS总谱对比完全一致(图 3b), 结合图 1 SEM谱图结果, 说明N-C/ZrO2为ZrO2基底N-C均匀掺杂结构.
图 3(Fig. 3)
 |
| 图 3 N-C/ZrO2 XPS全谱图(a)及刻蚀不同深度及反应后N-C/ZrO2 XPS全谱图(b) Fig. 3XPS survey of N-C/ZrO2(a) and XPS survey spectra of N-C/ZrO2 at different depths and after use(b) |
表 1(Table 1)
| 表 1 不同尿素比例合成的催化剂中各元素原子量比 Table 1 The percent of Zr, C, O and N of catalyst synthesized with different urea ratio | ||||||||||||||||||||||||
表 1 不同尿素比例合成的催化剂中各元素原子量比 Table 1 The percent of Zr, C, O and N of catalyst synthesized with different urea ratio
| ||||||||||||||||||||||||
3.1.4 FTIR光谱分析为了探究碳氮掺杂的N-C/ZrO2表面官能团和化学键结构, 采用FTIR对纳米N-C/ZrO2进行表征, 结果如图 4所示. 3427 cm-1处为O—H(Brindha et al., 2017);1616 cm-1处的峰是材料表面羧基及C=O特征伸缩振动峰(王莹等, 2017);在1460 cm-1处出现较微弱的C—N伸缩振动峰, 充分证明氮原子的掺杂结构(Zhang et al., 2017).在738 cm-1处尖锐的峰是m-ZrO2特征峰, 在510 cm-1处的宽带峰和584 cm-1处的肩峰归因于t-ZrO2的Zr—O振动(Hu et al., 2020), 这与XRD谱图(图 2a)中分析得到的两种氧化锆的结果是吻合的.
图 4(Fig. 4)
 |
| 图 4 使用前后N-C/ZrO2的FTIR图 Fig. 4FTIR spectra of N-C/ZrO2 before and after use |
3.2 N-C/ZrO2的催化性能3.2.1 N掺杂量的影响分别采用不同质量比例(3∶0、3∶1、3∶2、3∶3、3∶4)ZrCl4和尿素混合制备催化剂, 并依据尿素比例分别标记为N-C/ZrO2-0、N-C/ZrO2-1、N-C/ZrO2-2、N-C/ZrO2-3、N-C/ZrO2-4.当ZrCl4-尿素比为3∶0和3∶1时, 制备的催化剂对苯酚的去除率较低(图 5a).可能是因为较低的尿素浓度使得制备的催化剂N掺杂含量较低, 导致催化效果下降.而当尿素比例过高(3∶4)时, 由表 1可知, N-C/ZrO2-4中N的占比达9.54%, 而图 5a中N-C/ZrO2-4对苯酚的降解率为52.4%, 表明过高的N掺杂量不会带来更好的催化效果, 可能是由于过量的N掺杂破坏共价石墨电子体系的电荷平衡和电荷再分配造成的.ZrCl4与尿素的比例为3∶2与3∶3时, 30 min时苯酚的降解率均达99%以上.为节省原料, 后续实验所用催化剂均为ZrCl4-尿素质量比3∶3合成的催化剂.另外, 还将葡萄糖代替尿素与ZrCl4以2∶3质量比混合制备的催化剂作为无N掺杂对比.从XRD图(图 2a)可以发现, 葡萄糖-ZrCl4制备的催化剂为碳掺杂m-ZrO2基底复合材料.该催化剂对苯酚的降解效果如图 5d所示, 60 min时苯酚去除率仅为23.2%, 而N-C/ZrO2-0和N-C/ZrO2-1对苯酚的去除率分别仅为10.0%和6.5%, 可能是因为葡萄糖-ZrCl4(2∶3)中碳掺杂量高于N-C/ZrO2-0和N-C/ZrO2-1两种材料导致.因此, 一方面C掺杂有着不可替代的催化性能;另一方面, N掺杂对材料结构及催化性能有进一步的提升作用, 但催化性能的提升与N掺杂量并非成正比, 当催化剂中N掺杂量为3.65%~4.90%时催化剂催化性能最好.
图 5(Fig. 5)
 |
| 图 5 ZrCl4-尿素比例(a)、pH缓冲体系(b)、通O2和N2(c)对苯酚降解的影响和商用ZrC、ZrN、ZrO2及同族元素含Ti和Hf催化剂、葡萄糖替代尿素制备催化剂对比(d), 以及N-C/HfO2和N-C/TiO2 XRD光谱图(e)及降解不同浓度苯酚效果(f) ([苯酚]0=20 mg·L-1(a~d);[催化剂]0=250 mg·L-1(a~d, f);[PMS]0=200 mg·L-1(a~d, f);pH=6.5(a, c, d, f);25 ℃) Fig. 5Effect of different ZrCl4-urea radio(a), pH buffer system(b) and bubbling with O2 or N2 on degradation of phenol(c), comparison of commercial ZrC, ZrN, ZrO2 and Ti, Hf as congeners, and preparation with glucose instead of urea (d), XRD patterns of N-C/HfO2 and N-C/TiO2(e) and degradation of different concentration of phenol(f) |
3.2.2 反应体系pH的影响随着PMS的投入, 反应体系酸化, 在无缓冲体系实验中发现反应前后pH由6.8左右降至2.8左右.考虑到反应体系的稳定性与实验条件可控性, 利用20 mmol·L-1醋酸盐调节pH至4、5或用硼酸盐调节pH至6.0、6.5、7.0、8.0和8.5.本文中pH值均为缓冲体系加入PMS后体系初始pH. 经检测反应结束后溶液体系pH变化微弱.
如图 5b和图 6a所示, 在反应体系pH=4.0~8.5范围内, pH=5.0~7.0条件下能达到很好的降解效果, 苯酚去除速率常数为kobs=0.0845~0.1243 min-1.当pH < 5.0时降解效果略有下降, pH=4.0时, 苯酚去除速率常数为kobs=0.0530 min-1, 60 min时苯酚降解率为98.4%.而当pH>7.0为碱性环境时反应效果则有所下降, pH=8.5时, 苯酚去除速率常数下降至kobs=0.0114 min-1, 60 min时苯酚的降解率为82.8%.尽管1O2更倾向于在碱性条件下与酚类阴离子反应(Miller et al., 2005), 但N-C/ZrO2与PMS之间可能存在排斥力阻碍着相互作用, 抑制1O2的产生.此外, 将材料置于1 mol·L-1的HCl溶液中振荡1 h, 从振荡前后的XRD图(图 2a)和XPS图(图 6b)可见, N-C/ZrO2结构无任何变化.因此, 当pH较低时, 酸性条件下自由基与过硫酸氢根离子发生反应, 消耗了PMS导致降解效果有所下降(Deng et al., 2016).综上, 溶液pH对N-C/ZrO2/PMS体系是综合作用, 在pH=4.0~7.0范围内都有着不错的降解效果, 而碱性(pH>7.0)条件下降解效果有所下降.
图 6(Fig. 6)
 |
| 图 6 不同pH条件下苯酚降解动力学线性拟合图(a)、1 mol·L-1 HCl处理后N-C/ZrO2 XPS总谱图(b)、TOC去除效果(c)及重复实验苯酚降解率(d) ([苯酚]0=20 mg·L-1, [催化剂]0=250 mg·L-1, [PMS]0=200 mg·L-1, 25 ℃) Fig. 6Linear fitting diagram of phenol degradation kinetics under different pH(a), XPS survey of N-C/ZrO2 washing with 1 mol·L-1 HCl(b), removal effect of TOC(c) and catalytic stability test of N-C/ZrO2(d) |
3.2.3 氧含量的影响为了探究氧气是否为该体系起作用的必备条件, 反应中以曝氮气来维持无氧条件, 反应前通氮气10 min, 且反应中持续曝气, 制造无氧环境;另一组则在相同反应条件下一直通氧气;对照组为正常实验, 在不通气体条件下进行.两种条件下苯酚的降解结果如图 5c所示, 有无氧气的降解过程同对照组几乎相同, 反应常数差别极小, 表明N-C/ZrO2/PMS体系降解苯酚不受氧气影响, 反应在有氧和无氧条件下均能正常进行.
3.2.4 与其他催化材料比较本文还考察了几种含锆药剂的活化性能, 选择商用ZrO2、ZrC、ZrN作为含Zr元素物质对比, 发现相同条件下纯ZrO2几乎不能活化PMS降解苯酚, 苯酚降解率只有2.3%.这可能是因为一方面商用ZrO2为非纳米材料, 其比表面积远低于纳米材料;另一方面, 非纳米ZrO2活化活性位点非常少.而ZrN和ZrC对苯酚的降解率分别为10.4%和30.6%(图 5d).Zr的同族元素有常见的Ti和Hf, 因此, 将TiCl4和HfCl4按照2.2节纳米N-C/ZrO2催化剂制备中的方法合成N-C/TiO2和N-C/HfO2, 合成材料的XRD谱图如图 5e所示, 并与标准二氧化钛的XRD标准卡片(JCPD No.78-0050)和标准二氧化铪(JCPD No. 84-1285)进行对比, 可见N-C/TiO2和N-C/HfO2分别是以TiO2和HfO2为基底, 与N-C/ZrO2的掺杂方式类似.使用相同浓度的催化剂降解苯酚, 降解效果排序为N-C/ZrO2> N-C/HfO2> N-C/TiO2(图 5d).综上说明在同族元素中Zr元素的活化效果最理想, 而在含Zr材料中N-C/ZrO2的催化性能远优于其他材料.
3.2.5 N-C/ZrO2催化剂的稳定性从反应前后的XRD图(图 2a)和FTIR图(图 4), 以及1 mol·L-1 HCl振荡1 h实验中材料前后对比(图 2a和图 6b)发现, 材料结构及表面基团均无明显变化, 表明N-C/ZrO2具有较好的稳定性及耐酸性. pH=6.5的硼酸盐缓冲体系下, 当苯酚浓度为20 mg·L-1, N-C/ZrO2浓度为250 mg·L-1, PMS浓度为200 mg·L-1时, 25 min时TOC去除率为54.7%, 45 min时的去除率为58.3%, 最终60 min时的去除率为60.3%(图 6c).利用ICP-MS测得反应结束后Zr4+浸出浓度为0.245 μg·L-1, 说明N-C/ZrO2的使用不会对水体造成二次污染.而在改变污染物浓度负荷实验中(图 5f), 当污染物浓度降低为10 mg·L-1时, 15 min后苯酚完全去除;当苯酚浓度为30 mg·L-1和40 mg·L-1时, 60 min后苯酚去除率分别达到90.4%和70.3%.说明N-C/ZrO2/PMS体系对污染物浓度负荷的改变具有一定的承受能力.为了模拟真实污水处理环境, 每次反应结束采用直接向反应体系补充一定量缓冲液及催化剂, 随后重新投加苯酚和PMS的方法进行重复实验.图 6d中, 反应第2次苯酚去除效率为90.2%, 可能是由于降解产物附着在催化剂表面导致催化剂活性位点减少造成的;随后几次降解效率下降较少, 使用5次后苯酚降解率仍可达86.2%, 可能是因为N-C/ZrO2多次反应及PMS强氧化性使得材料表面碳结构被破坏, 导致活化性能降低.
3.3 N-C/ZrO2催化机理3.3.1 自由基分析在N-C/ZrO2/PMS体系中, 通过淬灭实验检测活性氧(ROS)的生成机理.淬灭剂甲醇(MeOH, k·OH=1.2×109~2.8×109 L·mol-1·s-1, kSO4·-=1.6×109~7.8×107 L·mol-1·s-1)(Neta et al., 1988)、叔丁醇(TBA, k·OH=3.8×109~7.6×109 L·mol-1·s-1, kSO4·-=4×109~8.1×107 L·mol-1·s-1)(Anipsitakis et al., 2004)对羟基自由基和硫酸根自由基有很好的淬灭作用. 从图 7a可以看出, 当甲醇和叔丁醇浓度为0.65 mol·L-1 ([MeOH/TBA]∶[PMS]=1000∶1)时, 对苯酚的降解几乎无影响.由此可以初步判断·OH和SO4·-非主要降解途径.
图 7(Fig. 7)
 |
| 图 7 MeOH/TBA(a)、FFA(b)、p-BQ(c)和NaN3(d)作为淬灭剂对N-C/ZrO2/PMS体系降解苯酚的影响 ([苯酚]0=20 mg·L-1, [催化剂]0=250 mg·L-1, [PMS]0=200 mg·L-1, pH=6.5, 25 ℃) Fig. 7Effect of MeOH and TBA(a), FFA (b), p-BQ (c) and NaN3 (d) as scavenger on the degradation of phenol by N-C/ZrO2/PMS system |
1O2作为一种温和氧化剂, 对酚类具有较强的氧化活性, 而与醇类基本不发生反应.为了鉴定体系中是否产生了1O2, 以叠氮化钠(NaN3)和糠醇(FFA)作为淬灭剂进行淬灭实验.从图 7d可以看到, 叠氮化钠浓度为1 mmol·L-1, 仅为PMS浓度的1.5倍, 苯酚降解率为36.7%;当NaN3浓度增加为1.5 mmol·L-1和2 mmol·L-1, 60 min时苯酚降解率分别为26.4%和16.2%.糠醇的淬灭速度低于叠氮化钠, 因此, 选择5、10和20 mmol·L-1 3种浓度进行实验, 3种浓度下苯酚的降解率依次为86.8%、77.2%和56.2%(图 7b). 由表 2可见, 除了浓度较高的2 mmol·L-1 NaN3实验组, 几种淬灭实验中苯酚降解速率常数的拟合度R2都较高, 综合说明N-C/ZrO2/PMS体系在降解苯酚过程中产生了大量单线态氧.
表 2(Table 2)
| 表 2 不同浓度FFA、p-BQ和NaN3条件下苯酚降解动力学速率常数 Table 2 Reaction rate constant of phenol degradation at different concentrations of FFA, p-BQ and NaN3 | ||||||||||||||||||||||||||||||||||||||||
表 2 不同浓度FFA、p-BQ和NaN3条件下苯酚降解动力学速率常数 Table 2 Reaction rate constant of phenol degradation at different concentrations of FFA, p-BQ and NaN3
| ||||||||||||||||||||||||||||||||||||||||
对苯醌(p-BQ)能在室温条件下有效淬灭O2·-(kO2·-=0.9~1.0×109 L·mol-1·s-1)(Rao et al., 1975), 因此, 本文将p-BQ用于O2·-的淬灭.如图 7c所示, 当p-BQ浓度为1 mmol·L-1时, 苯酚的降解动力学常数kp-BQ=0.0269 min-1, 说明p-BQ对苯酚的降解有明显抑制作用, 并且当p-BQ浓度增加到10 mmol·L-1时, 60 min时苯酚降解率小于30%.表 2中p-BQ的表观速率常数有着较好的拟合度, 且随着p-BQ浓度增加表观速率常数kobs降低, 说明O2·-参与了1O2的产生和活化降解过程, 因此, 当淬灭O2·-后苯酚降解效果出现明显下降.有研究表明, 催化剂活化过硫酸盐生成O2·-, 随后与水结合生成1O2(Zhu et al., 2019).从图 8可以看出, 当反应体系中只有PMS和苯酚时, 碱性、中性和酸性条件下苯酚几乎无降解, 但相比中性和酸性条件, 碱性条件下的苯酚最终去除率还是会稍高一点.这由于碱性条件下会产生少量·OH, 而·OH对苯酚的降解效率低于基于1O2的氧化过程, 因为富电子的酚类分子更易与单线态氧反应(Furman et al., 2010).
图 8(Fig. 8)
 |
| 图 8 不同pH条件对催化剂体系降解苯酚效果的影响([苯酚]0=20 mg·L-1, [催化剂]0=250 mg·L-1, [PMS]0=200 mg·L-1, 25 ℃) Fig. 8Effect of catalyst in different pH on the degradation of phenol by N-C/ZrO2/PMS system |
苯甲酸(BA)常被用作活性自由基指示剂来指示反应过程中生成的·OH和SO4·-.图 9a中起始浓度为20 mg·L-1时BA几乎无降解, 进一步说明N-C/ZrO2活化PMS过程中没有产生·OH和SO4·-.对羟基苯甲酸60 min时的降解率为45.5%, Liang等(2015)指出在对羟基苯甲酸的降解中, 1O2而非·OH和SO4·-起主要作用.双酚A(BPA)也是常被用作检测氧化能力的污染物, 由图 9a可见, 60 min时BPA降解完全.对于这些污染物的降解效果, 在之前关于单线态氧的研究中也有着相似的结果(Lee et al., 2011;Bokare et al., 2015;Zhu et al., 2019).综合苯甲酸、对羟基苯甲酸和双酚A 3种污染物的降解情况, 再次验证了1O2为主要活性物.
图 9(Fig. 9)
 |
| 图 9 N-C/ZrO2/PMS体系对不同污染物的降解效果(a)和N-C/ZrO2/PMS体系加捕获剂DMPO(b)、TEMP(c)的EPR谱图及不同条件下PMS的降解动力学图(d)([苯酚]0=20 mg·L-1, [催化剂]0=250 mg·L-1, [PMS]0=200 mg·L-1, pH=6.5, 25 ℃) Fig. 9The different pollutants degradation in N-C/ZrO2/PMS system(a), DMPO(b), TEMP(c) trapped EPR spectra of N-C/ZrO2/PMS system and the kinetics diagram of PMS consumption under different conditions(d) |
3.3.2 EPR分析为了进一步确定活性物种的种类, 以DMPO作为·OH和SO4·-捕获剂, TEMP作为1O2捕获剂, 采用EPR进行检测.如图 9b所示, N-C/ZrO2/PMS体系中没有检测到DMPO-OH和DMPO-SO4特征峰, 说明反应体系中没有·OH和SO4·-这两种自由基产生.同时不难看处, 图中特征峰(◆)是超精细分裂特征峰1∶2∶1∶2∶1∶2∶1, 属于5, 5-二甲基-1-吡咯烷酮-2-2氧基(DMPOX, αN=7.3 G, αH=3.9 G), 这个特征峰由1O2直接氧化DMPO产生(Bilski et al., 1996).此外, 如EPR图谱(图 9c)所示, 3条清晰的TEMPN加合物的特征峰(●)(1∶1∶1) (α=16.9G), 再一次证明反应体系中存在1O2. 其中, 只有PMS情况产生微弱的TEMPN峰, 这是PMS发生自分解产生少量·OH(图 9b), ·OH同水作用产生1O2. 图 9c中N-C/ZrO2/PMS体系中TEMPN特征峰信号非常强烈, 而在加入FFA(10 mmol·L-1)之后, TEMPN被明显削弱, 这是由于产生的1O2被FFA消耗, 使得TEMPN信号变弱.综合EPR谱图、淬灭实验结果和其他污染物降解实验, 可以确定N-C/ZrO2在活化PMS过程中产生大量1O2.表 2中苯酚降解动力学常数kobs同样也随[FFA]0和[NaN3]0增加而减小.苯酚降解符合伪一级动力学而不是零级动力学表明1O2氧化通过电子转移发生在溶液中而不是仅在催化剂表面区域(Zhou et al., 2015).
3.3.3 N-C/ZrO2活化PMS降解机理图 9d为pH=6.5缓冲体系几种条件下PMS消耗情况.由图可见, 同时存在催化剂和苯酚的对照组中, PMS消耗速率要高于无苯酚的实验组, 并且降解曲线与相同条件下苯酚降解速率接近, 至60 min时PMS剩余3.2%, 说明反应体系对PMS有较高的利用率.而当体系中没有苯酚只有催化剂和PMS时, 60 min时PMS剩余量为25.8%, 从纳米N-C/ZrO2 XPS谱图(图 2c)也可以看到, 位于288.4 eV处的CO官能团能促进活化PMS分解(Wang et al., 2016).没有苯酚的情况下PMS消耗远低于有苯酚的情况, 说明苯酚降解不止1O2氧化这一条途径, 可能还存在催化剂表面电子直接转移(Huang et al., 2019), 即附着在催化剂表面的PMS直接从苯酚夺取电子, 进而降解苯酚.这可能与材料结构有密切关系, 大量强导电性的C掺杂(表 1)有着丰富氧空位的纳米ZrO2, 使PMS易于吸附在催化剂表面并发生电子转移, 同时也从侧面说明碱性条件下PMS与N-C/ZrO2之间的排斥力导致PMS无法附着在催化剂表面进而影响直接电子转移途径降解苯酚.当体系中只有苯酚和PMS时, PMS也会有少量消耗, 这是由于PMS的不稳定性在水中自分解所造成, 如图 9b中当水中仅存在PMS时有微弱的·OH特征峰(1∶2∶2∶1)产生, 也说明了PMS的自分解特性.
图 10所示为N-C/ZrO2在中性环境下降解苯酚存在的降解途径与机理, 溶解于水中的PMS与催化剂N-C/ZrO2接触后, 在N-C/ZrO2/PMS体系中一部分PMS和水结合直接被转化成1O2(式(2)、(3)), 另一部分则先活化分解PMS产生O2·-, 随后O2·-同水结合进一步生成1O2(式(4)), 两种途径生成1O2的同时作用于苯酚;另一方面, 催化剂表面附着的PMS从靠近催化剂的苯酚分子处夺取电子, 使苯酚发生分解.因此, 苯酚的降解途径为1O2氧化和直接电子转移的非自由基途径.
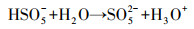 | (2) |
 | (3) |
 | (4) |
 |
| 图 10 N-C/ZrO2/PMS体系降解苯酚的机理图 Fig. 10Mechanism of phenol degradation by N-C/ZrO2/PMS |
4 结论(Conclusions)1) 利用纳米ZrO2良好的化学稳定和富氧空缺等性能, 以ZrCl4为前驱体, 采用尿素热解法制备出的N-C/ZrO2对PMS具有良好的活化性能, SEM、XPS、XRD和FTIR表征说明所合成的N-C/ZrO2为N-C均匀掺杂ZrO2且直径小于50 nm的纳米颗粒.
2) N-C/ZrO2材料的研究为锆基纳米材料在活化过硫酸盐降解有机污染物领域提供了参考价值.本研究中材料最佳合成方法为质量比例为ZrCl4∶尿素(3∶3), N-C/ZrO2/PMS体系最佳反应条件为:pH=6~7, N-C/ZrO2投加量为250 mg·L-1, PMS投加量为200 mg·L-1, 在此条件下反应30 min后苯酚去除率可达99%以上.最佳反应条件下PMS在实验中的利用率达96.8%, 反应后水中Zr4+浸出浓度仅为0.245 μg·L-1.对反应前后材料的表征结果表明, 催化剂主要成分及表面基团无明显变化, 证明材料具有较强的稳定性.重复实验结果表明, 重复5遍后仍有84.8%的苯酚被去除.
3) N-C/ZrO2/PMS体系降解苯酚为表面反应, 以1O2为主要氧化活性物和直接电子转移的非自由基机制, 反应过程没有·OH和SO4·-的参与.
参考文献
| Anipsitakis G P, Dionysiou D D. 2004. Radical generation by the interaction of transition metals with common oxidants[J]. Environmental Science & Technology, 38(13): 3705-3712. |
| Anipsitakis G P, Stathatos E, Dionysiou D D. 2005. Heterogeneous activation of oxone using Co3O4[J]. The Journal of Physical Chemistry B, 109(27): 13052-13055. DOI:10.1021/jp052166y |
| Bilski P, Reszka K, Bilska M, et al. 1996. Oxidation of the spin trap 5, 5-Dimethyl-1-pyrroline N-oxide by singlet oxygen in aqueous solution[J]. Journal of the American Chemical Society, 118(6): 1330-1338. DOI:10.1021/ja952140s |
| Bokare A D, Choi W. 2015. Singlet-oxygen generation in alkaline periodate solution[J]. Environmental Science & Technology, 49(24): 14392-14400. |
| Brindha A, Sivakumar T. 2017. Visible active N, S co-doped TiO2/graphene photocatalysts for the degradation of hazardous dyes[J]. Journal of Photochemistry and Photobiology A: Chemistry, 340: 146-156. DOI:10.1016/j.jphotochem.2017.03.010 |
| Deng J, Feng S, Ma X, et al. 2016. Heterogeneous degradation of Orange II with peroxymonosulfate activated by ordered mesoporous MnFe2O4[J]. Separation and Purification Technology, 167: 181-189. DOI:10.1016/j.seppur.2016.04.035 |
| Ding D, Yang S, Qian X, et al. 2020. Nitrogen-doping positively whilst sulfur-doping negatively affect the catalytic activity of biochar for the degradation of organic contaminant[J]. Applied Catalysis B: Environmental, 263: 118348. DOI:10.1016/j.apcatb.2019.118348 |
| Furman O S, Teel A L, Watts R J. 2010. Mechanism of base activation of persulfate[J]. Environmental Science & Technology, 44(16): 6423-6428. |
| Guo D, Shibuya R, Akiba C, et al. 2016. Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts[J]. Science, 351(6271): 361. DOI:10.1126/science.aad0832 |
| Hu X, Wang Y, Wu R, et al. 2020. Effects of zirconia crystal phases on the catalytic decomposition of N2O over Co3O4/ZrO2 catalysts[J]. Applied Surface Science, 514: 145892. DOI:10.1016/j.apsusc.2020.145892 |
| Huang K Z, Zhang H. 2019. Direct electron-transfer-based peroxymonosulfate activation by iron-doped manganese oxide (delta-MnO2) and the development of galvanic oxidation processes (GOPs)[J]. Environmental Science Technology, 53(21): 12610-12620. DOI:10.1021/acs.est.9b03648 |
| Jeong H K, Lee Y P, Lahaye R J W E, et al. 2008. Evidence of graphitic AB stacking order of graphite oxides[J]. Journal of the American Chemical Society, 130(4): 1362-1366. DOI:10.1021/ja076473o |
| Lee J, Hong S, Mackeyev Y, et al. 2011. Photosensitized oxidation of emerging organic pollutants by tetrakis C60 aminofullerene-derivatized silica under visible light irradiation[J]. Environmental Science & Technology, 45(24): 10598-10604. |
| Lei Y, Chen C S, Tu Y J, et al. 2015. Heterogeneous degradation of organic pollutants by persulfate activated by CuO-Fe3O4: Mechanism, stability, and effects of pH and bicarbonate ions[J]. Environmental Science & Technology, 49(11): 6838-6845. |
| Liang P, Zhang C, Duan X, et al. 2017. N-doped graphene from metal-organic frameworks for catalytic oxidation of p-hydroxylbenzoic acid: N-functionality and mechanism[J]. ACS Sustainable Chemistry & Engineering, 5(3): 2693-2701. |
| Liu H, Sun X, Yin C, et al. 2008. Removal of phosphate by mesoporous ZrO2[J]. Journal of Hazardous Materials, 151(2): 616-622. |
| Ma J. 2011. Preparation and characterization of ZrO2 nanoparticles capped by trioctylphosphine oxide (TOPO)[J]. Journal of Wuhan University of Technology-Mater Sci Ed, 26(4): 611-614. DOI:10.1007/s11595-011-0277-2 |
| Manna S, Ghoshal T, Deb A K, et al. 2010. Structural stability and optical properties of nanocrystalline zirconia[J]. Journal of Applied Crystallography, 43(4): 780-789. DOI:10.1107/S0021889810013488 |
| Miller J S. 2005. Rose bengal-sensitized photooxidation of 2-chlorophenol in water using solar simulated light[J]. Water Research, 39(2): 412-422. |
| Neta P, Huie R E, Ross A B. 1988. Rate constants for reactions of inorganic radicals in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 17(3): 1027-1284. DOI:10.1063/1.555808 |
| Poungchan G, Ksapabutr B, Panapoy M. 2016. One-step synthesis of flower-like carbon-doped ZrO2 for visible-light-responsive photocatalyst[J]. Materials & Design, 89: 137-145. |
| Rao P S, Hayon E. 1975. Redox potentials of free radicals. IV.Superoxide and hydroperoxy radicals.O2- and.HO2[J]. The Journal of Physical Chemistry, 79(4): 397-402. DOI:10.1021/j100571a021 |
| Shao P, Ding L, Luo J, et al. 2019. Lattice-defect-enhanced adsorption of arsenic on zirconia nanospheres: A combined experimental and theoretical study[J]. ACS Applied Materials & Interfaces, 11(33): 29736-29745. |
| Sheng Z H, Shao L, Chen J J, et al. 2011. Catalyst-free synthesis of nitrogen-doped graphene via thermal annealing graphite oxide with melamine and its excellent electrocatalysis[J]. ACS Nano, 5(6): 4350-4358. DOI:10.1021/nn103584t |
| Sun G X, Liu F T, Bi J Q, et al. 2015. Electrospun zirconia nanofibers and corresponding formation mechanism study[J]. Journal of Alloys and Compounds, 649: 788-792. DOI:10.1016/j.jallcom.2015.03.068 |
| Wang H, Guo W, Liu B, et al. 2019. Edge-nitrogenated biochar for efficient peroxydisulfate activation: An electron transfer mechanism[J]. Water Research, 160: 405-414. DOI:10.1016/j.watres.2019.05.059 |
| Wang Y, Ao Z, Sun H, et al. 2016. Activation of peroxymonosulfate by carbonaceous oxygen groups: experimental and density functional theory calculations[J]. Applied Catalysis B: Environmental, 198: 295-302. DOI:10.1016/j.apcatb.2016.05.075 |
| Wu B, Liu C, Fu C, et al. 2019. Selective separation of Cr(VI) and V(V) from solution by simple pH controlled two-step adsorption/desorption process with ZrO2[J]. Chemical Engineering Journal, 373: 1030-1041. DOI:10.1016/j.cej.2019.05.131 |
| 王莹, 魏成耀, 黄天寅, 等. 2017. 氮掺杂碳纳米管活化过一硫酸盐降解酸性橙AO7[J]. 中国环境科学, 37(7): 2583-2590. DOI:10.3969/j.issn.1000-6923.2017.07.021 |
| Xu F, Minniti M, Barone P, et al. 2008. Nitrogen doping of single walled carbon nanotubes by low energy N2+ ion implantation[J]. Carbon, 46(11): 1489-1496. DOI:10.1016/j.carbon.2008.06.047 |
| 许芬, 陈家斌, 张书源, 等. 2018. 丙酮活化过一硫酸盐性能及非自由基机制[J]. 环境科学学报, 38(11): 4333-4339. |
| Yu Y, Zhou Z, Ding Z, et al. 2019. Simultaneous arsenic and fluoride removal using {201}TiO2-ZrO2: Fabrication, characterization, and mechanism[J]. Journal of Hazardous Materials, 377: 267-273. DOI:10.1016/j.jhazmat.2019.05.060 |
| Zhang T, Li C, Gu Y, et al. 2017. Fabrication of novel metal-free "graphene alloy" for the highly efficient electrocatalytic reduction of H2O2[J]. Talanta, 165: 143-151. DOI:10.1016/j.talanta.2016.12.018 |
| Zhang T, Zhu H, Croué J-P. 2013. Production of sulfate radical from peroxymonosulfate induced by a magnetically separable CuFe2O4 spinel in water: Efficiency, stability, and mechanism[J]. Environmental Science & Technology, 47(6): 2784-2791. |
| Zhou Y, Jiang J, Gao Y, et al. 2015. Activation of peroxymonosulfate by benzoquinone: A novel nonradical oxidation process[J]. Environmental Science & Technology, 49(21): 12941-12950. |
| Zhu S, Li X, Kang J, et al. 2019. Persulfate activation on crystallographic manganese oxides: Mechanism of singlet oxygen evolution for nonradical selective degradation of aqueous contaminants[J]. Environmental Science Technology, 53(1): 307-315. DOI:10.1021/acs.est.8b04669 |
| Zhu Y, Chen S, Quan X, et al. 2013. Cobalt implanted TiO2 nanocatalyst for heterogeneous activation of peroxymonosulfate[J]. RSC Advances, 3(2): 520-525. DOI:10.1039/C2RA22039C |
| Zou J, Ma J, Chen L, et al. 2013. Rapid acceleration of ferrous iron/peroxymonosulfate oxidation of organic pollutants by promoting Fe(III)/Fe(II) cycle with hydroxylamine[J]. Environmental Science & Technology, 47(20): 11685-11691. |
| 赵浩迪, 张静, 王梅菁, 等. 2018. 预磁化强化ZnO@Fe3O4活化PMS降解AO7[J]. 环境科学学报, 38(12): 4663-4669. |
| 周雪君, 时鹏辉, 姚伟峰. 2016. 掺氮石墨烯的制备及其催化性能研究[J]. 上海电力学院学报, 32(4): 333-338. |
