 ,3,6, Chaozheng He,4, Hualong Tao5, Yonghao Xu3, Linghao Zhu1, Pengtao Wang1, Zhanying Zhang11Cultivating Base for Key Laboratory of Environment-Friendly Inorganic Materials in Henan Province, School of Materials Science and Engineering,
,3,6, Chaozheng He,4, Hualong Tao5, Yonghao Xu3, Linghao Zhu1, Pengtao Wang1, Zhanying Zhang11Cultivating Base for Key Laboratory of Environment-Friendly Inorganic Materials in Henan Province, School of Materials Science and Engineering, 2School of Mathematics and Informatics,
3School of Physics and Electronic Information Engineering,
4Institute of Environmental and Energy Catalysis, School of Materials Science and Chemical Engineering,
5Liaoning Key Materials Laboratory for Railway, School of Materials Science and Engineering,
First author contact:
Received:2019-10-18Revised:2019-12-18Accepted:2019-12-25Online:2020-02-21

Abstract
Keywords:
PDF (2749KB)MetadataMetricsRelated articlesExportEndNote|Ris|BibtexFavorite
Cite this article
Long Lin, Jingtao Huang, Weiyang Yu, Chaozheng He, Hualong Tao, Yonghao Xu, Linghao Zhu, Pengtao Wang, Zhanying Zhang. A periodic DFT study on adsorption of small molecules (CH4, CO, H2O, H2S, NH3) on the WO3 (001) surface-supported Au. Communications in Theoretical Physics, 2020, 72(3): 035501- doi:10.1088/1572-9494/ab690d
1. Introduction
In recent years, metal oxide semiconductor based sensor devices have been widely developed [1, 2]. Tungsten oxide (WO3) is one of the most intensively studied electrochromic materials because of its many technologically important electro-optic, electrochromic, ferroelectric, and semiconducting properties [3, 4]. The crystal structure of WO3 plays a crucial role in determining the physical properties such as electrical and surface topography. Therefore, the preparation of WO3 crystals with controllable size, size and crystal structure is of great significance for basic research as well as industrial and high-tech applications. So far, WO3 nano-materials have been successfully synthesized for various applications [5–7]. However, the semiconductor properties of WO3 are affected by many factors, including crystal form, octahedral tilt, surface hydrogenation, defects, etc [8–10]. WO3 and WO3.xH2O with nano-structures exhibit pronounced photochromism and that makes them to be a promising material used as gas sesors [11, 12], electrochromic device [13, 14], photo-electrochromic device [15], electrocatalyst [16, 17], photocatalyst [18], and so for. WO3 has important research value in gas-sensitive materials. Therefore, in the past few years, researches on the direction of WO3 gas-sensitivity have been endless. Tian et al [19] have studied the adsorption of CO on the oxygen deficient hexagonal of WO3(001) surface by using density functional theory (DFT), their results show that the existence of the oxygen vacancies decreased the sensitivity of the h-WO3(001) surface based gas sensor to CO gas. They also found that the lower defect density has the chance to decrease the sensitivity of the material substantially. Wang et al [20] have performed DFT calculations of the hydrogen adsorption on the monoclinic WO3(001) surface, and they found that dissociative adsorption of H2 is a weakly exothermic process at low H coverage but becomes weakly endothermic at high coverage. There have been many experimental and theoretical studies on adsorptions of small molecules on various surface planes of different WO3 phases.The mash gas is a toxic mixture of gases, mainly containing methane, carbon monoxide, hydrogen sulfide, water vapor, etc. Among them, methane (CH4) and CO etc gases are the most attractive and final candidates for future fuel and energy carriers. However, excessive inhalation of CH4 can cause nausea, fatigue, and increase heart rate. So detection of CH4 leakage is of great importance for wide application of CH4 energy. The doped sensitized WO3 nanoparticles are significantly more sensitive and selective than the pure WO3 nanoparticles [21]. This may be mainly attributed to the synergy between doped atom and WO3. It is expected that the sensitized WO3 nanoparticles thus prepared can also be used for research in other fields. WO3 nanostructures could be extensively applied in electrochromic and photochromic devices [22–25], lithium ion batteries [26, 18], photoelectrodes [27], photocatalysts [28–30], solar energy devices [31, 32], field electron emission [33, 34] and gas sensors [35–46] etc. As an important gas sensing material, the gas sensing properties can be adjusted due to the structure and morphology of the material, and more and more WO3 nanomaterials with novel structures or morphologies are synthesized. Experimentally, Gao et al [47] analyzed the structure changes during the hydrogen sensing coloring-bleaching cycles using both experimental and theoretical methods. Both the results show that the transition of WO3 from clusters into cross-linked networks will reduce the highest occupied molecular orbital-lowest unoccupied molecular orbital gap and increase the hydrogen desorption energy. The results also show that WO3/SiO2 film shows excellent reversibility and its structure keeps unchanged during the cycles. Hunge et al [48] synthesize WO3 and WO3/ZnO nanocomposite successfully by sonochemical route, the results show that the sonocatalytic degradation percentage of brilliant blue using WO3/ZnO nanocomposite has reached 90%, and a highest sonocatalytic degradation efficiency is achieved for WO3-ZnO nanocomposite than the WO3 nanoparticles. Theoretically, Oison et al [49] have clarified the microscopic mechanism of O2 and CO sensing on WO3 surfaces by a first principle study, the results show that CO is oxidized to CO2 on the WO3 surface, increasing the number of oxygen vacancies and the conductivity. Wijs et al [50] have studied several crystal structures of tungsten trioxide with a first-principles pseudo potential method. The results prove that the electronic band gap increases significantly with the distortion of the octahedra that are the building blocks of the various crystal structures. Su et al [51] use a model structure with twelve atomic layers to simulate WO3(010) surface, first-principles calculations further suggest that surface fluorination brings in an unoccupied impurity state in the band structure of WO3, which exhibits a strong correlation with the hydroxyl group of benzyalcohol and thus bridges the interaction between surfaces and alcohols.
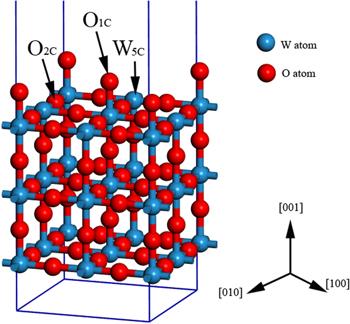
Up to now, a large number of literature have verified the effectiveness of first-principle calculation in the delineation of sensing mechanism on various surfaces [52, 53]. Although there have been many studies on the adsorption and dissociation of gas molecules (such as CH4, CO, H2O, H2S, NH3, etc) on metals, the problem of deposition on WO3 materials has never been considered. As seen in figure 2, we selected three adsorption sites as ${{\rm{O}}}_{1C}$, ${{\rm{O}}}_{2C}$, ${{\rm{W}}}_{5C}$ for the adsorption sites of intrinsic and Au doped WO3. In this paper, we performed the DFT calculations to reveal the sensing mechanism of WO3-based gas sensors. The geometric structures, stability, electronic and adsorptive properties of WO3 are calculated by using the first-principles calculation. Bulk and clean surface models of WO3 were firstly built and optimized. In addition, as far as we know, there are few studies on the electronic structures and adsorption properties of small molecules (CH4, CO, H2O, H2S, NH3) on the WO3 (001) surface-supported Au. How about the order of the surface adsorption energy for these gases? What is the mechanism of the adsorption of gas on the surface of WO3? Therefore, the electronic structures and adsorption properties of small molecules (CH4, CO, H2O, H2S, NH3) on the WO3 (001) surface-supported Au were systematically studied by first-principles method. The gas molecules (such as CH4, CO, H2O, H2S, NH3) adsorption on the most stable WO3 surface was then calculated. Dopants of different Au sites are tested, and alignments of pairs of dopants along different sites are examined. Both geometrical and electronic structures were analyzed to investigate the gas molecules sensing mechanism. In the research work of many scholars, Au is used to doped or load metal oxides to change the adsorption or catalytic performance of the system [54–61]. However, as far as we all know, few works have been used to study the gas-sensitivity properties of Au-doped WO3. In the study described in this paper, we have performed the DFT calculations to reveal the sensing mechanism of Au-doped WO3-based gas sensors.
2. Computational details
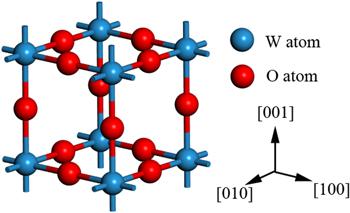
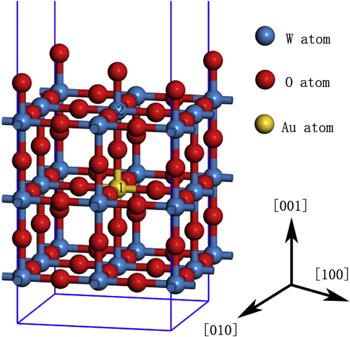
Bulk cubic phase WO3 was chosen as the starting point of our calculations, as presented in figure 1. Cubic WO3, with space group Pm3m, is an ideal model of WO3. It provides a simple and reasonable model to resemble most properties of other WO3 phases, since all WO3 polymorphs share a similar corner-sharing octahedral structure. The difference only lies in that the cubic WO3 ignores some details such as octahedral tilting and W atom displacement. Nevertheless it has been widely studied in many precious theoretical calculations for its simplicity [50]. All calculations are performed using periodic supercells, which consist in a slab of several atomic planes and a vacuum layer with a thickness of 15 Å. To test the reliability of the method and establish the reference, we have first performed a detailed calculation for the ideal WO3. Both experimental and theoretical studies have shown that the (001) surface is the most stable WO3 surface [62]. The unit cell of WO3 was optimized, and the corresponding lattice constants were used to build the 2×2 WO3(001) surface. The slab was fully relaxed and the effects caused by fixing the geometry of the slab will be discussed below. The adsorption model of the optimal adsorption sites for different gases are shown in figure 6.Figure 1.
 New window|Download| PPT slide
New window|Download| PPT slideFigure 1.Bulk model of the cubic WO3.
In this work, we use the first-principles plane-wave ultrasoft pseudopotential based on the DFT [63–65] system and Cambridge Sequential Total Energy Package [66] software package for calculations, and the generalized gradient approximation [67] with Perdew–Burke–Ernzerhof exchange correlation functional was used in all calculations.
Figure 2.
 New window|Download| PPT slide
New window|Download| PPT slideFigure 2.Surface reconstruction model of WO3(001) surface.
Population analysis of the resulting projected states is then performed using the Mulliken formalism. This technique is widely used in the analysis of electronic structure calculations performed with Linear Combination of Atomic Orbitals basis sets. In this study, we are concerned with differential charge density, density of states (DOS), band structures, etc. Therefore, the method is suitable for our calculations. At the same time, there are considerable literature reports using Mulliken population analysis calculation [68–71].
The smooth part of the wavefunctions was expanded in plane waves with a kinetic energy cutoff of 500 eV. A grid of 5×5×3 Monkhorst–Pack k-point mesh was used for surface simulations with a large supercell. The convergence criteria of the energy is set to 2×10−5 eV/atom. All atoms were relaxed until the largest component in the forces for each atom becomes smaller than 0.05 eV Å−1. The maximum displacement is 2×10−4 nm, and stresses are smaller than 0.1 GPa, respectively. During the surface structure optimization, atoms in all layers were allowed to relax with fixed cell size.
3. Results and discussion
3.1. Bulk properties
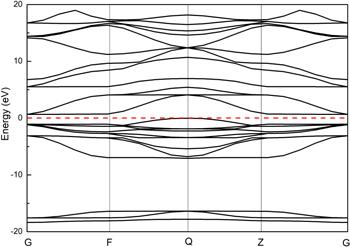
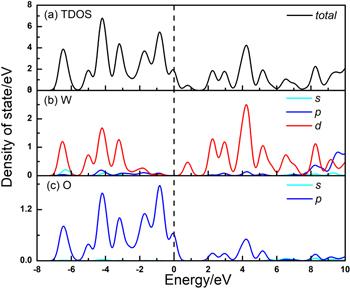
In the first part of the investigation, we performed a volume and atoms position relaxation for the cubic WO3 unit cell. In this way, we begin with a description of the bulk phases of WO3. The geometrical unit cell of cubic WO3 is represented in figure 1, which contains one six-fold coordinated W atom and three two-fold coordinated O atoms. The W atom is located at the center of an octahedron made of six O atoms, and each O atom bridges two W atoms with W–O bonds. During the synthesis and preparation of materials, various defects will occur. For example, in WO3, W vacancy or O vacancy are likely to be generated. Therefore, it is necessary to calculate the formation energy of the material to form vacancies. The forming energy of O vacancy and W vacancy are 6.84 eV and 18.09 eV, respectively. The calculation results prove that compared with W vacancy, O vacancy are easier to form.In condensed matter physics, the band theory of solids is the fundamental concept used in understanding the physical and electronic properties of materials, such as conductivity, thermal conductivity, infrared absorption, and the optical dielectric function. To study the adsorption properties of WO3, the electronic structure of the system was first studied. The electronic structure of WO3 is discussed by combining the band structure and the DOSs. Our calculated lattice parameters and bandgaps for the unit cell of bulk phases are listed in table 1, in comparison with previous theoretical calculation results. The results of a=3.82 Å and Eg=0.65 eV agree quite well with those of previous self-consistent calculations: a=3.73–3.84 Å and Eg=0.3−0.7 eV [74–76], as well as the experimental value of a=3.71–3.85 Å [77]. The band gap, however, is much smaller than the experimental value of 2.62 eV (for monoclinic phase) due to the well-known LDA error. Nevertheless, as an effective approximation method, the calculation results are very accurate and do not affect the analysis of the density states, which is well consistent with the previous results, as seen in table 1. The band structure and DOS are shown in figures 3 and 4. For W atom, the 5d and 6s states are treated as valence states; for O atom, the valence states are the 2s and 2p states. The PDOS results suggest that the valence band maximum (VBM) is dominated by O: 2p states (−2/0 eV), while the conduction band minimum has mainly W:5d states with a small mixing of the O:2p states(0/5 eV).
Figure 3.
 New window|Download| PPT slide
New window|Download| PPT slideFigure 3.Band structure of bulk cubic WO3, the Fermi level is set to 0 eV.
Figure 4.
 New window|Download| PPT slide
New window|Download| PPT slideFigure 4.Total and partial density of states for (a) cubic WO3 (b) W atom and (c) O atom. The Fermi level is set to 0 eV.
Table 1.
Table 1.Bulk properties of cubic WO3 in comparison with other calculations.
| a/Å | Eg/eV | |
|---|---|---|
| This work | 3.82 | 0.65 |
| Yakovkin et al [72] | 3.81 | 0.60 |
| Wijs et al [50] | 3.83 | 0.40 |
| Tian et al [73] | 3.82 | 0.59 |
New window|CSV
For the adsorption of gases molecules, we have examined three configurations, depending on the orientation of molecules with respect to the WO3 monolayer surface. To compare the surface stability of different reconstructions, surface energy σ is defined as:
Figure 5.
 New window|Download| PPT slide
New window|Download| PPT slideFigure 5.Different sites of Au stom doped WO3(001) surface.
3.2. Electronic structures and CH4 gas sensing mechanism
Adsorption reactions of CH4 on WO3 surface are of critical importance to understand the CH4 sensing mechanism. In this part, we consider three types of CH4 dissociative adsorption models on the WO3(001) surface as displayed in figure 2. As shown in figure 2, we selected three adsorption sites, which were labeled as ${{\rm{O}}}_{1C}$, ${{\rm{O}}}_{2C}$, and ${{\rm{W}}}_{5C}$, respectively. In order to find the most stable adsorption site, the following adsorption formula was used to calculate the adsorption energy. The adsorption energy formula of ${{\rm{O}}}_{1C}$ and ${{\rm{O}}}_{2C}$ is defined as follows:The adsorption energy formula of ${{\rm{W}}}_{5C}$ is defined as follows:
Figure 6.
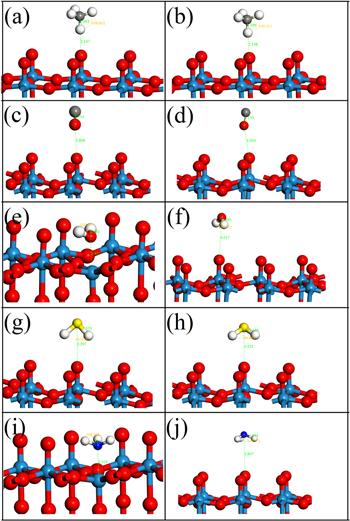 New window|Download| PPT slide
New window|Download| PPT slideFigure 6.Adsorption models of different gases on pure and Au-doped WO3(001) surfaces. Among them, (a), (c), (e), (g), (i) are the adsorption models of CH4, CO, H2O, H2S, NH3 gases on the pure WO3(001) surface, (b), (d), (f), (h), (j) are the adsorption models of CH4, CO, H2O, H2S, NH3 gases on Au-doped WO3(001) surface, respectively.
Table 2.
Table 2.Adsorption energies of different adsorption molecule(Eads in eV), Charge transfer from the WO3(001) surface to the adsorbed molecule (Q in e).
| Type of gas | Sites | EAds (eV) | Q | Style |
|---|---|---|---|---|
| CH4 | O1C | −0.57 | −0.48 | Acceptor |
| CO | O1C | −0.13 | −0.32 | Acceptor |
| H2O | W5C | −0.92 | 0.17 | Donor |
| H2S | O1C | −0.36 | −0.17 | Acceptor |
| NH3 | W5C | −1.04 | −0.13 | Acceptor |
New window|CSV
Table 3.
Table 3.Adsorption energies of different adsorption molecule(Eads in eV), Charge transfer from the Au–WO3 monolayer to the adsorbed molecule (Q in e).
| Type of gas | Sites | EAds (eV) | Q | Style |
|---|---|---|---|---|
| CH4 | O1C | −0.57 | −0.42 | Acceptor |
| CO | O1C | −0.37 | −0.35 | Acceptor |
| H2O | O1C | −0.76 | 0.19 | Donor |
| H2S | O1C | −0.61 | −0.12 | Acceptor |
| NH3 | O1C | −1.43 | −0.15 | Acceptor |
New window|CSV
CH4 adsorption not only affects the surface structure of WO3(001), but also changes the electronic structure substantially. To investigate the origin on the electronic and adsorptive properties of the instrinsic and Au-doping WO3(001) adsorptions of CH4, we calculated the total density of states (TDOS) of WO3(001) and Au doped WO3(001) adsorbs CH4 gas molecules. We also calculated the DOS of WO3 and CH4 for comparison, as shown in figure 7. The DOS/PDOS comparisons of the intrinsic WO3(001) surface before and after CH4 molecules adsorption are shown in figures 7(a) and (b), respectively.
Figure 7.
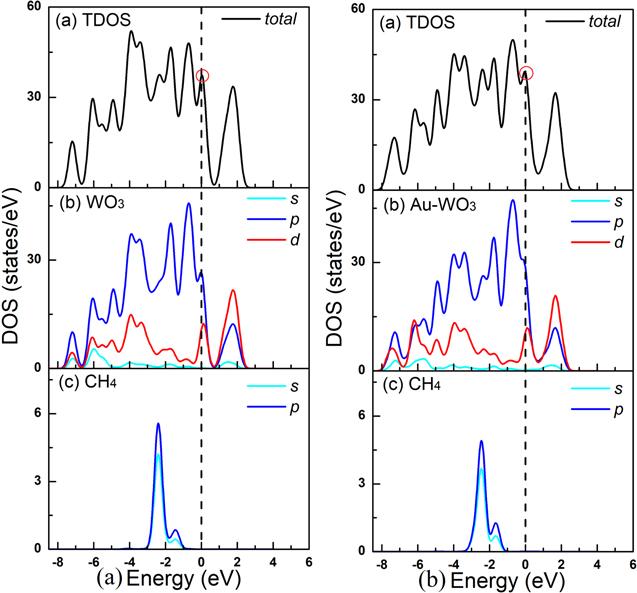 New window|Download| PPT slide
New window|Download| PPT slideFigure 7.Total and partial density of states of CH4 molecule adsorption on the pure WO3(001) surface (a) and CH4 molecule adsorption on the Au doped-WO3(001) surface (b).
As seen in figure 7, the electron levels of the conduction band (0/0.4 eV) that lies in the Fermi level is mainly composed of the O:2p states, and the electron levels of the conduction band (0.5/2.5 eV) that lies above the Fermi level is mainly composed of the W:5p states electron contributions from the tungsten atoms. As shown in figure 7, by comparing the TDOS before and after doping Au atoms, it can be seen that the TDOS curve near Fermi level moves towards high energy after doping Au atoms. The most obvious change is that all bands shift towards high energies after adsorption, leading to the partial filling of the VBM. PDOS analysis suggests that the new occupied electronic states (−3/−2) involve mainly 5d states of W with slight 2p states of O. Compare with the undoped system, the electron levels of the conduction band (0/2 eV) that lies above the Fermi level is mainly composed of the Au:5d states electron contributions from the Au atoms, and the electron levels of the conduction band (0/2 eV) that lies above the Fermi level is mainly composed of the Au:5d states electron contributions from the tungsten atoms. Comparing the TDOS before and after doping with Au CH4 molecules adsorption, we found that there is almost no much change in the peak near the Fermi level, which confirm that the doping of Au atoms has little effect on the adsorption of CH4 gas. For further research, we calculated the band structures. As shown in figures 13(a) and (b), comparing the band structure of CH4 adsorption before and after Au doping, we can see that there is not much change at the Fermi level, which is consistent with the DOS result calculated earlier.
In addition, in order to further understand the sensing mechanism of CH4, we have calculated the charge density difference of CH4 adsorption. It can be seen from the redistribution of charge after adsorbing CH4 from the WO3(001) face ${{\rm{O}}}_{1C}$ site that the charge difference is mainly concentrated on the CH4 gas molecule and the WO3 surface. However, when CH4 is adsorbed on the Au-doped WO3(001) plane, its charge distribution changes greatly. In addition, some electrons converge between the surface of CH4 and WO3, forming a spatial connection as a channel for charge transfer. This connection facilitates the accelerated flow of current, reducing the resistivity of the sensor and thereby increasing the output voltage. More importantly, these electrons stimulate electron mobility and redistribution near the surface, and the corresponding reaction increases the high energy properties of WO3(001) plane, promoting better binding of the substrate surface to more adsorbed CH4 gases, and ultimately leads to more high response, as observed from previous measurement in experimental part [21].
The charge differential density can intuitively reflect the charge transfer after molecular adsorption, which is defined as:
Then, we discuss the bond formation and electron structure changes after gas adsorption under these two conditions before and after doping. Figure 9 shows the charge differential density of adsorbed gases before and after doping Au. Yellow represents electron aggregation and blue represents electron absence. The results show that adsorption only affects the electron distribution of surface atoms. By charge differential density, we can see that the amount of electrons gained and lost in CH4 gas has little change after Au doping, which is confirmed by the calculation results of Mulliken charge arrangement number.
3.3. Electronic structures and CO gas sensing mechanism
To study the effect of WO3(001) surface on the gas sensing properties of CO molecule, we established an adsorption model for CO molecules adsorbed above the surface features of the WO3(001) surface, as shown in figure 2. Based on the surface model, a CO molecular model was established and the adsorption distance was set to 3.00 Å. We also set three adsorption sites for ${{\rm{O}}}_{1C}$, ${{\rm{O}}}_{2C}$ and ${{\rm{W}}}_{5C}$. After the optimization of the adsorption model geometry and the energy calculation, the adsorption energy of the CO adsorption on the adsorption sites of the WO3(001) plane can be obtained. By calculating the adsorption energy of CO molecule at different adsorption sites, we can conclude that the adsorption energy of CO molecule at the ${{\rm{O}}}_{1C}$ adsorption site is lower, indicating that CO molecule is more easily adsorbed at this site. As shown in tables 2 and 3, the adsorption energy of the CO molecule at the ${{\rm{O}}}_{1C}$ adsorption site was −0.13 eV, and the charge transfer amount was 0.32 e. Since the intrinsic WO3(001) surface adsorbs CO molecule, its surface and CO molecule have less charge transfer, so its sensitivity to CO molecule is lower. In order to improve its molecule sensitivity, some transition metals such as Cu, Pd, Au, etc, are often used to increase the sensitivity of metal oxide semiconductor materials to CO molecule. So, we further calculated the adsorption of CO molecules on the Au-doped WO3(001) surface, and the adsorption model chosen was consistent with the ones mentioned above. By calculating the adsorption energy of different adsorption sites, we found that the ${{\rm{O}}}_{1C}$ position is the most stable adsorption site on the Au-doped WO3(001) plane. As shown in table 2, the adsorption energy of the CO molecule at ${{\rm{O}}}_{1C}$ was 0.37 eV, and the charge transfer amount was 0.35 e. Compared with the intrinsic WO3, the adsorption energy of the CO molecule at ${{\rm{O}}}_{1C}$ of WO3(001) doped with Au atom on the plane is lower, indicating that the WO3 after doping Au is more likely to adsorb CO molecules than the intrinsic WO3.In order to analyze the mechanism of the influence of Au atom before and after doping on the adsorption performance of the system. We further calculated the DOSs of the intrinsic WO3 and Au-doped WO3 adsorbed CO molecule, as shown in figure 8. As can be seen in figures 8(a) and (b) represent the DOSs of CO molecule adsorption on the pure WO3(001) surface and CO molecule adsorption on the Au doped-WO3(001) surface. It can be seen from the figure 8 that the value of CO molecule adsorbed at the Fermi level of the WO3(001) plane system after Au doping (15.5 eV) is higher than that of the CO adsorbed intrinsic WO3(001) surface system (6.5 eV). There is a significant improvement, indicating that after the doping of the Au atom, the electron concentration at the Fermi level increases, making the system more active and improving the gas sensitivity of the material. It can be seen from figures 13(c) and (d) that in the adsorption system doped by Au atom, the band structures near the Fermi level is enhanced, which is consistent with the DOS results.
Figures 9(c) and (d) are charge differential density for two forms of adsorption, with yellow indicating electron aggregation and blue indicating electron loss. The calculation results show that the adsorption only affects the electron distribution of the surface atoms, so only the atoms on the surface are selected as the research object. By analyzing the charge differential density, we found that the surface electron loss of CO after Au doping is significantly stronger than that of the intact surface. It can be seen that the Au-doped surface is significantly more active than the intact surface. It can be confirmed by the calculation result of Mulliken charges layout that the charge transfer amount between the intrinsic WO3(001) surface and CH4 is 0.32 e, while the charge transfer amount between WO3(001) surface and CH4 after Au doping is 0.35 e. This also shows that the amount of charge transfer after doping with Au increases, and the adsorption effect on CO molecules is improved.
3.4. Electronic structures and H2O gas sensing mechanism
For the adsorption of H2O molecules, we have studied six configurations based on the orientation of the molecules relative to the surface of WO3(001) surface. In the intrinsic WO3(110) surface adsorption H2O molecular system, the most stable configuration is the ${{\rm{W}}}_{5C}$ position, as shown in figure 2. The calculated adsorption energy for this configuration is −0.92 eV, which is larger than that for the CO and CH4 adsorption by −0.13 and −0.57 eV. According to the charge transfer analysis, 0.17 e has provided electrons from the H2O molecule and thus acts as an electron donor, which is also true for the configuration, as shown in figure 2. Furthermore, we consider the effect of Au atom doped on the adsorption properties of the system. We found that due to the introduction of Au atom, the adsorption energy of the system was reduced to −0.76 eV. Compared with the un-doped Au atom system, the adsorption performance of the system on H2O molecules is reduced to some extent, which can also be reflected in Band structures, as shown in figures 13(e) and (f).In order to further understand the reasons for the change of adsorption performance of the system before and after Au doping, we further calculated the DOSs of H2O molecules adsorbed before and after Au doping, as shown in figure 10. From the TDOS, we can clearly see that near the Fermi energy level, the value of H2O molecular adsorbed at WO3(001) surface is about 14.5 eV. As we can see in figure 10(b), near the Fermi energy level, the value of TDOS of H2O molecule adsorption on the Au doped-WO3(001) surface is about 12.5 eV. Compared with intrinsic WO3, the peak value of TDOS in the system doped with Au atom decreases at the Fermi energy level, which is also corresponding to the previously calculated adsorption energy.
Figure 8.
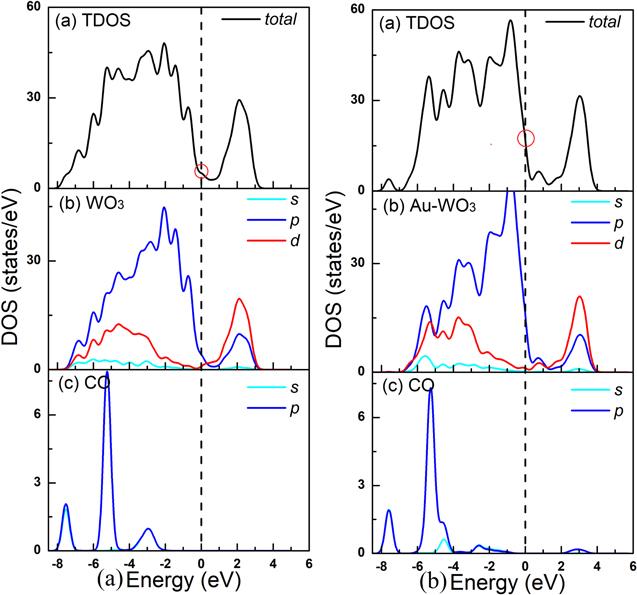 New window|Download| PPT slide
New window|Download| PPT slideFigure 8.Total and partial density of states of CO molecule adsorption on the pure WO3(001) surface (a) and CO molecule adsorption on the Au doped-WO3(001) surface (b).
3.5. Electronic structures and H2S gas sensing mechanism
In the process of chemical production, natural gas and mineral deposit survey, the safety of production is greatly affected by the presence of H2S gas. The development of a gas sensor capable of detecting trace H2S gas has very high economic value. In recent years, in order to improve the sensing performance of H2S gas, a series of new gas sensing materials such as SnO2 have been researched and developed. So far, little research has been done on the adsorption properties of H2S on metal oxides such as WO3. Here we study the adsorption mechanism of H2S adsorption on WO3 (001) surface.In order to determine the adsorption position of H2S on the WO3 (001) plane, we first calculated the adsorption energy of different models. The adsorption energy is defined as shown in formula (
Figure 9.
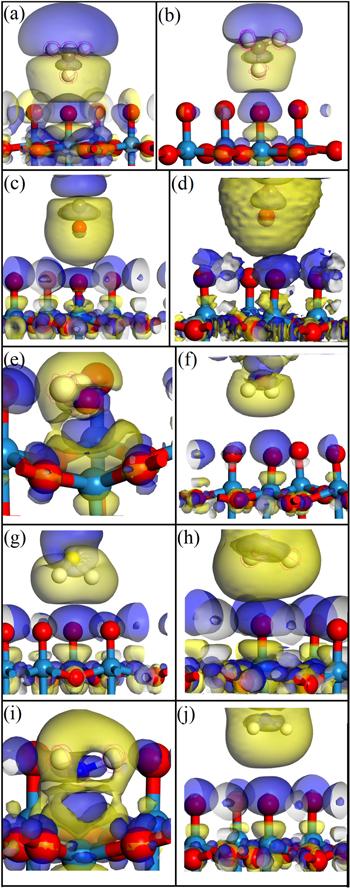 New window|Download| PPT slide
New window|Download| PPT slideFigure 9.Charge differential density of different gases on pure and Au-doped WO3(001) surfaces. Among them, (a), (c), (e), (g), (i) are the charge differential density of CH4, CO, H2O, H2S, NH3 gases on the pure WO3(001) surface, (b), (d), (f), (h), (j) are the charge differential density of CH4, CO, H2O, H2S, NH3 gases on Au-doped WO3(001) surface, respectively.
Figure 10.
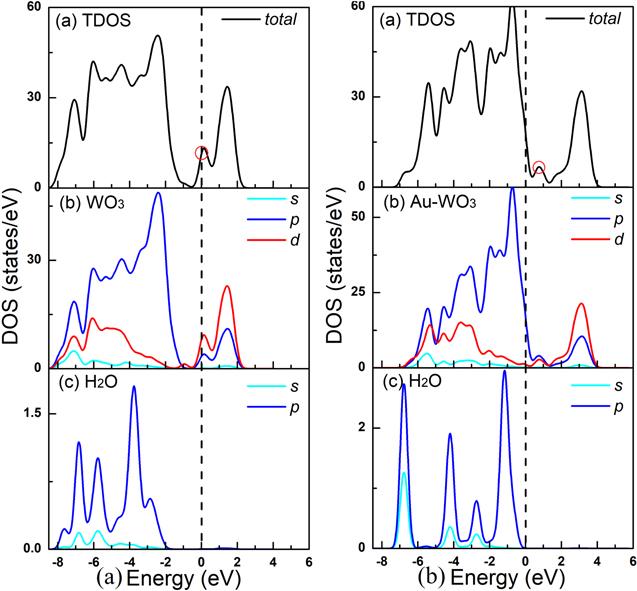 New window|Download| PPT slide
New window|Download| PPT slideFigure 10.Total and partial density of states of H2O molecule adsorption on the pure WO3(001) surface (a) and H2O molecule adsorption on the Au doped-WO3(001) surface (b).
As shown in figure 11, we calculated the DOSs before and after doping with Au. It can be clearly seen from the figure 11 that the TDOSs after doping with Au atom increases significantly at the peak near the Fermi level. An increase in the peak at the Fermi level means that the H2S gas is more readily adsorbed on the Au-doped WO3(001) surface than the un-doped WO3 (001) surface, which is consistent with figures 13(g) and (h).
Figure 11.
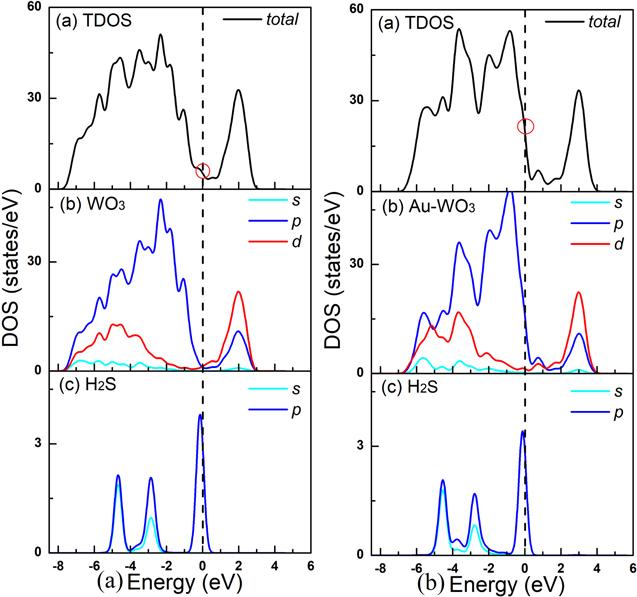 New window|Download| PPT slide
New window|Download| PPT slideFigure 11.Total and partial density of states of H2S molecule adsorption on the pure WO3(001) surface (a) and H2S molecule adsorption on the Au doped-WO3(001) surface (b).
3.6. Electronic structures and NH3 gas sensing mechanism
Finally, we consider the adsorption properties of the gas small molecule NH3 on the surface of WO3(001) surface. In the case of NH3 adsorption, six kinds of configurations have been tested with the adsorption energy determines the best adsorption site to the WO3(001) and Au-doped WO3(001) surface. The most stable configuration of NH3 molecule adsorption on the intrinsic WO3(001) surface and NH3 molecule adsorption on the Au-doped WO3(001) surface are ${{\rm{W}}}_{5C}$ and ${{\rm{O}}}_{1C}$ adsorption sites, as shown in figure 5, and the calculated data are listed in tables 2 and 3. As shown in table 2, the adsorption energy of NH3 molecule adsorption on the pure WO3(001) surface was −1.04 eV, and the charge transfer from WO3 is 0.13 e. Adsorption energy of Au doped system is −1.43 eV, and the charge transfer from Au-doped WO3 is 0.15 e. It can be seen from our calculation results that, in the system doped with Au atom, the adsorption performance of Au-doped WO3 on NH3 molecules is significantly improved, and the charge transfer is also significantly increased, which is conducive to the adsorption of NH3 in the system. Consistent with the calculation results, the band structure can also highlight this point. It can be seen from figures 13(i) and (j) that there can be a noticeably dense trend near the Fermi level.To clarify the nature of the effect of the best adsorption site to the WO3(001) and Au-doped WO3(001) surface on the electronic and adsorptive property of this system, we calculated the DOSs for NH3 molecule adsorption on the un-doped WO3(001) surface and NH3 molecule adsorption on the Au doped-WO3(001) surface, as shown in figure 12. As can be seen from the figure 12, the Fermi energy level changes greatly compared with the adsorption effect of NH3 and Au-doped WO3 before and after doping. As a result, it has a great influence on the TDOS. Compared with the un-doping system, the peak value of the TDOS near the Fermi level increases significantly, which is also an important reason for the enhancement of gas sensitivity performance. Our calculations provide an effective guide for the application of WO3 based gas sensing materials to NH3 gas adsorption.
Figure 12.
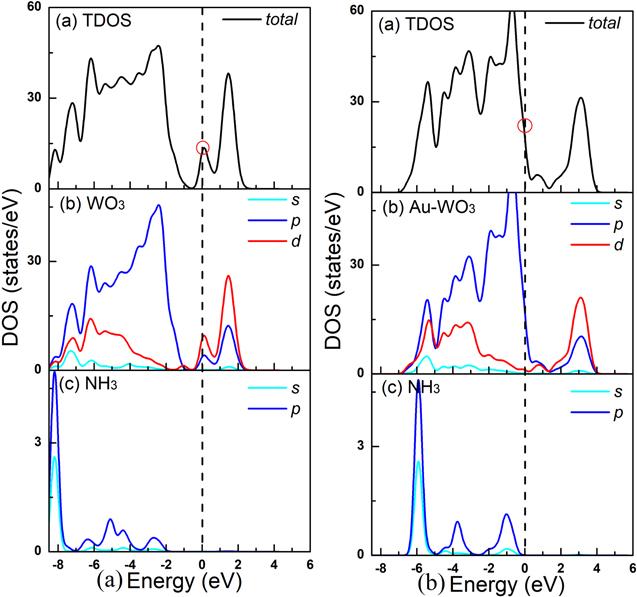 New window|Download| PPT slide
New window|Download| PPT slideFigure 12.Total and partial density of states of NH3 molecule adsorption on the pure WO3(001) surface (a) and NH3 molecule adsorption on the Au doped-WO3(001) surface (b).
Figure 13.
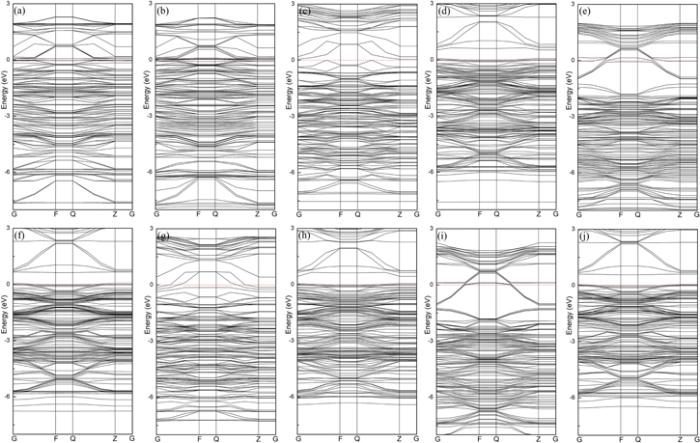 New window|Download| PPT slide
New window|Download| PPT slideFigure 13.Band structures of different gases on pure and Au-doped WO3(001) surfaces. Among them, (a), (c), (e), (g), (i) are the band structures of CH4, CO, H2O, H2S, NH3 gases on the pure WO3(001) surface, (b), (d), (f), (h), (j) are the band structures of CH4, CO, H2O, H2S, NH3 gases on Au-doped WO3(001) surface, respectively.
4. Conclusions
In summmary, we have presented the electronic structures and adsorptive property of Au-doped WO3 with adsorbents CH4, CO, H2O, H2S and NH3 molecules, using DFT method. Firstly, we performed a volume and atoms position relaxation for the cubic WO3 unit cell. The results indicates that WO3 is a gas sensitive material with the band gap 0.65 eV. After energy calculation of several low index surfaces, a WO3(001) surface with the lowest surface energy, and the most stable electronic structures have been calculated. The WO3(001) surface is examined for CH4, CO, H2O, H2S and NH3 molecules sensing mechanism due to its advantages of electronic stability and low energy. Comparing the adsorption of the same gas before and after doping with Au atoms, we found that there is a certain correspondence between the adsorption energy and TDOS near the Fermi level. For example, the adsorption energy of NH3, H2S, and CO gas increases after doping with Au. At the same time, TDOS near the Fermi level also increases to a certain extent. The adsorption energy of H2O gas decreases after doping with Au, and at the same time, the TDOS near the Fermi level also decreases. Our calculation results show that when the five kinds of gas molecules are adsorbed on the surface of the pure WO3 (001) surface, the order of surface adsorption performance is CO > H2S > CH4 > H2O > NH3. And the results show that NH3 is the most easily adsorbed gas among the four gases adsorbed on the pure WO3 (001) surface. We also calculated the five different gases on the Au-doped WO3(001) surface, and the order of adsorption energy was found to be different from the previous calculation: CO > CH4 > H2S > H2O > NH3. The results of adsorption energy, charge transfer, adsorption distance show that NH3 is the most easily adsorbed gas on the surface of WO3. The results provide a new route for the potential applications of Au doped WO3 in gas molecule adsorption system and conducible to the understanding of the sensing mechanism of WO3-based gas sensors.Acknowledgments
This work was supported by the Key Projects of National Natural Science Foundation of China (U1704255), the National Natural Science Foundation of China (11804081), the National Natural Science Foundation of China (Grant No. 21603109), the Henan Joint Fund of the National Natural Science Foundation of China (Grant No. U1404216), the Natural Science Foundation of Henan Province (182102210305), the Natural Science Foundation of Henan Province (19B430003, 20A430016, 182300410288), the Key Research Project for the Universities of Henan Province (19A140009), the Doctoral Foundation of Henan Polytechnic University (B2018-38), and the Open Project of Key Laboratory of Radio Frequency and Micro-Nano Electronics of Jiangsu Province (LRME201601). Computational resources have been provided by the Henan Polytechnic University high-performance grid computing platform.Reference By original order
By published year
By cited within times
By Impact factor
DOI:10.1021/jp0756350 [Cited within: 1]
DOI:10.1063/1.1543983 [Cited within: 1]
DOI:10.1364/AO.8.S1.000192 [Cited within: 1]
[Cited within: 1]
DOI:10.1007/s10562-012-0785-5 [Cited within: 1]
DOI:10.1039/c2jm15837j
DOI:10.1039/c000055h [Cited within: 1]
DOI:10.1103/PhysRevB.89.235106 [Cited within: 1]
DOI:10.1021/acs.jpcc.6b02707
DOI:10.1021/jp201057m [Cited within: 1]
DOI:10.1021/cr0681086 [Cited within: 1]
DOI:10.1023/A:1023436725457 [Cited within: 1]
DOI:10.1155/2013/785023 [Cited within: 1]
DOI:10.1021/la061299n [Cited within: 1]
DOI:10.1007/s12274-010-1031-3 [Cited within: 1]
DOI:10.1039/B918783A [Cited within: 1]
DOI:10.1088/0022-3727/41/15/155417 [Cited within: 1]
DOI:10.1007/s12274-012-0266-6 [Cited within: 2]
DOI:10.1016/j.apsusc.2014.05.069 [Cited within: 1]
DOI:10.1021/jp302210y [Cited within: 1]
DOI:10.1142/S1793604718500716 [Cited within: 2]
DOI:10.1021/cm0403816 [Cited within: 1]
DOI:10.1039/C2TA00545J
DOI:10.1002/adfm.201000261
DOI:10.1021/jp3090777 [Cited within: 1]
DOI:10.1016/j.apsusc.2009.10.085 [Cited within: 1]
DOI:10.1021/am200897n [Cited within: 1]
DOI:10.1016/j.apcatb.2009.11.003 [Cited within: 1]
DOI:10.1039/c0cc01237h
DOI:10.1016/S0926-860X(00)00796-1 [Cited within: 1]
DOI:10.1021/la103692y [Cited within: 1]
DOI:10.1039/c2jm16178h [Cited within: 1]
DOI:10.1021/cg100995f [Cited within: 1]
DOI:10.1002/smll.200400054 [Cited within: 1]
DOI:10.1016/j.snb.2009.08.031 [Cited within: 1]
DOI:10.1016/S0925-4005(03)00383-6
DOI:10.1016/j.jeurceramsoc.2007.09.001
DOI:10.1016/j.tsf.2008.10.021
DOI:10.1039/c2jm16709c
DOI:10.1016/j.snb.2010.06.055
DOI:10.1016/j.snb.2008.01.044
DOI:10.1088/0957-4484/21/3/035501
DOI:10.1016/j.snb.2004.05.015
DOI:10.1016/j.snb.2008.01.023
DOI:10.1016/j.snb.2010.11.001
DOI:10.1016/j.tsf.2010.10.026 [Cited within: 1]
DOI:10.1016/j.snb.2012.06.028 [Cited within: 1]
DOI:10.1016/j.ultsonch.2018.02.052 [Cited within: 1]
DOI:10.1016/j.snb.2011.08.018 [Cited within: 1]
DOI:10.1103/PhysRevB.59.2684 [Cited within: 3]
DOI:10.1016/j.apcatb.2017.05.075 [Cited within: 1]
DOI:10.1039/C6TA04291K [Cited within: 1]
DOI:10.1016/j.spmi.2014.11.027 [Cited within: 1]
DOI:10.1016/j.molcata.2012.07.014 [Cited within: 1]
DOI:10.1016/j.molcata.2006.02.058
DOI:10.1021/acscatal.7b00251
DOI:10.1023/A:1019038503569
DOI:10.1016/S0926-860X(02)00575-6
DOI:10.1006/jcat.1999.2619
DOI:10.1023/A:1018918017777
DOI:10.1023/A:1019037902776 [Cited within: 1]
DOI:10.1103/PhysRevB.52.R14392 [Cited within: 2]
DOI:10.1103/PhysRevLett.77.3865 [Cited within: 1]
DOI:10.1103/RevModPhys.61.689
DOI:10.1103/PhysRevB.45.13244 [Cited within: 1]
DOI:10.1088/0953-8984/14/11/301 [Cited within: 1]
DOI:10.1088/0953-8984/20/6/064201 [Cited within: 1]
DOI:10.1016/j.apsusc.2016.07.112 [Cited within: 1]
DOI:10.1142/S0217984918501397
DOI:10.1016/j.apsusc.2008.09.006
DOI:10.1002/advs.201500101 [Cited within: 1]
DOI:10.1016/j.susc.2007.01.013 [Cited within: 1]
DOI:10.3866/PKU.WHXB201203021 [Cited within: 2]
DOI:10.1103/PhysRevB.35.8246 [Cited within: 1]
DOI:10.1103/PhysRevB.54.2436
DOI:10.1103/PhysRevB.56.983 [Cited within: 1]
DOI:10.1016/S0167-2738(01)00806-2 [Cited within: 1]
DOI:10.1088/1674-1056/20/10/102101 [Cited within: 1]
DOI:10.1088/1674-1056/20/8/082101 [Cited within: 1]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}