 , 刘欢1, 王樱1, 付名利1,2,3, 吴军良1,2,3, 黄皓旻1,2,3, 叶代启1,2,3, 陈礼敏1,2,3
, 刘欢1, 王樱1, 付名利1,2,3, 吴军良1,2,3, 黄皓旻1,2,3, 叶代启1,2,3, 陈礼敏1,2,3
1. 华南理工大学环境与能源学院, 广州 510006;
2. 挥发性有机物污染治理技术与装备国家工程实验室, 广州 510006;
3. 广东省大气环境与污染控制重点实验室, 广州 510006
收稿日期: 2021-04-10; 修回日期: 2021-05-17; 录用日期: 2021-05-17
基金项目: 国家自然科学基金(No.21976059,91645119,51878292);广东省自然科学基金(No.2019A1515011849);国家留学基金委奖学金项目(No.201906155006);广州市科技计划项目(No.201607010095);中央高校基本科研业务费专项资金资助项目(No.2017ZD076);厦门大学固体表面物理化学国家重点实验室项目(No.201602)
作者简介: 钟雯(1995-), 女, E-mail: 382655191@qq.com
通讯作者(责任作者): 陈礼敏, 女, 博士, 副教授, 博士生/硕士生导师.主要研究方向为大气污染控制理论与技术, 主要集中于温室气体资源化、有毒有害大气污染物控制及多功能环境纳米材料的研究等.在国内外著名刊物上发表论文50余篇, 近年来主持科研项目10余项. E-mail: liminchen@scut.edu.cn
摘要:采用共沉淀-浸渍法制备了不同载体结构的Co/Al2O3、Co/MgO(25)-Al2O3(75)(Co/M1A3-Spinel)、Co/MgO(75)-Al2O3(25)(Co/M3A1-Solid)和Co/MgO催化剂,用于CO2氧化乙烷脱氢(ODEC)和乙烷干重整(DRE)反应.XRD、Raman、H2-TPR、XPS表征结果表明,载体结构显著影响催化剂上Co物种的落位、结构和还原性能.Co3+和Co2+在C2H6/CO2反应气氛中可相互转化,促进C2H6和CO2活化;尖晶石结构中的Co物种比固溶体中的Co物种可还原性强,在高温ODEC反应条件下,催化剂表面的Co物种会被还原成Co0,协同Co3+/Co2+导致C—C键和/或C=C键断裂,使得反应路径向DRE转变.MgO基固溶体结构中Co物种与载体相互作用更强,在高温ODEC反应中避免了被大量还原为低价态的Co0,ODEC反应可稳定进行.
关键词:CO2乙烷乙烷氧化脱氢乙烷干重整Co基催化剂
The influences of support structure on the performance of Co/MgO-Al2O3 catalyst for C2H6/CO2 co-conversion
ZHONG Wen1
, LIU Huan1, WANG Ying1, FU Mingli1,2,3, WU Junliang1,2,3, HUANG Haomin1,2,3, YE Daiqi1,2,3, CHEN Limin1,2,31. School of Environment and Energy, South China University of Technology, Guangzhou 510006;
2. National Engineering Laboratory for VOCs Pollution Control Technology and Equipment, Guangzhou 510006;
3. Guangdong Provincial Key Laboratory of Atmospheric Environment and Pollution Control (SCUT), Guangzhou 510006
Received 10 April 2021; received in revised from 17 May 2021; accepted 17 May 2021
Abstract: Co/Al2O3, Co/MgO(25)-Al2O3(75) (Co/M1A3-Spinel), Co/MgO(75)-Al2O3(25) (Co/M3A1-Solid) and Co/MgO were prepared through co-precipitation combined impregnation method and applied to oxidative dehydrogenation of ethane in the presence of CO2 (ODEC) and dry reforming of ethane (DRE). XRD, Raman, H2-TPR and XPS results showed the properties of supports significantly affected the location, structure and reducibility of Co species. The reversible conversion between Co3+ and Co2+ can be detected in C2H6/CO2 atmosphere which can promote the activation of C2H6 and CO2; Co species in M1A3-Spinel are more reducible than those in M3A1-Solid, Co0 reduced from Co species on the surface of spinel at high temperature in ODEC reaction condition, which shows synergetic effects with Co3+/Co2+ and contributes to the cleavage of C—C and/or C=C bonds, resulting in the reaction routs shift from ODEC to DRE. The structure of Co species in M3A1-Solid have stronger interactions with support and are more difficult to be reduced, which inhibits to generate mass metal Co0 at high temperature in ODEC reaction, then the steady ODEC reaction can be kept.
Keywords: CO2ethaneoxidative dehydrogenation of ethanedry reforming of ethaneCo-based catalysts
1 引言(Introduction)乙烷(C2H6)是一种气态的碳氢化合物, 也是空气中挥发性有机化合物(VOCs)的一种, 可与氮氧化物(NOx)反应生成臭氧(O3)等二次污染物(Etiope et al., 2009; Li et al., 2015).据统计, 全球乙烷年排放量约为11.3 Tg, 主要来源于化工行业产生的蒸汽、生物质的燃烧, 以及少量海洋、生物和地质源的排放(Simpson et al., 2012; Mo et al., 2015).二氧化碳(CO2)是主要的温室气体, 根据美国国家海洋大气局(NOAA)的气候状况报告, 2020年大气中CO2平均水平为413.3 ppm, 并将持续上升.联合国政府间气候变化专门委员会(IPCC)提出了“碳中和”的目标(王勇等, 2017), 为此中国政府提出力争于2030年前CO2排放达到峰值, 2060年前实现碳中和.
在化学合成中充分利用无毒、廉价、资源丰富的CO2是减少C2H6排放的可行方法之一(杨淑英等, 1991; Braunstein et al., 1998; 陈明亮, 2000; 张法智等, 2002; Gomez et al., 2019).为了实现CO2减排并解决C2H6泄露逃逸的污染问题, C2H6氧化脱氢(ODEC)和C2H6干重整(DRE)作为两种解决问题的化学合成方法, 逐渐成为研究热点.同时, C2H6/CO2共转化可以生产基础化工原料乙烯(C2H4)和合成气(CO和H2), 具有巨大的商业价值.然而, 实现这一具有应用前景的化学合成需要设计高效、稳定、廉价的催化剂.
目前, ODEC领域研究最广泛的是具有毒性的Cr基催化剂(蒋冰等, 2014), 而低毒、廉价的Co基催化剂具有接近Cr基催化剂的ODEC活性, 应用前景十分广阔.研究发现, Co基催化剂采用具有较多强酸性位点的ZrO2、TiO2和TiO2-ZrO2载体时, 催化剂活性高, 但乙烯选择性低(Koirala et al., 2016);采用碱性BaCO3和MgAl2O4载体, 催化剂展现出较优秀的催化性能(Zhang et al., 2007; 2009), 并可增强CO2吸附活化.但目前有关Co基催化剂的ODEC反应机理尚未有定论, 如何精准控制反应路径避免DRE反应的发生, 还有待进一步研究.
Al2O3和MgO可提供酸碱位点, 二者通常以尖晶石和MgO基固溶体形式存在(Guo et al., 2004; Madduluri et al., 2018).Hu等(2002)研究表明, MgO基固溶体的干重整催化性能主要取决于催化剂的结构、制备方法、碱性和与活性金属的相互作用;对于尖晶石, 金属离子的化学反应性由于占据不同位置导致几何和电子结构不同从而影响催化活性(Markov et al., 1991).对载体结构进行调控会最终影响活性金属的结构及C2H6/CO2反应的催化性能, 因此, 研究不同结构催化剂的催化性能对于设计具有高选择性Co基催化剂具有指导性意义.
基于此, 本研究通过共沉淀方法制备尖晶石和固溶体结构的MgO-Al2O3载体用于担载活性金属Co, 进行ODEC和DRE反应的活性评价, 并研究MgO-Al2O3载体性质对催化剂上Co存在形式、还原性能、价态分布和催化剂催化性能的影响.同时, 对比研究单一氧化物载体MgO和Al2O3上Co的结构, 以揭示活性金属的存在形式及与载体MgO和Al2O3组分相互作用的关系, 并提出不同价态Co物种对C2H6/CO2反应的路径调控作用.
2 材料与方法(Materials and methods)2.1 材料制备MgO、MgO(75)-Al2O3(25)固溶体(Mg/Al原子比=3/1, M3A1-Solid)、MgO(25)-Al2O3(75)尖晶石(Mg/Al原子比=1/3, M1A3-Spinel)和Al2O3载体的合成参考文献(Xia et al., 2015)中采用的共沉淀法.依次称量总物质的量为0.1 mol的Mg(NO3)2·6H2O和/或Al(NO3)3·9H2O, 分别溶解于100 mL去离子水, 配置成混合溶液A.称量对应量的NaOH和Na2CO3(n(OH-)/(n(Mg2+)+n(Al3+))=2/1, n(CO32-)/n(Al3+)=2/1), 溶解于100 mL去离子水, 配置成混合溶液B.将溶液A、溶液B通过蠕动泵并逐滴滴加至20 mL去离子水中, 沉淀过程控制溶液温度60 ℃、pH=9.5.共沉淀结束后, 悬浊液在室温下老化18 h, 再经抽滤、干燥、研磨后得到所需的前驱体.然后将此前驱体置于管式炉中650 ℃焙烧5 h.通过浸渍法担载Co, 称取0.49 g的Co(NO3)2·6H2O置于100 mL烧杯, 溶解搅拌, 再加入0.50 g载体搅拌浸渍, 待水分几乎完全挥发置于烘箱中80 ℃干燥12 h, 再将干燥样品置于管式炉中500 ℃焙烧4 h, 最终得到Co/MgO-Al2O3催化剂(分别记为Co/MgO、Co/M1A3-Spinel、Co/M3A1-Solid、Co/Al2O3), 通过ICP测试, 样品的实际Co担载量均接近理论值15.7%.
2.2 材料表征采用美国Thermo fisher 7200电感耦合等离子体发射光谱(ICP-OES)测试样品的Co担载量.X射线衍射(XRD)测试使用Bruker D8 Advance型XRD仪, 装配Cu-Kα线, 步长为0.013°, 扫描范围2θ=10°~80°, 扫描速度为4°·min-1.拉曼(Raman)光谱的采集在HORIBA LabRAMHR Evolution拉曼光谱仪上进行, 激发光波长为532 nm, 拉曼光谱的测试范围为100~800 cm-1, 采集时间为60 s, 累计次数为3.比表面积及孔结构采用Micromeritics ASAP 2460测定仪测定, 采用Brunauer-Emmett-Teller (BET)法计算比表面积, 并根据脱附支采用Barrett-Joyner-Halenda (BJH)法计算孔径;测试前, 样品先在300 ℃下脱气4 h.扫描电子显微图(SEM)通过ZEISS Merlin观察.H2程序升温还原(H2-TPR)在装配TCD检测器的DAS-7000化学吸附仪上进行, 测试前通Ar气在500 ℃下吹扫30 min, 然后冷却至50 ℃, 待基线平稳;然后切换10% H2/Ar(30 mL·min-1), 以10 ℃·min-1的升温速率将温度从50 ℃升至950 ℃.X射线光电子能谱(XPS)在Thermo Scientific EscaLab Xi+上测试, 搭载Mg Kα射线(1253.6 eV), 采集Co2p、Mg1s、Al2p、O1s和C1s信号, 284.8 eV作为C1s校准结合能.紫外可见吸收光谱(UV-Vis)采用Lambda 650 S检测, 测试时将样品与BaSO4均匀混合后压片, 并转入原位处理池中, 采集范围为200~800 nm.漫反射傅立叶变换红外光谱(Drifts)采用装配MCT检测器的Nicolet 6700仪测试, 该红外光谱仪的检测器为MCT, 测试参数:分辨率为4 cm-1, 采谱次数为32次;将样品压片, 并转入原位处理池中.还原处理的Co/M3A1-Solid首先在固定床反应器通入5% H2/Ar, 680 ℃还原2 h后, 再转移到反应池原位通入20% H2/Ar, 550 ℃还原30 min.红外测试前通入10% C2H6/Ar, 500 ℃吸附活化.350 ℃采集Ar吹扫后和通入2% CO2气氛下保持0.2 min、3 min、15 min的原位红外谱图.
2.3 催化性能评价使用自制的“固定床连续流动反应器-GC组合系统”评价催化剂催化C2H6/CO2反应的性能.催化剂装填在内径4 mm的U型管中, 粒径为40~60目, 催化剂用量为200 mg, 原料气体积比为V(C2H6)∶V(CO2)∶V(N2)=1∶2∶17, 质量空速WHSV采用文献报道(Zhao et al., 2006; Talati et al., 2016)常用的9000 mL·g-1·h-1.ODEC反应催化性能评价前, 先将催化剂在Ar气氛下预处理, 流速为40 mL·min-1, 500 ℃保持30 min, 再进行程序升温反应.反应条件如下:反应温度控制在625、650、675、700 ℃4个温度段, 升温速率为2 ℃·min-1.DRE反应催化性能评价前, 先将催化剂在5% H2/Ar气氛下还原, 流速为40 mL·min-1, 680 ℃保持2 h, 再进行程序升温反应, 升温速率为2 ℃·min-1.通过GC218型气相色谱仪(上海凡伟)对反应物及产物进行在线检测分析.色谱仪配置两个检测通道, TCD检测器连接碳分子筛柱和2 m的5A分子筛色谱柱;FID检测器采用Porapak Q毛细管柱, 柱长30 m, 使用Ar作为载气, 采用5A分子筛填充柱分离CH4、CO2、H2、N2及CO, 采用Porapak Q毛细管柱分离烷烃类物质如CH4、C2H4、C2H6.反应物转化率(Xi)和各产物收率(Yj)计算公式如下:
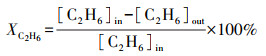 | (1) |
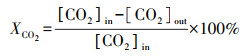 | (2) |
 | (3) |
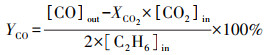 | (4) |
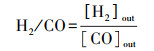 | (5) |
3 结果(Results)3.1 结构特性合成的Al2O3、M1A3-Spinel、M3A1-Solid和MgO载体的XRD衍射谱图如图 1a所示, MgO在2θ为36.8°、42.8°、62.1°、74.5°和78.4°处为方镁石(111)、(200)、(220)、(311)和(222)晶面的特征衍射(JCPDS 89-7746), M3A1-Solid与方镁石在相同位置具有同MgO的衍射峰, 但峰型加宽, 强度明显减弱, 表明MgO基固溶体的形成(Tan et al., 2014), Al2O3载体在2θ为19.5°、31.9°、37.6°、39.5°、45.9°、60.9°、67.0°、85.0°处为氧化铝(111)、(220)、(311)、(222)、(400)、(511)、(440)、(444)晶面的特征衍射(JCPDS 10-0425), M1A3-Spinel载体在2θ为19.0°、31.3°、36.9°、38.5°、44.8°、55.7°、59.4°、65.2°、68.6°处呈现MgAl2O4尖晶石(111)、(220)、(311)、(222)、(400)、(422)、(511)、(440)、(531)晶面的特征衍射(JCPDS 21-1152), 表明不同晶型的MgO-Al2O3载体成功合成.
图 1(Fig. 1)
 |
| 图 1 载体(a)、焙烧后催化剂(b)和还原后催化剂(c)的XRD谱图 Fig. 1XRD patterns of the supports(a), catalysts after calcination(b) and catalysts after reduction(c) |
焙烧后催化剂的XRD谱图如图 1b所示, 4种催化剂具有不同的晶型结构.焙烧后Co/Al2O3的XRD谱图仅在2θ为19.5°、31.9°、39.5°、45.9°、60.9°、67.0°处呈现微弱的Al2O3尖晶石衍射峰, 可能是焙烧后Al2O3或CoAl2O4结晶度较低;Co/M1A3-Spinel特征峰与M1A3-Spinel载体相似, 归属为尖晶石结构, Co3O4同样具有尖晶石结构, 与MgAl2O4尖晶石特征峰位置一致, 因而在XRD谱图中难以区分(Jiang et al., 2010);Co/M3A1-Solid和Co/MgO特征峰与各自催化剂载体相似, 而强度明显减弱, MgO晶格中可能掺入了Al3+、Co3+或Co2+, 导致XRD谱图同没有明显的Co3O4特征峰(Li et al., 2018).
如图 1c所示, 还原的Co/Al2O3在2θ为22.1°、36.6°、43.1°、59.3°和65.7°处显示出明显的衍射峰, 归属为Co氧化物和/或CoAl2O4(Ding et al., 2001), 以及在2θ为32.5°、34.1°、46.0°、59.8°和65.6°附近的衍射峰, 归属为缺陷尖晶石Al2O3的衍射(Alabi et al., 2020).680 ℃还原后形成这种晶体主要是由于高温可促进铝组分的相变(Zhou et al., 1991);Co/M1A3-Spinel、Co/M3A1-Solid和Co/MgO还原后保持了相同的晶型, 但衍射强度降低, 可能由于Co离子取代了Mg2+进入到MgO晶格中, 导致MgO的晶格畸变(Alabi et al., 2020), 同时, Co/Al2O3、Co/M1A3-Spinel催化剂在2θ为51.5°和75.8°处有与金属钴Co相关的衍射峰(JCPDS 15-0806), 说明Al组分可提高催化剂还原性能.此外, 所有催化剂在2θ为36.6°处的峰归属为被掩盖的混合尖晶石(MgCo2O4、CoAl2O4、Co3O4)(Madduluri et al., 2018)衍射峰, 在还原处理后强度降低, 可能是因为尖晶石结构中的Co被还原.
由于MgO和CoO具有相似的晶格参数, 可以以任意物质的量比形成固溶体, 两者在XRD中难以区分.因此, 通过Raman光谱判断Co在催化剂中的结构特征.如图 2所示, 197、483、523、623和690 cm-1处的拉曼信号, 分别对应于Co3O4的F2g(1)、E2g、F2g(2)、F2g(3)和A1g的振动模式(Zhao et al., 2016), 其中, 197 cm-1处的信号归属于四面体位置Co(CoO4)的特征振动, 而690 cm-1处的信号归属于八面体位置Co(CoO6)的特征振动(Wang et al., 2017).如图 2a所示, Co/Al2O3上具有最强的Co3O4信号, 表明催化剂表面被Co3O4颗粒覆盖, Co/M1A3-Spinel和Co/MgO则具有少量的Co3O4颗粒, Co/M3A1-Solid的Raman谱图呈现宽峰, 这表明M3A1-Solid载体上的Co3O4相结晶度较低.催化剂还原后具有显著改变, 如图 2b所示, 所有催化剂上, 690 cm-1处的特征振动信号移动至675 cm-1处, 表明配位环境的变化(Zhang et al., 2005), 可能是由于八面体位置的Co3+已被还原和/或在还原处理过程中通过取代Mg2+, 更多的Co2+占据八面体位置;Co/Al2O3和Co/M1A3-Spinel上存在部分Co3+取代Al3+占据八面体位置;Co/M3A1-Solid样品还原后在197 cm-1处出现一个尖锐的峰, 表明Co2+进入了分散良好的Al2O3晶格的四面体位置(Wang et al., 2017);Co/MgO仍具有483、523 cm-1处Co尖晶石的特征振动模式, 表明MgCo2O4的存在.综上所示, 不同的载体性质显著影响Co的结构特征, 并在还原处理后仍然保持了部分高价态的Co.
图 2(Fig. 2)
 |
| 图 2 焙烧后(a)和还原后(b)催化剂的拉曼谱图 Fig. 2Raman spectra of the catalysts after calcination(a) and after reduction(b) |
焙烧后催化剂的孔结构性质如表 1所示, 单金属氧化物载体担载Co催化剂(Co/Al2O3、Co/MgO)具有较低的比表面积, 而双金属固溶体载体担载Co催化剂(Co/M3A1-Solid)具有较大的比表面积;载体的比表面积排序为:Al2O3(42.6 m2·g-1) < MgO(61.3 m2·g-1) < M1A3-Spinel(69.7 m2·g-1) < M3A1-Solid(181.8 m2·g-1);负载Co后催化剂的比表面积数值较载体均发生了一定程度的下降, 这可能是Co的负载堵塞了部分孔道所致;各催化剂比表面积的下降幅度顺序为:Co/MgO < Co/M3A1-Solid < Co/M1A3-Spinel < Co/Al2O3, 这是由于在浸渍过程中, MgO表面的碱性位点会促进Co2+的分散(Yang et al., 2017), 缓解孔道堵塞.
表 1(Table 1)
| 表 1 催化剂孔结构性质 Table 1 Pore structure properties of catalysts | ||||||||||||||||||||
表 1 催化剂孔结构性质 Table 1 Pore structure properties of catalysts
| ||||||||||||||||||||
3.2 催化剂形貌焙烧后催化剂的FE-SEM如图 3所示, Co/Al2O3和Co/MgO催化剂分别呈现出片状颗粒堆积圆盘和球形颗粒堆积圆盘的形貌;Co/M1A3-Spinel和Co/M3A1-Solid则分别呈现出片状颗粒组装小球和纳米薄片交错组装纳米花球的形貌.形貌的差异与孔结构性质表征结果吻合, 双金属氧化物载体有利于实现片状形貌向球形形貌的转变, 增大催化剂比表面积, 进而提高活性金属的分散度.
图 3(Fig. 3)
 |
| 图 3 Co/Al2O3 (a)、Co/M1A3-Spinel (b)、Co/M3A1-Solid (c)和Co/MgO (d)的FE-SEM图像 Fig. 3FE-SEM images of Co/Al2O3(a), Co/M1A3-Spinel(b), Co/M3A1-Solid(c) and Co/MgO catalysts(d) |
3.3 催化剂还原性质催化剂的H2-TPR曲线如图 4所示,低温300~450 ℃(峰Ⅰ)处较大宽峰与表面Co物种的还原有关,可归属为Co3O4的两步还原,首先还原为CoO,然后还原为Co0(Zheng et al., 2020);Co/MgO催化剂直到950 ℃高温仍未出现其他还原峰,表明MgO载体上更多的Co物种进入体相;Co/M1A3-Solid催化剂在高温600~800 ℃(峰Ⅱ)附近的还原峰归属为与载体形成的难还原结构的Co物种,如MgCo2O4或Mg(Co, Al)O固溶体(Sewell et al., 1996; Li et al., 2017);Co/M1A3-Spinel和Co/Al2O3催化剂上高温还原峰的温度较低,与形成CoAl2O4尖晶石结构中Co物种的还原有关,并随着载体中Mg含量的升高,还原温度升高(Souza et al., 2016).
图 4(Fig. 4)
 |
| 图 4 焙烧后催化剂的H2-TPR曲线 Fig. 4H2-TPR profiles of calcined samples |
为了探究Co物种在各载体上的存在形式,假设峰Ⅰ对应的Co3O4颗粒完全还原为金属态,根据式(6)~(7)计算了在不同催化剂上Co3O4颗粒中Co物种的摩尔浓度(C(I, Co3O4))及其占Co担载量的百分比(P(Co3O4)),并列于表 2.
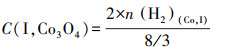 | (6) |
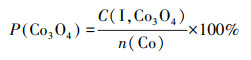 | (7) |
| 表 2 不同催化剂的H2-TPR曲线计算结果 Table 2 Calculated results from H2-TPR patterns of different catalysts | ||||||||||||||||||||||
表 2 不同催化剂的H2-TPR曲线计算结果 Table 2 Calculated results from H2-TPR patterns of different catalysts
| ||||||||||||||||||||||
式中,n(H2)(Co, I)为H2-TPR中峰Ⅰ对应的H2消耗量;n(Co)为Co担载量,取值为2.66 mmol·g-1.
所有催化剂的H2消耗总量(< 950 ℃)低于总担载Co物种完全还原为金属所需的量(假设催化剂中的Co以Co3O4的形式存在,理论值3.54 mmol·g-1),这一结果表明即使是950 ℃还原仍然有部分Co物种未被还原,可能是由于一部分Co2+取代了M3A1-Solid载体中的Mg2+或是Co物种进入载体体相中(Li et al., 2017).此外,Co/MgO催化剂上H2消耗总量最低,随着催化剂结构逐渐从MgO基固溶体结构转变为尖晶石结构,H2消耗总量显著提高,进一步说明载体性质显著影响Co物种的结构和还原性能.根据上述峰Ⅰ和峰Ⅱ的归属,Co/Al2O3表面的Co3O4颗粒占Co担载量的69.2%,而Co/MgO表面Co3O4量显著降低,仅占Co担载量的12.4%,说明Co物种与MgO载体具有更强的相互作用导致更多的Co物种进入载体,进一步证实了Raman表征的结论.
3.4 XPS结果为了探究反应过程中催化剂表面组成和Co物种的变化, 进行了XPS测试.Co、Mg、Al 3种元素的原子含量百分比见表 3, 从计算结果发现ODEC反应后催化剂表面的Co/(Co+Mg+Al)占比高于反应前, 说明ODEC反应过程中更多的Co物种富集到催化剂表面.同时, Co原子含量百分比的提升顺序为:Co/Al2O3 (6%)>Co/M1A3-Spinel(5%)>Co/M3A1-Solid(3%)≥Co/MgO(3%), 说明ODEC反应过程中尖晶石结构Co/Al2O3和Co/M1A3-Spinel催化剂的Co物种更多地迁移到催化剂表面.
表 3(Table 3)
| 表 3 ODEC反应前后催化剂表面各元素原子含量百分比 Table 3 Surface atom percent for catalysts before and after ODEC reaction | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
表 3 ODEC反应前后催化剂表面各元素原子含量百分比 Table 3 Surface atom percent for catalysts before and after ODEC reaction
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
焙烧后、ODEC反应后及5% H2/Ar还原后Co/Al2O3、Co/M1A3-Spinel、Co/M3A1-Solid、Co/MgO催化剂的Co 2p3/2轨道XPS谱图如图 5所示.Co 2p3/2轨道的光电子能谱可分为5个峰, 778.0、780.0和782.2 eV处的3个峰分别归属为Co0、Co3+和Co2+(Yang et al., 2009; Sun et al., 2019), 而在784.0和788.0 eV处的峰分别归属为Co2+和Co3+的卫星峰(Zheng et al., 2020).根据相应的分峰结果, 各Co价态的分布及变化趋势如图 5e所示, 焙烧后尖晶石结构的Co/Al2O3和Co/M1A3-Spinel催化剂表面, 更多Co物种以Co3+形式存在, 而固溶体结构的Co/ M3A1-Solid和Co/MgO催化剂表面, 更多Co物种以Co2+形式存在, 在ODEC反应后及5% H2/Ar还原后, 尖晶石结构的催化剂表面Co物种的价态分布较固溶体结构的催化剂具有更显著的变化, 表明尖晶石结构中的Co物种还原性能更高, 与H2-TPR结果一致;Co2+在ODEC反应后的变化趋势一致, 在4个催化剂表面均增加, 然而对比5% H2/Ar还原后的催化剂, Co/Al2O3和Co/M1A3-Spinel催化剂上Co2+含量低于ODEC反应后表面的含量, 而Co/M3A1-Solid、Co/MgO催化剂上还原后Co2+含量高于ODEC反应后表面的含量.这是由于MgO基固溶体结构更利于维持Co2+不被还原为Co0, 除Co/MgO外, 其余催化剂表面有少量Co0生成, 同样证实了这一点.ODEC反应后催化剂表面Co3+含量减少, 而Co2+含量增加, 说明Co3+/Co2+在ODEC反应气氛下可能发生价态的相互转化.
图 5(Fig. 5)
 |
| 图 5 焙烧后、ODEC反应后、5%H2/Ar还原2 h后Co/Al2O3 (a)、Co/M1A3-Spinel (b)、Co/M3A1-Solid (c)和Co/MgO样品(d)的XPS谱图及Co价态分布趋势图(e) Fig. 5XPS spectra of the Co/Al2O3(a), Co/M1A3-Spinel(b), Co/M3A1-Solid(c) and Co/MgO(d) after calcined ODEC reacted 680 ℃, 5%H2/Ar, 2 h reduced and valence distribution trend of cobalt(e) |
3.5 原位UV-Vis原位处理后催化剂的UV-Vis谱图如图 6所示, Co/Al2O3、Co/M1A3-Spinel、Co/M3A1-Solid、Co/MgO催化剂的紫外光谱具有431、490和695 nm处的3个峰, 分别归属于Co3+的1A1g→1T1g荷移跃迁(Lima et al., 2020)、八面体配位的Co2+及八面体配位的Co3+(Gómez et al., 2011).由于载体结构从尖晶石的Co/Al2O3和Co/M1A3-Spinel变为MgO基固溶体结构的Co/M3A1-Solid和Co/MgO, 431 nm处Co3+的吸收峰逐渐减弱, 表明催化剂表面Co3O4颗粒减少, 更多Co物种与载体具有强相互作用形成固溶体, 这与H2-TPR结果一致.除Co/MgO样品表面Co3O4颗粒少, 紫外吸收信号弱, 导致变化趋势不明显外, 其余样品在原位处理后的UV-Vis谱图具有类似的变化趋势, 通入10% C2H6/Ar气氛处理, 温度从500 ℃逐渐升高到580 ℃, 490 nm处Co2+的紫外吸收峰峰强逐渐增强, 而695 nm处Co3+吸收峰则呈现相反的趋势逐渐减弱;当处理气氛切换为20% CO2/Ar, 温度从500 ℃升至550 ℃时, 490 nm处Co2+紫外吸收峰峰强逐渐减弱, 而695 nm处Co3+峰强逐渐增强.这一结果表明催化剂表面Co物种在C2H6气氛下由高价态Co3+转化为低价态的Co2+, CO2气氛下可逆地由低价态Co2+转化为高价态Co3+, 意味着Co3+/Co2+可以在C2H6/CO2气氛下发生相互转化, 这与XPS结果吻合.
图 6(Fig. 6)
 |
| 图 6 Co/Al2O3 (a)、Co/M1A3-Spinel (b)、Co/M3A1-Solid (c)及Co/MgO (d)催化剂在不同气氛处理下的原位紫外谱图 Fig. 6In situ UV-Vis spectra of Co/Al2O3(a), Co/M1A3-Spinel (b), Co/M3A1-Solid(c) and Co/MgO(d) under different treatment condition |
3.6 ODEC与DRE反应催化活性图 7展示了Co/Al2O3、Co/M1A3-Spinel、Co/M3A1-Solid、Co/MgO催化剂的ODEC反应活性, 具有相近的C2H6转化率, 除Co/Al2O3催化剂在700 ℃时C2H6转化率急剧升高外, 其它催化剂的C2H6转化率均随着温度的升高而逐渐升高.值得注意的是, Co/MgO在低温区间具有略高的C2H6转化率, 同时, C2H4选择性较低且H2/CO仅达到0.13, 这意味着625 ℃时Co/MgO同时发生C2H6裂化(C2H6+2CO2 → CH4+3CO+H2O)形成积碳(C2H6 +H2→2CH4, CH4→2H2+C)和逆Boudouard反应(CO2+C→2CO)消除积碳生成大量CO.与此同时, 催化剂展现出截然不同的C2H4收率及H2/CO, Co/M3A1-Solid具有稳定较高的C2H4选择性, 700 ℃时C2H4收率达到41%, 同时生成H2且H2/CO接近1.0, 意味着Co/M3A1-Solid催化剂上同时发生C2H6直接脱氢耦合逆水煤气反应(RWGS, CO2+H2→CO+H2O)和C2H6氧化脱氢, 这也解释了在反应温度升高至675~700 ℃时, C2H6转化率与CO2转化率比值接近4而不是较低温(625~650 ℃)时的氧化脱氢理论值2.Co/Al2O3和Co/M1A3-Spinel催化剂具有较为类似的催化行为, 随着温度的升高, C2H6转化率升高, 而C2H4选择性逐渐下降, 同时H2/CO也逐渐减小, 说明随着温度的升高, 催化剂上RWGS增强.这与Co/Al2O3和Co/M1A3-Spinel催化剂还原能力较强有关, 随着Co3+被还原为Co2+, 促进RWGS反应的发生, 更高温度下Co0的产生导致反应路径转变为DRE.Al2O3载体上的Co物种具有更高的可还原能性, 700 ℃时, Co/Al2O3催化剂上C2H6转化率与CO2转化率比值接近1, 说明Co/Al2O3上反应路径完全转向DRE;Co/M1A3-Spinel催化剂上C2H6转化率与CO2转化率比值仍接近4, 表明温度升高对Co/M1A3-Spinel上的直接脱氢更具促进效果.反应路径的选择与反应条件下不同载体上Co物种的结构和价态密切相关, 揭示不同Co物种对C2H6和CO2的活化作用, 对研究C2H6/CO2共转化反应至关重要.
图 7(Fig. 7)
 |
| 图 7 不同催化剂的ODEC活性 (a.乙烷转化率, b.C2H6与CO2转化率之比, c.乙烯选择性, d.H2/CO) Fig. 7Ethane conversion(a), ratio of C2H6 to CO2 conversion(b), C2H4 selectivity (c) and H2/CO(d) molar ratio for different catalysts (C2H6∶CO2∶N2=1∶2∶17, WHSV=9000 h-1, mcatalyst=200 mg) |
为了揭示不同Co物种与反应路径的关系, 评价了Co/Al2O3、Co/M1A3-Spinel、Co/M3A1-Solid、Co/MgO催化剂上DRE反应活性, 反应前, 催化剂在5% H2/Ar气氛中于680 ℃下还原2 h.DRE活性评价结果如图 8所示, C2H6和CO2转化率随着温度的升高而升高, 升至最高温650 ℃时, 不同催化剂上C2H6和CO2转化率相近, C2H6转化率近乎100%, CO2转化率略低于C2H6, 但也达到80%, 这可能与C2H6裂化有关.根据H2-TPR结果, Co/Al2O3、Co/M1A3-Spinel表面具有较多的Co0, 而Co3+/Co2+含量较低, 二者具有较低的C2H6和CO2转化率, 意味着Co3+/Co2+对C2H6和CO2的活化至关重要, 同时, Co/Al2O3具有较高的CO选择性, 说明Co0有效地断裂C—C和C=C键生成CO;Co/M3A1-Solid具有最高的C2H6转化率和CO收率, 表明形成Mg(Co, Al)O固溶体具有合适的还原性质, 可有效促进Co0和Co3+/Co2+的协同作用;Co/MgO催化剂上的CO选择性最低, 而CH4选择性较高, 这是Co/MgO催化剂上C2H6裂化为甲烷, 进而形成少量积碳导致的.
图 8(Fig. 8)
 |
| 图 8 不同催化剂的DRE活性 (a.乙烷转化率, b.CO2转化率, c.CO收率, d.产物选择性) Fig. 8Ethane conversion(a), CO2 conversion(b), CO yield(c) and product selectivity(d) for different catalysts (C2H6∶CO2∶N2=1∶2∶17, WHSV=9000 h-1, mcatalyst=200 mg) |
4 讨论(Discussion)XRD和Raman结果证实了不同载体担载的Co基催化剂形成了不同结构, Co/Al2O3表面具有较多Co3O4颗粒, 并有部分Co2+/Co3+进入载体形成CoAl尖晶石相;Co/M1A3-Spinel表面同样具有孤立的Co3O4颗粒, 体相则以Co-Mg-Al混合尖晶石相为主;Co/M3A1-Solid上Co2+和Co3+掺入Mg(Al)O载体, 在焙烧和还原过程中形成Mg(Co, Al)O固溶体;Co/MgO表面具有少量Co3O4颗粒, 大量Co2+取代Mg2+形成类MgO固溶体.根据H2-TPR和XPS结果, Al2O3和M1A3-Spinel载体上尖晶石结构中的Co在ODEC反应条件下部分还原为Co0, 导致反应路径由ODEC转向DRE反应, 并且Co0含量为Co/Al2O3>Co/M1A3-Spinel, 导致Co/Al2O3催化剂上C2H4的收率低于Co/M1A3-Spinel;固溶体M3A1-Solid载体上更多的Co进入载体, 在反应条件下可维持Co3+/Co2+的相互转化, 保证ODEC反应在高温下稳定进行, 高温反应条件下生成的Co0是C—C键断裂导致反应路径转变的关键.催化剂表面Co2+含量按照以下顺序减少:Co/MgO>Co/M3A1-Solid>Co/M1A3-Spinel>Co/Al2O3, 与ODEC较低温(625~650 ℃)下展现的C2H6转化率高低顺序一致, 表明Co2+可能与C2H6活化有关, 考虑到UV-Vis的结果, 可相互转化的Co3+/Co2+对C2H6和CO2的活化至关重要;DRE反应中Co2+对于C2H6转化率的影响略有偏差, 可能由于Co0位点的增加和相关副反应的发生, 以及Co/MgO催化剂上裂化副反应增加导致Co3+/Co2+被覆盖.DRE反应中, Co0的含量与催化活性并不呈对应关系, 说明DRE反应同样需要Co3+/Co2+的转化协同反应物活化.
为进一步研究Co0与C2H6/CO2反应体系中ODEC和DRE路径的关系, 进行漫反射傅立叶变换红外(DRIFT)表征.经还原处理的Co/M3A1-Solid(R-Co/M3A1-Solid)和未经还原处理的Co/M3A1-Solid催化剂在C2H6活化、吸附、吹扫后通入CO2的中间物种信号如图 9所示.乙烷吸附经Ar吹扫后R-Co/M3A1-Solid和Co/M3A1-Solid催化剂表面同样具有1575、1465 cm-1处的两个峰, 归属为乙酸(νas(COO)和νs(COO))(Shee et al., 2010), 考虑到CO2氧化乙烷脱氢反应体系中的氧化还原机理(Mukherjee et al., 2016), 含氧物种的出现可能与催化剂表面的晶格氧有关, 说明ODEC和DRE反应中C2H6活化的路径基本一致.随着CO2的通入, 催化剂表面原本的乙酸信号迅速消失, 产生新的信号;Co/M3A1-Solid催化剂表面立即在1615 cm-1处出现较强的信号并伴随1654 cm-1处峰的出现, 而且随着CO2持续通入信号增强, 可归属为ODEC反应的中间产物烯醇(ν(CH2)和ν(C=C))(Sanchez et al., 1990;Raskó et al., 2005).值得注意的是, R-Co/M3A1-Solid上同样出现烯醇信号, 但信号较弱, 同时在1595 cm-1处出现甲酸(νa(OCO))(Meunier et al., 2000)及992、709 cm-1处稳定的碳酸盐(Xie et al., 2019)的信号, 说明无论ODEC还是DRE反应路径, C2H6在CO2协助下都会发生C—H的断裂, 生成烯醇, 而DRE反应中C=C进一步断裂生成甲酸等C1物种, 通入的CO2将快速吸附形成大量的碳酸盐. 此外, R-Co/M3A1-Solid和Co/M3A1-Solid催化剂表面在1675、1337、1302、1214 cm-1处同样具有较强的信号, 并随着CO2的通入逐渐增强, 这些波段的信号归属为乙醛(ν(C=O)、νa(C—C)、δaCH3和δsCH3)(Shee et al., 2019), 说明乙醛同样是ODEC和DRE反应路径中C—H断裂的中间产物, 这与Porosoff等的研究认为C2H6在Pt(111)上的活化, 优先氧化形成CH3CH2O*再连续经历两次脱氢形成CH3CO*具有一致性(Porosoff et al., 2015); 剩余R-Co/M3A1-Solid和Co/M3A1-Solid催化剂表面同样具有的1522、1376、1030 cm-1处的红外信号归属为CO2-在Co3+的吸附(Li et al., 1989), 随着CO2通入时间增加, Co3+吸附的CO2-信号逐渐增强, 说明CO2活化后导致Co物种转化为Co3+, 结合UV-Vis结果, 进一步证实了可相互转化的Co3+/Co2+是C2H6和CO2的活化位点;3600~3800 cm-1为羟基振动区间(Guo et al., 2021), Co/M3A1-Solid催化剂表面的—OH随CO2通入先增加后略微减弱, 而R-Co/M3A1-Solid催化剂表面的—OH信号先增强后明显减弱, 可能与DRE反应路径更快消耗C2H6活性形成的含氧物种, 产物H2O也随之减少有关.综上所述, ODEC和DRE反应中C2H6的活化路径和位点相似, 烯醇和醛作为二者共同的反应中间产物, 需要CO2的协助产生, R-Co/M3A1-Solid经还原处理, 是导致ODEC路径转向DRE反应路径的关键原因, 还原导致表面具有Co0, 协助C2H4的C=C键进一步断裂产生含氧C1物种, 进而生成CO.
图 9(Fig. 9)
 |
| 图 9 Co/M3A1-Solid (a) 及经还原处理的Co/M3A1-Solid (b)的漫反射傅立叶变换红外谱图 Fig. 9DRIFT spectra obtained on Co/M3A1-Solid(a) and reduced Co/M3A1-Solid(b) |
5 结论(Conclusions)1) 采用共沉淀法制备了不同载体的Co/MgO-Al2O3催化剂, 并用于C2H6/CO2共转化反应中主要的ODEC和DRE反应.结果表明, 载体性质会显著影响催化剂的还原性能和Co物种的存在形式, Co/M3A1-Solid在不同的预处理条件下, 分别展现出较好的ODEC反应活性(700 ℃, 41%的C2H4收率)和DRE(600 ℃, 83%的CO收率)反应活性.
2) 可相互转化的Co3+/Co2+对C2H6和CO2的活化起重要作用, DRE反应需要Co3+/Co2+/Co0的协同, 尖晶石结构的Co/M1A3-Spinel在高温ODEC反应条件下生成较多Co0导致反应路径转向DRE, 而固溶体结构的Co/M3A1-Solid具有较合适的金属-载体相互作用, 维持Co3+/Co2+可相互转化的同时, 可避免Co0物种的生成, 在ODEC反应中避免DRE副反应的发生.
参考文献
| Alabi W O, Sulaiman K O, Wang H. 2020. Sensitivity of the properties and performance of Co catalyst to the nature of support for CO2 reforming of CH4[J]. Chemical Engineering Journal, 390: 124486. DOI:10.1016/j.cej.2020.124486 |
| Braunstein P, Matt D, Nobel D. 1988. Reactions of carbon dioxide with carbon-carbon bond formation catalyzed by transition-metal complexes[J]. Chemical Reviews, 88(5): 747-764. DOI:10.1021/cr00087a003 |
| 陈明亮. 2000. 以CO2为原料的反应与合成[J]. 有机化学, 20(4): 591-596. DOI:10.3321/j.issn:0253-2786.2000.04.030 |
| Ding R G, Yan Z F. 2001. Structure characterization of the Co and Ni catalysts for carbon dioxide reforming of methane[J]. Catalysis Today, 68(1): 135-143. |
| Etiope G, Ciccioli P. 2009. Earth's degassing: A missing ethane and propane source[J]. Science, 323(5913): 478. DOI:10.1126/science.1165904 |
| Gomez E, Yan B, Kattel S, et al. 2019. Carbon dioxide reduction in tandem with light-alkane dehydrogenation[J]. Nature Reviews Chemistry, 3(11): 638-649. DOI:10.1038/s41570-019-0128-9 |
| Gómez L E, Tiscornia I S, Boix A V, et al. 2011. Co/ZrO2 catalysts coated on cordierite monoliths for CO preferential oxidation[J]. Applied Catalysis A: General, 401(1/2): 124-133. |
| Guo H Y, Miao C X, Hua W M, et al. 2021. Enhanced catalytic performance of Cr/MOR for ethane dehydrogenation through dealumination[J]. Catalysis Letters, 151: 1499-1507. DOI:10.1007/s10562-020-03421-7 |
| Guo J J, Lou H, Zhao H, et al. 2004. Dry reforming of methane over nickel catalysts supported on magnesium aluminate spinels[J]. Applied Catalysis A: General, 273(1/2): 75-82. |
| Hu Y H, Ruckenstein E. 2002. Binary MgO-based solid solution catalysts for methane conversion to syngas[J]. Catalysis Reviews-Science and Engineering, 44(3): 423-453. DOI:10.1081/CR-120005742 |
| 蒋冰, 王国清, 张利军, 等. 2014. 二氧化碳氧化乙烷脱氢催化剂研究进展[J]. 乙烯工业, (3): 59-64. DOI:10.3969/j.issn.1671-7120.2014.03.019 |
| Jiang Z, Yu J J, Cheng J, et al. 2010. Catalytic combustion of methane over mixed oxides derived from Co-Mg/Al ternary hydrotalcites[J]. Fuel Processing Technology, 91: 97-102. DOI:10.1016/j.fuproc.2009.08.023 |
| Koirala R, Buechel R, Pratsinis S E, et al. 2016. Silica is preferred over various single and mixed oxides as support for CO2-assisted cobalt-catalyzed oxidative dehydrogenation of ethane[J]. Applied Catalysis A: General, 527: 96-108. DOI:10.1016/j.apcata.2016.08.032 |
| Li C, Sakata Y, Arai T, et al. 1989. Carbon monoxide and carbon dioxide adsorption on cerium oxide studied by Fourier-transform Infrared Spectroscopy[J]. Journal of the Chemical Society Faraday Transactions, 85(4): 1451-1461. |
| Li D L, Lu M M., Xu S P, et al. 2017. Preparation of supported Co catalysts from Co-Mg-Al layered double hydroxides for carbon dioxide reforming of methane[J]. International Journal of Hydrogen Energy, 42(8): 5063-5071. DOI:10.1016/j.ijhydene.2016.10.114 |
| Li D L, Xu S P, Song K, et al. 2018. Hydrotalcite-derived Co/Mg(Al)O as a stable and coke-resistant catalyst for low-temperature carbon dioxide reforming of methane[J]. Applied Catalysis A: General, 552: 21-29. DOI:10.1016/j.apcata.2017.12.022 |
| Li L Y, Xie S D, Zeng L M, et al. 2015. Characteristics of volatile organic compounds and their role in ground-level ozone formation in the Beijing-Tianjin-Hebei region, China[J]. Atmospheric Environment, 113: 247-254. DOI:10.1016/j.atmosenv.2015.05.021 |
| Lima T M, Macedo V, Silva D S A, et al. 2020. Molybdenum-promoted cobalt supported on SBA-15:Steam and sulfur dioxide stable catalyst for CO oxidation[J]. Applied Catalysis B: Environmental, 277: 119248. DOI:10.1016/j.apcatb.2020.119248 |
| Madduluri V R, Nagaiah P, Prathap C, et al. 2018. Synergistic interface between Co3O4 and MgAl2O4 in CO2 assisted continuous vapour phase oxidative dehydrogenation of ethylbenzene to styrene monomer[J]. Arabian Journal of Chemistry, 13(1): 2883-2896. |
| Markov L, Lyubchova A. 1991. Precursor control of the inversion degree of magnesium-cobalt spinels[J]. Journal of Materials Science Letters, 10(9): 512-514. DOI:10.1007/BF00726922 |
| Meunier F C, Zuzaniuk V, Breen J P, et al. 2000. Mechanistic differences in the selective reduction of NO by propene over cobalt- and silver-promoted alumina catalysts: Kinetic and in situ DRIFTS study[J]. Catalysis today, 59(3): 287-304. |
| Mo Z W, Shao M, Lu S H, et al. 2015. Process-specific emission characteristics of volatile organic compounds (VOCs) from petrochemical facilities in the Yangtze River Delta, China[J]. Science of the Total Environment, 533: 422-431. DOI:10.1016/j.scitotenv.2015.06.089 |
| Mukherjee D, Park S E, Reddy B M. 2016. CO2 as a soft oxidant for oxidative dehydrogenation reaction: An eco benign process for industry[J]. Journal of CO2 Utilization, 16: 301-312. DOI:10.1016/j.jcou.2016.08.005 |
| Porosoff M D, Myint M N Z, Kattel S, et al. 2015. Identifying different types of catalysts for CO2 reduction by ethane through dry reforming and oxidative dehydrogenation[J]. Angewandte Chemie International Edition, 54(51): 15501-15505. DOI:10.1002/anie.201508128 |
| Raskó J, Kiss J. 2005. Adsorption and surface reactions of acetaldehyde on TiO2, CeO2 and Al2O3[J]. Applied Catalysis A: General, 287(2): 252-260. DOI:10.1016/j.apcata.2005.04.003 |
| Sanchez E V, Busca G, Lorenzelli V. 1990. Fourier transform infrared spectroscopic studies of the reactivity of vanadia-titanla catalysts toward olefins[J]. Journal of Chemical Physics, 94(26): 8945-8950. DOI:10.1021/j100389a018 |
| Sewell G S, Steen E, O'Connor C T. 1996. Use of TPR/TPO for characterization of supported cobalt catalysts[J]. Catalysis Letters, 37: 255-260. DOI:10.1007/BF00807763 |
| Shee D, Deo G. 2019. In situ DRIFT studies of alkane adsorption on vanadia supported titania-doped catalysts[J]. Catalysis Today, 325: 25-32. DOI:10.1016/j.cattod.2018.06.003 |
| Shee D, Sayari A. 2010. Light alkane dehydrogenation over mesoporous Cr2O3/Al2O3 catalysts[J]. Applied Catalysis A: General, 389(1/2): 155-164. |
| Simpson I J, Andersen M S, Meinardi S, et al. 2012. Long-term decline of global atmospheric ethane concentrations and implications for methane[J]. Nature, 488(7412): 490-494. DOI:10.1038/nature11342 |
| Souza G D, Ruoso C, Marcilio N R, et al. 2016. Dry reforming of methane over Mg-Co-Al mixed-oxides catalysts: Effect of Mg content and reduction conditions[J]. Chemical Engineering Communications, 203(6): 783-790. |
| Sun Y H, Zhang G J, Xu Y, et al. 2019. Catalytic performance of dioxide reforming of methane over Co/AC-N catalysts: Effect of nitrogen doping content and calcination temperature[J]. International Journal of Hydrogen Energy, 44(31): 16424-16435. DOI:10.1016/j.ijhydene.2019.04.250 |
| Talati A, Haghighi M, Rahmani F. 2016. Oxidative dehydrogenation of ethane to ethylene by carbon dioxide over Cr/TiO2-ZrO2 nanocatalyst: Effect of active phase and support composition on catalytic properties and performance[J]. Advanced Powder Technology, 27(4): 1195-1206. DOI:10.1016/j.apt.2016.04.003 |
| Tan P J, Gao Z H, Shen C F, et al. 2014. Ni-Mg-Al solid basic layered double oxide catalysts prepared using surfactant-assisted coprecipitation method for CO2 reforming of CH4[J]. Chinese Journal of Catalysis, 35(12): 1955-1971. DOI:10.1016/S1872-2067(14)60171-6 |
| Wang X Y, Liu Y, Zhang T H, et al. 2017. Geometrical-site-dependent catalytic activity of ordered mesoporous Co-based spinel for benzene oxidation: in situ DRIFTS study coupled with Raman and XAFS spectroscopy[J]. ACS Catalysis, 7(3): 1626-1636. DOI:10.1021/acscatal.6b03547 |
| 王勇, 贾雯, 毕莹. 2017. 效率视角下中国2030年二氧化碳排放峰值目标的省区分解-基于零和收益DEA模型的研究[J]. 环境科学学报, 37(11): 4399-4408. |
| Xia K, Lang W Z, Li P P, et al. 2015. Analysis of the catalytic activity induction and deactivation of PtIn/Mg(Al)O catalysts for propane dehydrogenation reaction[J]. RSC Advances, 5(79): 64689-64695. DOI:10.1039/C5RA11284B |
| Xie Z H, Yan B H, Lee J H, et al. 2019. Effects of oxide supports on the CO2 reforming of ethane over Pt-Ni bimetallic catalysts[J]. Applied Catalysis B: Environmental, 245: 376-388. DOI:10.1016/j.apcatb.2018.12.070 |
| Yang J, Liu H W, Martens W N, et al. 2009. Synthesis and characterization of cobalt hydroxide, cobalt oxyhydroxide, and cobalt oxide nanodiscs[J]. The Journal of Physical Chemistry C, 114(1): 111-119. |
| Yang R Y, Cui Y H, Yan Q H, et al. 2017. Design of highly efficient NOx storage-reduction catalysts from layered double hydroxides for NOx emission control from naphtha cracker flue gases[J]. Chemical Engineering Journal, 326: 656-666. DOI:10.1016/j.cej.2017.06.016 |
| 杨淑英, 陈立班. 1991. 我国的二氧化碳化学研究现状[J]. 化学进展, (1): 20-27. |
| 张法智, 徐柏庆. 2002. CO2选择氧化低碳烷烃的研究进展[J]. 化学进展, (1): 56-60. DOI:10.3321/j.issn:1005-281X.2002.01.008 |
| Zhao S, Hu F Y, Li J H. 2016. Hierarchical Core-shell Al2O3@Pd-CoAlO microspheres for low-temperature toluene combustion[J]. ACS Catalysis, 6(6): 3433-3441. DOI:10.1021/acscatal.6b00144 |
| Zhao X H, Wang X L. 2006. Oxidative dehydrogenation of ethane to ethylene by carbon dioxide over Cr/TS-1 catalysts[J]. Catalysis Communications, 7(9): 633-638. DOI:10.1016/j.catcom.2006.02.005 |
| Zhang X, Ye Q, Xu B Q, et al. 2007. Oxidative dehydrogenation of ethane over Co-BaCO3 catalysts using CO2 as oxidant: effects of Co promoter[J]. Catalysis Letters, 117(3/4): 140-145. DOI:10.1007/s10562-007-9122-9 |
| Zhang X. 2009. Hydrothermal synthesis and catalytic performance of high-surface-area mesoporous nanocrystallite MgAl2O4 as catalyst support[J]. Materials Chemistry and Physics, 116(2/3): 415-420. |
| Zhang Y H, Xiong H F, Liew K Y, et al. 2005. Effect of magnesia on alumina-supported cobalt Fischer-Tropsch synthesis catalysts[J]. Journal of Molecular Catalysis A: Chemical, 237(1/2): 172-181. |
| Zheng Y F, Yu Y Q, Zhou H, et al. 2020. Combustion of lean methane over Co3O4 catalysts prepared with different cobalt precursors[J]. RSC Advances, 10(8): 4490-4498. |
| Zhou R S, Snyder R L. 1991. Structures and transformation mechanisms of the eta, gamma and theta transition aluminas[J]. Acta Crystallographica, B47: 617-630. |
