全文HTML
--> --> -->随着计算材料科学的发展, 材料的理论研究有助于材料成分设计及其性能的提高[17-19]. 固体与分子经验电子理论在材料的电子结构和性能研究方面, 具有丰富的研究成果, 涉及材料的力学性能、热性能、电磁性能和光特性等方面. 性能的理论计算与实验相符[20-22]. 其优势在于: 理论模型简单, 没有涉及复杂的微积分及数理方程计算, 而且计算参数少.
本文应用固体与分子经验电子理论研究液态金属电池Na||Sb-Pb-Sn, 并通过价电子结构的分析和热、电性能(电极熔点, 结合能, 电势, 开路电压)计算, 揭示电池性能与电子结构的关联性.
2
2.1.价电子结构的三种假设
32.1.1.双态的假设
电子结构存在两种基本的状态: 能量低的稳态(h态)及能量高的激发态(t态), 双态分布着各种各样的电子, 如: s, p, d轨道上的价电子ns, np, nd; 共价电子nc; 晶格电子nl; 哑对电子nY; 磁电子m3d及总电子nT.3
2.1.2.杂化态叠加的假设
依据量子力学态叠加原理, 物质的电子结构是由h态和t态成分比例分数Ch和Ct的叠加. 其比例分数Ch和Ct是不连续的, 呈现量子化. 其计算公式为
3
2.1.3.键距方程
对于晶胞内任何两个相邻的u和v原子所形成的u—v键, 其键矩可以根据Pauling提供的键距计算公式:


2
2.2.键距差方法[20,21]:
第1条键:












2
2.3.性能计算模型
根据价电子结构参数, 建立结合能理论模型. 能量由四项组成: 键能、晶格电子电位能、磁能及内壳层电子的内聚能. 结合能









熔化模型: 热声子能量随着温度的升高增加. 当达到温度达到临界值(熔点





开路电压V: 对于液态金属电池而言, 开路电压等于阳、阴电极的电势差. 应用阳、阴电极的电位能差除以其总的晶格电子数

3.1.阴极的价电子结构与热电性能计算
为了降低电池阴极的熔点, 在阴极Na金属中添加熔点低的IA族碱金属元素K, Rb, Cs形成Na1–xIAx合金电极(x ≤ 0.05). Na具有体心立方结构, 空间群为











| Na1–xIAx | $ {I}_{\alpha } $ | $ {D}_{\mathrm{uv}}\left({n}_{\alpha }\right) $/? | ${\bar{D}}_{\mathrm{uv}}\left({n}_{\alpha }\right) $/? | $ {n}_{\rm{A}}$ | $ { I}_{\alpha } $ | $ {D}_{\mathrm{uv}}\left({n}_{\alpha }\right) $/${ \text{?} }$ | ${\bar{D} }_{\mathrm{uv} }\left({n}_{\alpha }\right)/{ \text{?} }$ | $ {n}_{\alpha } $ | |?D|/${\text{?} }$ |
| Na | 8 | 3.7296 | 3.7502 | 0.05160 | 6 | 4.3004 | 4.3210 | 0.00810 | 0.0206 |
| Na0.99K0.01 | 8 | 3.7381 | 3.7538 | 0.05220 | 6 | 4.3103 | 4.3260 | 0.00820 | 0.0157 |
| Na0.99Rb0.01 | 8 | 3.7430 | 3.7572 | 0.05220 | 6 | 4.3157 | 4.3300 | 0.00820 | 0.0143 |
| Na0.99Cs0.01 | 8 | 3.7454 | 3.7628 | 0.05220 | 6 | 4.3187 | 4.3361 | 0.00810 | 0.0175 |
| Na0.98K0.02 | 8 | 3.7465 | 3.7574 | 0.05290 | 6 | 4.3202 | 4.3310 | 0.00823 | 0.0108 |
| Na0.98Rb0.02 | 8 | 3.7564 | 3.7643 | 0.05290 | 6 | 4.3310 | 4.3389 | 0.00820 | 0.0079 |
| Na0.98Cs0.02 | 8 | 3.7611 | 3.7755 | 0.05290 | 6 | 4.3370 | 4.3513 | 0.00817 | 0.0143 |
| Na0.97K0.03 | 8 | 3.7550 | 3.7610 | 0.05348 | 6 | 4.3301 | 4.3361 | 0.00829 | 0.0060 |
| Na0.97Rb0.03 | 8 | 3.7697 | 3.7713 | 0.05351 | 6 | 4.3463 | 4.3479 | 0.00825 | 0.0016 |
| Na0.97Cs0.03 | 8 | 3.7769 | 3.7881 | 0.05354 | 6 | 4.3552 | 4.3665 | 0.00821 | 0.0113 |
| Na0.96K0.04 | 8 | 3.7635 | 3.7647 | 0.05411 | 6 | 4.3399 | 4.3411 | 0.00834 | 0.0012 |
| Na0.96Rb0.04 | 8 | 3.7831 | 3.7785 | 0.05415 | 6 | 4.3616 | 4.3570 | 0.00829 | 0.0047 |
| Na0.96Cs0.04 | 8 | 3.7926 | 3.8009 | 0.05419 | 6 | 4.3735 | 4.3818 | 0.00824 | 0.0082 |
| Na0.95K0.05 | 8 | 3.7719 | 3.7684 | 0.05474 | 6 | 4.3498 | 4.3463 | 0.00840 | 0.0036 |
| Na0.95Rb0.05 | 8 | 3.7965 | 3.7856 | 0.05479 | 6 | 4.3769 | 4.3661 | 0.00834 | 0.0109 |
| Na0.95Cs0.05 | 8 | 3.8084 | 3.8136 | 0.05484 | 6 | 4.3918 | 4.3971 | 0.00827 | 0.0052 |
表1Na1–xIAx合金键距计算
Table1.Calculation of bond distances of Na1–xIAx alloy.
Na1–xIAx合金的价电子结构计算结果如表2所示. 基体金属Na取第2杂阶, 掺杂金属K, Rb, Cs取第4杂阶. 随着IA族元素的掺杂量的增加, 共价电子数nc, s与p价电子数ns与np趋于增加, 晶格电子数nl减少. 将这些价电子结构参数代入计算性能模型, 计算合金的热、电性能. 计算结果见表3.
| Na1–xIAx | nc | ns | np | nl | R(1) |
| Na | 0.4614 | 0.4606 | 0.0008 | 0.5386 | 1.4181 |
| Na0.99K0.01 | 0.4668 | 0.4660 | 0.0008 | 0.5332 | 1.4217 |
| Na0.98K0.02 | 0.4722 | 0.4713 | 0.0008 | 0.5278 | 1.4254 |
| Na0.97K0.03 | 0.4776 | 0.4767 | 0.0009 | 0.5224 | 1.4290 |
| Na0.96K0.04 | 0.4830 | 0.4821 | 0.0009 | 0.5170 | 1.4327 |
| Na0.95K0.05 | 0.4884 | 0.4875 | 0.0009 | 0.5116 | 1.4363 |
| Na0.99Rb0.01 | 0.4668 | 0.4660 | 0.0008 | 0.5332 | 1.4235 |
| Na0.98Rb0.02 | 0.4722 | 0.4713 | 0.0008 | 0.5278 | 1.4289 |
| Na0.97Rb0.03 | 0.4776 | 0.4767 | 0.0009 | 0.5224 | 1.4343 |
| Na0.96Rb0.04 | 0.4830 | 0.4821 | 0.0009 | 0.5170 | 1.4397 |
| Na0.95Rb0.05 | 0.4884 | 0.4875 | 0.0009 | 0.5116 | 1.4451 |
| Na0.99Cs0.01 | 0.4668 | 0.4660 | 0.0008 | 0.5332 | 1.4263 |
| Na0.98Cs0.02 | 0.4722 | 0.4713 | 0.0008 | 0.5278 | 1.4345 |
| Na0.97Cs0.03 | 0.4776 | 0.4767 | 0.0009 | 0.5224 | 1.4428 |
| Na0.96Cs0.04 | 0.4830 | 0.4821 | 0.0009 | 0.5170 | 1.4510 |
| Na0.95Cs0.05 | 0.4884 | 0.4875 | 0.0009 | 0.5116 | 1.4592 |
表2Na阴极合金的价电子结构
Table2.Valence electron structures of cathode Na based alloy.
| 掺杂量x | 原子 | 杂阶 | 掺杂 | 杂阶 | $ \bar{T}_{\rm{m}} $/K | $ {E}_{\mathrm{c}} $/(eV·atom–1) | $ {\bar{E}}_{\mathrm{c}} $/(eV·atom–1) | $\left| { {\Delta E}_{\mathrm{c} } }/{ {E}_{\mathrm{c} } }\right|/{\%}$ | 电势/V |
| 0 | Na | 3 | — | — | 336.76 | 1.113 | 1.165 | 4.67 | 0.1482 |
| 0.01 | Na | 2 | K | 4 | 336.64 | 1.111 | 1.164 | 4.77 | 0.1481 |
| 0.01 | Na | 2 | Rb | 4 | 336.45 | 1.110 | 1.163 | 4.77 | 0.1480 |
| 0.01 | Na | 2 | Cs | 4 | 336.02 | 1.103 | 1.161 | 5.26 | 0.1478 |
| 0.02 | Na | 2 | K | 4 | 336.64 | 1.109 | 1.163 | 4.39 | 0.1481 |
| 0.02 | Na | 2 | Rb | 4 | 336.14 | 1.108 | 1.161 | 4.78 | 0.1478 |
| 0.02 | Na | 2 | Cs | 4 | 335.29 | 1.110 | 1.158 | 4.32 | 0.1475 |
| 0.03 | Na | 2 | K | 4 | 336.60 | 1.103 | 1.162 | 5.35 | 0.1480 |
| 0.03 | Na | 2 | Rb | 4 | 335.85 | 1.109 | 1.159 | 4.51 | 0.1476 |
| 0.03 | Na | 2 | Cs | 4 | 334.58 | 1.108 | 1.154 | 4.15 | 0.1471 |
| 0.04 | Na | 2 | K | 4 | 336.57 | 1.107 | 1.162 | 4.97 | 0.1479 |
| 0.04 | Na | 2 | Rb | 4 | 335.57 | 1.103 | 1.157 | 4.90 | 0.1474 |
| 0.04 | Na | 2 | Cs | 4 | 333.88 | 1.109 | 1.151 | 3.79 | 0.1467 |
| 0.05 | Na | 2 | K | 4 | 336.55 | 1.108 | 1.161 | 4.78 | 0.1478 |
| 0.05 | Na | 2 | Rb | 4 | 335.30 | 1.107 | 1.156 | 4.43 | 0.1472 |
| 0.05 | Na | 2 | Cs | 4 | 333.20 | 1.108 | 1.147 | 3.52 | 0.1463 |
表3阴极Na1–xIAx合金的熔点、结合能与电势
Table3.Melting point, cohesive energy, and electric potentials of cathode Na1–xIAx alloy.
计算结果表明: 阴极合金Na1–xIAx的熔点与结合能随着IA族原子掺杂量x的增加趋于降低; Cs原子掺杂降低更显著. 其电位能和熔点变化与结合能变化情况相似, 随着K, Rb, Cs原子掺杂量的增加而降低. 当掺杂量相同时, Cs的Ec最低, K时Ec最高. 阴极合金的电势变化情况与熔点和结合能一致. 计算结合能与实验值相符. 实验值由Na和IA族元素的单质的内聚能[32]按成分比例平均获得:
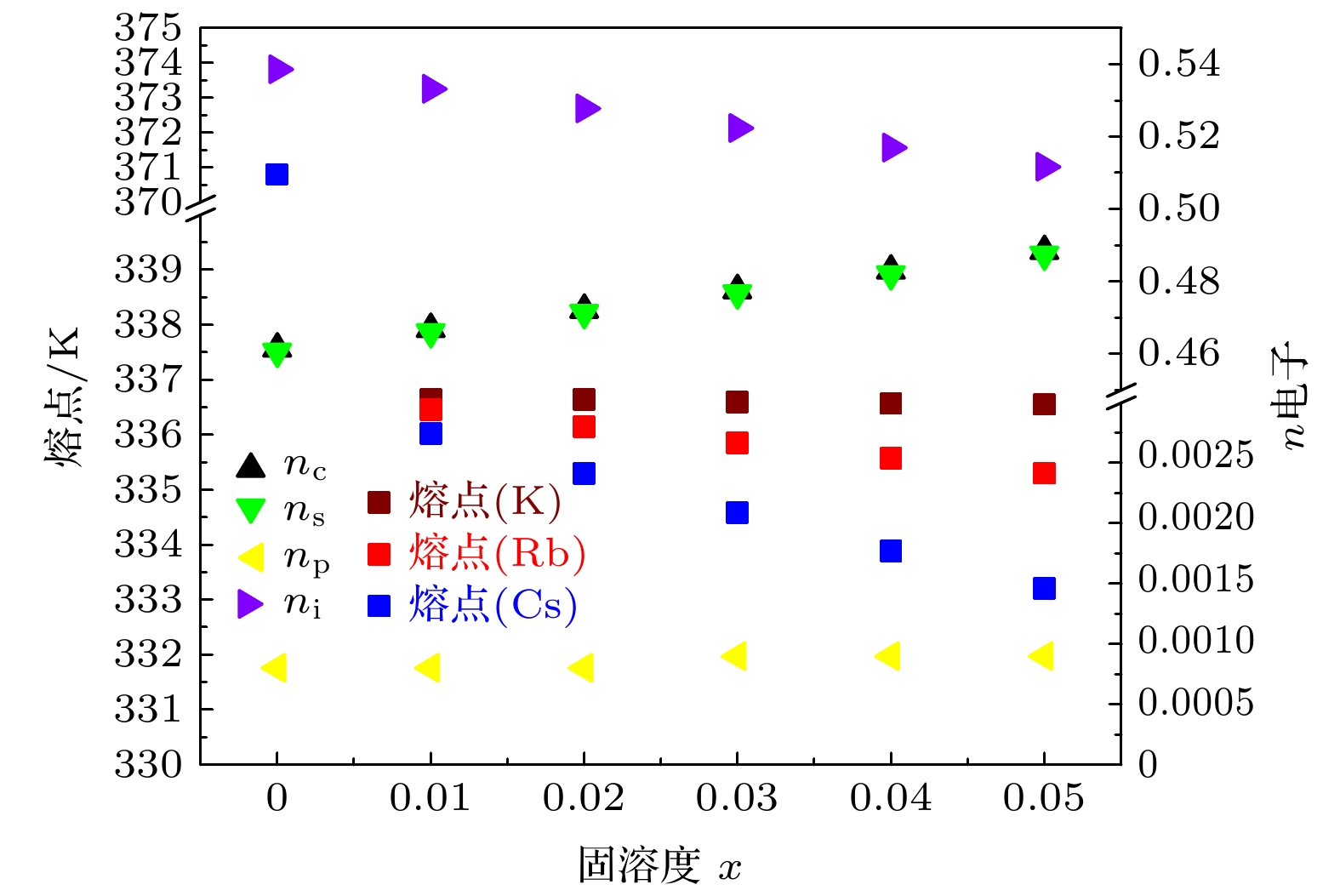 图 1 阴极Na1–xIAx合金的n电子数与熔点的关联图
图 1 阴极Na1–xIAx合金的n电子数与熔点的关联图Figure1. Correlations between various electron numbers and melting points of Na1–xIAx cathode alloys.
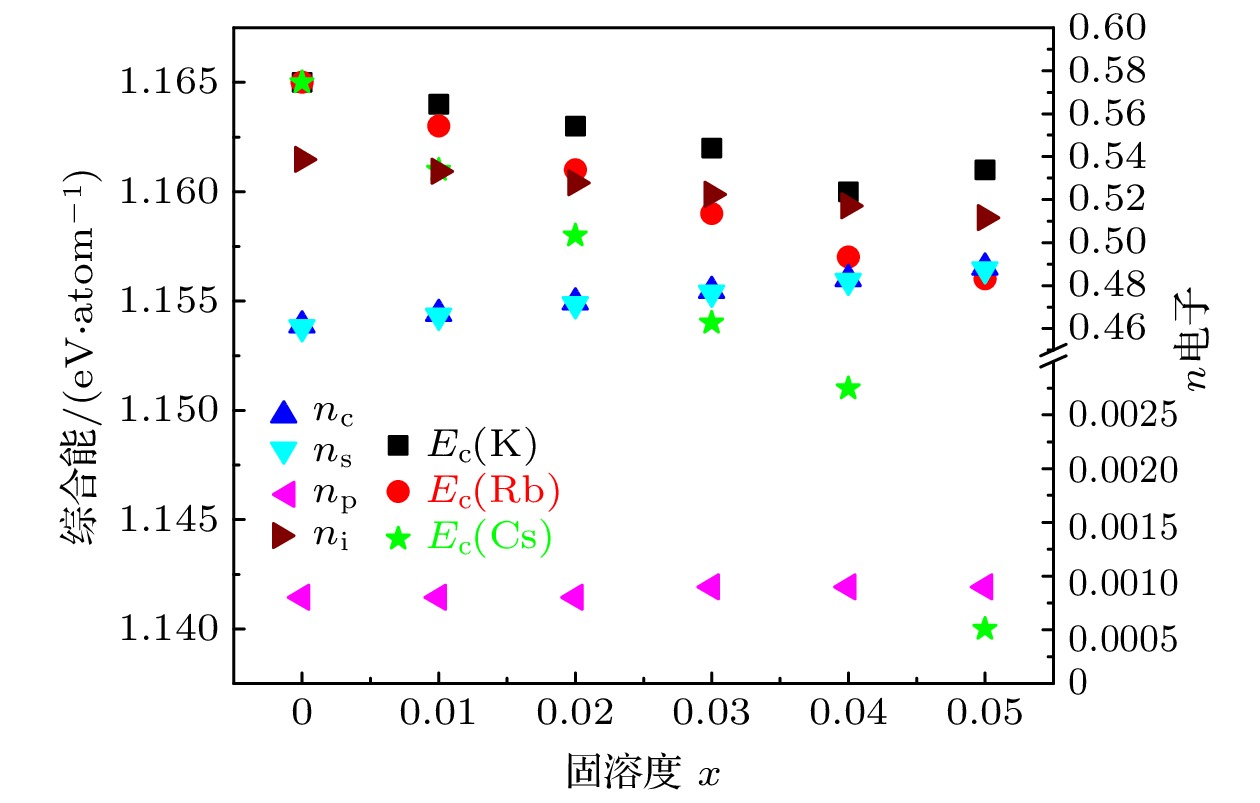 图 2 阴极Na1–xIAx合金的各种电子数与结合能的关联图
图 2 阴极Na1–xIAx合金的各种电子数与结合能的关联图Figure2. Correlations between various electrons and cohesive energy of Na1–xIAx cathode alloys.
 图 3 阴极Na1–xIAx合金的各种电子数与电势的关联图
图 3 阴极Na1–xIAx合金的各种电子数与电势的关联图Figure3. Correlations of various electrons and electric potentials for Na1–xIAx anode alloys.
2
3.2.正极合金价电子结构与热、电性能计算
本文选择Pb, Sb, Sn三种元素来设计液态金属电池的阳极合金; Sn, Pb, Sb相对于其他金属, 具有较高的电负性, 适合作为液态金属电池正极材料[33-34]. Na离子输运到阳极, 与阳极金属Pb, Sb, Sn反应形成阳极产物(合金化合物): Na3Sb, NaSn, Na15Sn4, NaPb. 四种合金化合物的晶体结构, 见表4.| 合金 | 空间群 | a/$\text{?}$ | b/$\text{?}$ | c/$\text{?}$ | 原子 | 占位 | x | y | z |
| Sb | 2c | 0.3333 | 0.6666 | 0.2500 | |||||
| Na3Sb | P63mmc (194) | 5.355 | 5.355 | 9.496 | Na1 | 2b | 0 | 0 | 0.2500 |
| Na2 | 4f | 0.3333 | 0.6666 | 0.5830 | |||||
| NaSn | I41/acd (142) | 10.460 | 10.460 | 17.390 | Sn | 32g | 0.0696 | 0.1260 | 0.9362 |
| Na1 | 16f | 0.6258 | 0.8758 | 0.1250 | |||||
| Na2 | 16e | 0.8724 | 0 | 0.2500 | |||||
| Sn | 16c | 0.2083 | 0.2083 | 0.2083 | |||||
| Na15Sn4 | I43d (220) | 13.140 | 13.140 | 13.140 | Na1 | 12a | 0.3750 | 0 | 0.2500 |
| Na2 | 48e | 0.1270 | 0.1548 | 0.9670 | |||||
| Pb | 32g | 0.0696 | 0.1186 | 0.9383 | |||||
| NaPb | I41/acd (142) | 10.580 | 10.580 | 17.746 | Na1 | 16e | 0.2500 | 0.1250 | 0.5000 |
| Na2 | 16f | 0.1250 | 0.3750 | 0.6250 |
表4正极合金的晶体结构
Table4.Crystal structures of anode alloys.
应用键距差方法计算这四种合金化合物中的所有键距, 计算值与实验相符, 最大键距差都满足判据: |ΔD| ≤ 0.05 ?. 计算结果见表5.
| 合金 | 键序 | 成键原子 | $ {I}_{\alpha } $ | $ {D}_{\mathrm{uv}}\left({n}_{\alpha }\right) $/$\text{?}$ | $ {\bar{D}}_{\mathrm{uv}}\left({n}_{\alpha }\right)/$$\text{?}$ | $ {n}_{\alpha } $ | |ΔD|/$\text{?}$ |
| Na3Sb | 1 | Sb-Na2 | 6 | 3.0975 | 3.0910 | 0.39055 | 0.0065 |
| 2 | Sb-Na1 | 4 | 3.1685 | 3.1620 | 0.19416 | 0.0065 | |
| 3 | Na1-Na2 | 4 | 3.1780 | 3.1715 | 0.18030 | 0.0065 | |
| 4 | Na1-Na1 | 6 | 3.4769 | 3.4704 | 0.03738 | 0.0065 | |
| 5 | Sb-Na1 | 12 | 3.4813 | 3.4748 | 0.05846 | 0.0065 | |
| 6 | Na2-Na1 | 12 | 3.4813 | 3.4748 | 0.05630 | 0.0065 | |
| 7 | Na2-Na1 | 12 | 4.4310 | 4.4245 | 0.00147 | 0.0065 | |
| 8 | Na2-Na2 | 2 | 4.7575 | 4.7510 | 0.00064 | 0.0065 | |
| NaSn | 1 | Sn-Sn | 2 | 2.9748 | 3.0201 | 0.42650 | 0.0453 |
| 2 | Sn-Sn | 4 | 2.9925 | 3.0378 | 0.39849 | 0.0453 | |
| 3 | Na1-Sn | 4 | 3.3355 | 3.3808 | 0.07506 | 0.0453 | |
| 4 | Na1-Sn | 4 | 3.3592 | 3.4045 | 0.06854 | 0.0453 | |
| 5 | Na2-Sn | 4 | 3.3974 | 3.4427 | 0.13064 | 0.0453 | |
| 6 | Na2-Sn | 4 | 3.4231 | 3.4684 | 0.11837 | 0.0453 | |
| 7 | Na1-Sn | 4 | 3.4870 | 3.5323 | 0.04197 | 0.0453 | |
| 8 | Na2-Sn | 2 | 3.5225 | 3.5678 | 0.08083 | 0.0453 | |
| 9 | Na2-Sn | 4 | 3.5482 | 3.5935 | 0.07324 | 0.0453 | |
| 10 | Na1-Na2 | 4 | 3.6148 | 3.6601 | 0.03985 | 0.0453 | |
| 11 | Na1-Na2 | 4 | 3.6658 | 3.7111 | 0.03277 | 0.0453 | |
| 12 | Na1-Na1 | 1 | 3.7218 | 3.7671 | 0.01197 | 0.0453 | |
| 13 | Sn-Sn | 2 | 3.7406 | 3.7859 | 0.02257 | 0.0453 | |
| 14 | Sn-Sn | 2 | 4.3780 | 4.4233 | 0.00196 | 0.0453 | |
| 15 | Na1-Na2 | 4 | 4.4919 | 4.5372 | 0.00138 | 0.0453 | |
| 16 | Na1-Sn | 4 | 4.6674 | 4.7127 | 0.00045 | 0.0453 | |
| 17 | Na2-Na2 | 1 | 4.7095 | 4.7548 | 0.00132 | 0.0453 | |
| Na15Sn4 | 1 | Sn-Na2 | 24 | 3.2378 | 3.2854 | 0.20850 | 0.0476 |
| 2 | Na2-Na2 | 24 | 3.2624 | 3.3100 | 0.19499 | 0.0476 | |
| 3 | Na1-Na2 | 24 | 3.3425 | 3.3901 | 0.11017 | 0.0476 | |
| 4 | Na2-Na2 | 12 | 3.3468 | 3.3944 | 0.14830 | 0.0476 | |
| 5 | Sn-Na2 | 24 | 3.4049 | 3.4525 | 0.12127 | 0.0476 | |
| 6 | Sn-Na2 | 24 | 3.4189 | 3.4665 | 0.11589 | 0.0476 | |
| 7 | Na1-Na2 | 24 | 3.5026 | 3.5502 | 0.06555 | 0.0476 | |
| 8 | Sn-Na1 | 24 | 3.5482 | 3.5958 | 0.05582 | 0.0476 | |
| 9 | Na2-Na2 | 24 | 3.8138 | 3.8614 | 0.03261 | 0.047 | |
| 10 | Na2-Na2 | 24 | 3.9794 | 4.0270 | 0.01906 | 0.0476 | |
| 11 | Na2-Na2 | 12 | 4.1712 | 4.2188 | 0.01023 | 0.0476 | |
| NaPb | 1 | Pb-Pb | 2 | 3.1464 | 3.1452 | 0.33477 | 0.0013 |
| 2 | Pb-Pb | 4 | 3.1618 | 3.1606 | 0.31556 | 0.0013 | |
| 3 | Pb-Na2 | 4 | 3.3653 | 3.3641 | 0.19895 | 0.0013 | |
| 4 | Pb-Na1 | 4 | 3.3888 | 3.3876 | 0.08237 | 0.0013 | |
| 5 | Pb-Na2 | 4 | 3.4215 | 3.4203 | 0.16035 | 0.0013 | |
| 6 | Pb-Na2 | 4 | 3.4847 | 3.4835 | 0.12582 | 0.0013 | |
| 7 | Pb-Na1 | 4 | 3.4929 | 3.4917 | 0.05524 | 0.0013 | |
| 8 | Pb-Na1 | 4 | 3.5549 | 3.5537 | 0.04354 | 0.0013 | |
| 9 | Pb-Na1 | 4 | 3.6172 | 3.6160 | 0.03428 | 0.0013 | |
| 10 | Pb-Pb | 2 | 3.6418 | 3.6406 | 0.05001 | 0.0013 | |
| 11 | Na1-Na2 | 8 | 3.6967 | 3.6955 | 0.03479 | 0.0013 | |
| 12 | Na2-Na2 | 1 | 3.7406 | 3.7394 | 0.06488 | 0.0013 | |
| 13 | Pb-Pb | 2 | 4.4008 | 4.3996 | 0.00272 | 0.0013 | |
| 14 | Na1-Na2 | 4 | 4.5455 | 4.5443 | 0.00134 | 0.0013 | |
| 15 | Pb-Na2 | 4 | 4.7513 | 4.7501 | 0.00097 | 0.0013 |
表5阳极合金的键距
Table5.Bond distances of the anode alloy.
四种阳极合金的价电子结构计算结果如表6所示. Na3Sb合金化合物: 2c晶位的Sb取第2杂阶, 只提供价电子; 2b晶位的Na取4杂阶, 只提供价电子; 4f晶位的Na取第2杂阶, 提供晶格电子. NaSn合金化合物: 晶格电子由16e晶位的Na及32g晶位的Sn所提供. Sn也提供价电子, 然而位于16f晶位的Na只提供价电子. Na15Sn4合金化合物: 晶格电子由16c晶位的Sn及48e晶位的Na所提供, 位于12a晶位的Na只提供价电子. NaPb合金化合物: 位于32g晶位的Pb提供价电子和晶格电子; 16e晶位的Na只提供价电子; 位于16f晶位的Na只提晶格电子.
| 合金 | 原子 | 杂阶 | nc | ns | np | nl | R(1) |
| Na3Sb | Sb | 2 | 3.0000 | 0.5694 | 2.4306 | 0 | 1.4279 |
| Na1 | 4 | 1.0000 | 0.9982 | 0.0018 | 0 | 1.3070 | |
| Na2 | 2 | 0.4614 | 0.4606 | 0.0008 | 0.5386 | 1.4181 | |
| NaSn | Sn | 1 | 2.0000 | 0 | 2.0000 | 2.0000 | 1.3990 |
| Na1 | 1 | 1.0000 | 0.9982 | 0.0018 | 0 | 1.3070 | |
| Na2 | 4 | 0 | 0 | 0 | 1.0000 | 1.5133 | |
| Na15Sn4 | Sn | 4 | 3.6638 | 0.8319 | 2.8319 | 0.3362 | 1.3990 |
| Na1 | 4 | 1.0000 | 0.9982 | 0.0018 | 0 | 1.3070 | |
| Na2 | 3 | 0.5350 | 0.5340 | 0.0010 | 0.4650 | 1.4029 | |
| NaPb | Pb | 2 | 2.0962 | 0.0481 | 2.0481 | 1.9038 | 1.4300 |
| Na1 | 4 | 1.0000 | 0.9982 | 0.0018 | 0 | 1.3070 | |
| Na2 | 1 | 0 | 0 | 0 | 1.0000 | 1.5133 |
表6阳极产物的价电子结构
Table6.Valence electron structures of anode products
依据电子结构参数, 计算阳极合金化合物的热、电性能, 理论熔点与实验相符. 计算结果由表7所示. 计算结果表明: 四种阳极合金化合物的熔点的范围在604.89—1142.96 K之间, NaPb的熔点最低, Na3Sb的熔点最高. 然而电势, Na3Sb最高1.152 V, NaSn的最低0.7343 V.
| 合金 | Tm/K [35] | $ \bar{T}_{\rm{m}} $/K | |${\Delta {T}_{\mathrm{m} } }/{ {T}_{\mathrm{m} } }$|/% | 电势/V | n | β | Ec/(eV·atom–1) |
| Na3Sb | 1129 | 1142.96 | 1.2 | 1.1520 | 4 | 0.60 | 1.766 |
| NaSn | 851 | 813.16 | 4.4 | 0.7343 | 5 | 0.60 | 2.103 |
| Na15Sn4 | 681 | 746.16 | 9.6 | 0.9074 | 3 | 0.71 | 1.318 |
| NaPb | 645 | 630.68 | 2.2 | 0.8263 | 6 | 0.60 | 1.559 |
表7正极合金的熔点、结合能与电势
Table7.Melting point, cohesive energy, and electric potentials of anode alloy.
2
3.3.液态金属电池的开路电压
应用开路电压(18)式系统地计算阴、阳电极之间的开路电压. 表8列出金属电极及其开路电压值. 计算结果表明: Na||Sb电极之间的开路电压最高略高于1 V. Na||Sn电极的开路电压低, 约为前者的一半. 开路电压与平均每原子的晶格电子数成反比, 平均晶格电子数最低的Na3Sb(0.2693), 其开路电压最高; 而平均晶格电子最高的NaSn, 其开路电压则最低.| Na1–xIAx | 开路电压/V | |||
| Na3Sb | NaSn | Na15Sn4 | NaPb | |
| Na | 1.0038 | 0.5861 | 0.7592 | 0.6781 |
| Na0.09K0.01 | 1.0039 | 0.5862 | 0.7593 | 0.6782 |
| Na0.98K0.02 | 1.0039 | 0.5862 | 0.7593 | 0.6782 |
| Na0.97K0.03 | 1.0040 | 0.5863 | 0.7594 | 0.6783 |
| Na0.96K0.04 | 1.0041 | 0.5864 | 0.7595 | 0.6784 |
| Na0.95K0.05 | 1.0042 | 0.5865 | 0.7596 | 0.6785 |
| Na0.99Rb0.01 | 1.0040 | 0.5863 | 0.7594 | 0.6783 |
| Na0.98Rb0.02 | 1.0042 | 0.5865 | 0.7596 | 0.6785 |
| Na0.97Rb0.03 | 1.0044 | 0.5867 | 0.7598 | 0.6787 |
| Na0.96Rb0.04 | 1.0046 | 0.5869 | 0.7600 | 0.6789 |
| Na0.95Rb0.05 | 1.0048 | 0.5871 | 0.7602 | 0.6791 |
| Na0.99Cs0.01 | 1.0042 | 0.5865 | 0.7596 | 0.6785 |
| Na0.98Cs0.02 | 1.0045 | 0.5868 | 0.7599 | 0.6788 |
| Na0.97Cs0.03 | 1.0049 | 0.5872 | 0.7603 | 0.6792 |
| Na0.96Cs0.04 | 1.0053 | 0.5876 | 0.7607 | 0.6796 |
| Na0.95Cs0.05 | 1.0057 | 0.5880 | 0.7611 | 0.6800 |
| nl/atom | 0.2693 | 1.2500 | 0.3645 | 1.0682 |
表8电池的开路电压
Table8.Open gate voltages of the battery.
| σ | 1 | 2 | 3 | 4 | |
| Chσ | 1 | 0.5386 | 0.4650 | 0 | |
| Ctσ | 0 | 0.4616 | 0.5350 | 1 | |
| nTσ | 1 | 1 | 1 | 1 | |
| nlσ | 1 | 0.5386 | 0.4650 | 0 | |
| ncσ | 0 | 0.4616 | 0.5350 | 1 | |
| Rσ(1) | H | 0.3708 | 0.3289 | 0.3222 | 0.2800 |
| Li | 1.3260 | 1.2089 | 1.1440 | 0.9860 | |
| Na | 1.5133 | 1.4551 | 1.4308 | 1.3070 | |
| K | 1.9628 | 1.8794 | 1.8601 | 1.7820 | |
| Rb | 2.0870 | 2.0270 | 2.0175 | 1.9570 | |
| Cs | 2.2140 | 2.2260 | 2.2279 | 2.2400 | |
| 注: $ l, \; m, \;n, \; \tau $: 1 0 0 0 $l{'}, \; m{'}, \;n{'}, \; \tau {'}$: 0.9982 0.0018 0 0 | |||||
表A1IA族元素的乙种杂化表
TableA1.B type hybrid table of IA group
| σ | 1 | 2 | 3 | 4 | |
| Chσ | 1 | 0.5694 | 0.1983 | 0 | |
| Ctσ | 0 | 0.4306 | 0.8017 | 1 | |
| nTσ | 3 or 5 | 3 or 5 | 3 or 5 | 3 or 5 | |
| nlσ | 0 | 0 | 0 | 0 | |
| ncσ | 3 or 5 | 3 or 5 | 3 or 5 | 3 or 5 | |
| Rσ (1) | N | 0.7000 | 0.7517 | 0.7973 | 0.8200 |
| P | 1.0980 | 1.1173 | 1.1343 | 1.1428 | |
| As | 1.1800 | 1.2390 | 1.2911 | 1.3170 | |
| Sb | 1.3560 | 1.4279 | 1.4919 | 1.5230 | |
| Bi | 1.3990 | 1.4455 | 1.5044 | 1.5290 | |
| 注: $ l, \; m, \; n, \; \tau $: 1 2 0 1; $ l{'}, \; m{'}, \; n{'}, \; \tau {'} $: 0 3 0 1 | |||||
表A2VA族元素的甲种杂化表
TableA2.A type hybrid table of VA group
| σ | 1 | 2 | 3 | 4 | 5 | 6 | |
| Chσ | 1 | 0.9502 | 0.8320 | 0.1681 | 0.0481 | 0 | |
| Ctσ | 0 | 0.0498 | 0.1680 | 0.8319 | 0.9519 | 1 | |
| nTσ | 4 | 4 | 4 | 4 | 4 | 4 | |
| nlσ | 2 | 1.9040 | 1.6640 | 0.3360 | 0.0960 | 0 | |
| ncσ | 2 | 2.0960 | 2.3360 | 3.6640 | 3.9040 | 4 | |
| Rσ(1) | C | 0.7630 | 0.7630 | 0.7630 | 0.7630 | 0.7630 | 0.7630 |
| Si | 1.1700 | 1.1700 | 1.1700 | 1.1700 | 1.1700 | 1.1700 | |
| Ge | 1.2230 | 1.2230 | 1.2230 | 1.2230 | 1.2230 | 1.2230 | |
| Sn | 1.3990 | 1.3990 | 1.3990 | 1.3990 | 1.3990 | 1.3990 | |
| Pb | 1.4300 | 1.4300 | 1.4300 | 1.4300 | 1.4300 | 1.4300 | |
| 注: $ l, \; m, \; n, \; \tau $; 2 2 0 0; $ l{'}, \; m{'}, \; n{'}, \; \tau {'}; $ 1 3 0 1 | |||||||
表A3IVA族元素的甲种杂化表
TableA3.A type hybrid table of IVA group
