摘要: 随着计算机技术的快速发展, 计算研究在探究材料体系微结构演化方面展示出巨大的优势. 作为材料动力学的一种计算研究方法, 相场模型不仅可以避免复杂的界面追踪, 而且便于处理各类外场因素, 因而受到广泛关注. 藉此本文介绍了相场模型的理论框架以及目前主流的多元多相系相场模型: Carter模型, Steinbach模型和Chen模型, 并从相场变量的解释、耦合热力学数据库的方式、体系自由能密度的构建方式以及演化方程等方面对上述三个模型进行了系统地概括和比较. 进一步, 聚焦于相场模型在各向异性输运和相分离、弹性和塑性变形、裂纹扩展和断裂、枝晶生长机制等方面的应用, 系统展示了相场模型在描述电化学储能材料微结构演化以及改进其性能方面的巨大潜力. 最后, 从相场模型的理论改进和应用拓展两个方面, 讨论并展望了电化学储能材料相场模拟的未来发展方向和亟待解决的关键问题.
关键词: 相场模型 /
电化学储能材料 /
应力演化 /
塑性变形 /
枝晶生长 English Abstract Phase-field model and its application in electrochemical energy storage materials Zhang Geng 1 ,Wang Qiao 2 ,Sha Li-Ting 3 ,Li Ya-Jie 3 ,Wang Da 3 ,Shi Si-Qi 2,3 1.Physical Science and Engineering Division (PSE), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia Fund Project: Project supported by the National Natural Science Foundation of China (Grant No. 11874254) and Shanghai Pujiang Talents Program, China (Grant No. 2019PJD016)Received Date: 26 August 2020Accepted Date: 21 October 2020Available Online: 20 November 2020Published Online: 20 November 2020Abstract: With the rapid progress of computer technology, computational research exhibits significant advantages in investigating microstructure evolution of material systems. As a computational research method of material dynamics, increasing attention has been paid to the phase-field model because of its avoidance of complicated interface tracking and convenience of dealing with applied fields. Theoretical framework of the phase-field model and three current phase-field models for multicomponent multiphase systems (the Carter, Steinbach, and Chen models) are introduced and reviewed in terms of interpretation of phase-field variables, way of coupling thermodynamic database, way of constructing the free energy density, and evolution equations. This review only focuses on the application of the phase-field model in electrochemical energy storage materials, and introduces its existing phase-field simulation results, which demonstrates that the phase-field model has tremendous potential in describing the microstructure evolution (anisotropic transport and phase separation, elastic and plastic deformation, crack propagation and fracture, dendrite growth, etc) and improving the performance of electrochemical energy storage materials. Finally, from two aspects of improving phase-field theory and extending application, future development trend and problems to be solved of phase-field simulations in electrochemical energy storage materials are discussed and looked ahead.Keywords: phase-field model /electrochemical energy storage materials /stress evolution /plastic deformation /dendrite growth 全文HTML --> --> --> 1.引 言 在能源危机和环境污染日益严重的背景下, 发展绿色可再生能源是社会发展的必然趋势. 太阳能、风能等可再生能源发电具有不稳定、不连续、不可控等特点. 大规模储能系统可有效进行可再生能源发电的调幅调频与平滑输出, 实现能源跨越时间和空间的分配调节, 对可再生能源的生产和消费具有革命性意义. 电化学储能是当前主要的储能技术之一, 主要的电化学储能设备包括锂离子电池、铅酸电池和超级电容器等, 具有使用方便、环境污染少、不受地域限制和能量转化效率高等特点. 提高电化学储能材料的能量/功率密度、循环寿命和安全性能, 是当前主要的研究方向, 而建立能够反映电化学储能体系基本物理化学机理的模型, 对开发高效安全的电化学储能材料具有重要意义[1 ] . 随着计算机技术的飞速发展, 模拟研究越来越多地受到科研工作者的青睐. 在建立可靠的模型并设计优良的数值算法的前提下, 可以节约人力物力地高效开展研究. 模拟研究不仅有助于了解材料的微观组织结构演化, 还可以根据预测结果设计材料、探索材料组分、加工方式等与材料性能之间的联系. 到目前为止, 模拟研究方法已经涵盖了广泛的时空尺度, 包括密度泛函理论(density functional theory, DFT)[2 ,3 ] 、分子动力学(molecular dynamics, MD)[4 ] 、蒙特卡洛(Monte-Carlo, MC)[5 ] 、有限元分析(finite element analysis, FEA)[6 ] 和相场模型(phase-field model, PFM)[7 ] 等.[7 ] . 在热力学中, 相是性质均匀稳定的空间结构, 场是时空变量的函数, 相场即为用时空变量的函数来描述材料的微结构演化. 相场模型的一个重要特点是扩散界面, 斯洛文尼亚物理学家Stefan在19世纪80年代提出处理相边界问题的尖锐界面方法并广泛用于解释扩散相变动力学[8 ] . 求解尖锐界面模型需要相边界处的衔接条件, 这需要对相边界进行追踪. 但在实际问题中, 相边界的形状和运动往往十分复杂, 这导致求解尖锐界面模型的演化方程极其困难. 相场通过引入扩散界面模拟解决了这一问题, 图1 显示相变界问题中的尖锐界面和扩散界面的对比. 相场模型引入相场变量来标度相态, 其演化直接反映相变过程, 无需追踪界面, 这大大降低了问题的复杂性. 相场模型的另一特点是处理外场的便利性. 若体系除了相变以外还有浓度、温度、外加电磁场等参与, 则还需引入其他场变量来描述体系的状态. 所有描述体系的场变量构成完备场变量集, 它们可以构建出体系的自由能泛函. 场变量的演化方程可依据自由能最小化原理[9 ] 和局域平衡原理[10 ] 导出, 它们通常为非线性偏微分方程. 相场模型中的外场被视为对总自由能的一部分贡献, 其对体系微结构演化的影响可以通过其对自由能的影响体现出来. 通过数值求解演化方程得到场变量在模拟区域和时间节点上的值, 便可得出材料体系的微结构演化. 可以看出, 相场模型对材料体系的描述方法与Maxwell理论对电磁场的描述方法完全一致, 所以本质上相场模拟是经典场论思想在材料科学中的应用. 然而目前的相场模型也存在不足, 对于二相系, 大部分模型都相当成功, 但拓展到多相系时, 目前的相场模型具有数学处理不严谨或物理描述不自洽等问题. 过去的三十年中, 相场模型在模拟材料微结构演化方面发挥了重要作用. 最初的相场模型用来研究二元合金的凝固[11 ] , 发展到今天, 涉及材料力学和热力学性能的大部分微观过程都由此得到了比较深入的研究, 如晶粒生长[12 ] 、枝晶生长[13 ] 、形核[14 ,15 ] 、析出[16 ,17 ] 、烧结[18 ,19 ] 、位错[20 ,21 ] 、弹性[22 ,23 ] 、塑性[24 ,25 ] 和断裂[26 ,27 ] 等.图 1 相变界问题的尖锐界面和扩散界面的对比Figure1. Comparison between sharp and diffusive interfaces for phase boundary problems.2.相场理论框架及关于目前主要相场模型的评述 22.1.相场模型的理论框架 2.1.相场模型的理论框架 本文中约定: 当相指标和组分指标都出现时, 相指标为上标, 组分指标为下标; 当相指标和组分指标单独出现时, 它们均为下标. 在相场模型中, 最基本的场变量是描述相态的相场变量${\phi _\alpha }{\rm{ }}(\alpha = 1, \cdots, n)$ . 在不同的模型中相场变量的含义不尽相同, 但是由于相可以生成或消失, 相场变量都被当作非守恒变量, 它们遵循Allen-Cahn方程[28 ] :${c_i}{\rm{ }}(i = 1, \cdots, m)$ . 在没有化学反应的前提下, 浓度变量被当作守恒变量. 它们遵循Cahn-Hilliard方程[29 ] :1 )和方程(2 )中, ${({{{L}}_{\alpha \beta }})_{n \times n}}$ 为界面迁移率矩阵, ${({{{M}}_{ij}})_{m \times m}}$ 为组分的移动性参数矩阵, 它们均为半正定矩阵, $f$ 为体系的自由能密度, $\dfrac{{\delta f}}{{\delta {\phi _\beta }}} = \dfrac{{\partial f}}{{\partial {\phi _\beta }}} - \nabla \cdot\dfrac{{\partial f}}{{\partial \nabla {\phi _\beta }}}$ 和$\dfrac{{\delta f}}{{\delta {c_j}}} = \dfrac{{\partial f}}{{\partial {c_j}}} - \nabla \cdot \dfrac{{\partial f}}{{\partial \nabla {c_j}}}$ 称为变分导数. 与普通导数相比, 变分导数还包含了场变量梯度的贡献.$\{ {u_i}, \nabla {u_i}\} $ , 则体系的自由能泛函可以表达为1 )和方程(2 )分别代入上述方程得到:$\dfrac{{{\rm{d}}F}}{{{\rm{d}}t}}$ 的正负性的判断, 用到了${{L}}$ 和${{M}}$ 的半正定性. 它们表明了浓度变量和相场变量的演化均满足自由能最小化原理. 本质上, 浓度方程是物质守恒的反映, 所以若无化学反应, 浓度方程(2 )与组分演化的物理过程完全一致. 相比而言, 相场方程(1 )未必与相变的物理过程完全一致, 因为它只是自由能最小化的一种最简单的表达, 不是从更基本、更深刻的物理规律导出的. 尽管目前的相场模型都采用关于浓度变量变分导数的线性项来表达自由能最小化, 但完全可以构造包含浓度变量的变分导数的非线性项来体现自由能的最小化. 虽然自由能最小化的所遵循的规律仍是未知的, 但大量的相场模拟的文献表明, 反映自由能最小化趋势的方程(1 )可以很好地描述材料体系的微结构演化.1 )和方程(2 )是相场模型的基础, 不同的相场模型是指浓度描述方式和总自由能密度表达方式的差异.2.2.常见相场模型及其特点 -->2.2.常见相场模型及其特点 相场模拟通常分为三个层次: 第一层次描述材料体系的微结构演化, 是定性、半定量、定量研究的混合; 第二层次分析材料的微结构和宏观性能的关联, 根据研究体系的模拟结果预测其性能, 是半定量、定量研究的混合; 第三层次消除模拟中可调参数, 通过理论计算和实验测量确定所有模拟参数, 进一步通过调控体系的物理环境改进和控制体系的性能, 是完全的定量研究. 不管是哪种层次的模拟, 都需要精准的热力学数据库来识别所研究的体系. 在计算热力学中, 体系的热力学数据库是由相浓度(相摩尔分数, 本文用$y$ 表达)来构造的, 但是扩散动力学只表达总浓度(总摩尔分数, 本文用$c$ 表达)的演化方程——Fick扩散方程. 所以, 要实现热力学数据库与相场模拟的耦合主要有以下两种方式: 1) 采用总浓度的演化方程, 将求解得到的总浓度以一定的方式分解为相浓度与热力学数据库对接, 代表为KKS模型[30 ] ; 2) 直接构造相浓度的演化方程与热力学数据库对接, 代表为Steinbach模型[31 ,32 ] . 耦合热力学数据库的方式, 相场变量的解释, 体系自由能密度的构建方式以及演化方程等要素一起区分了不同的相场模型. 由于二相系模型是多相系模型的特例, 故这里只讨论多相系(两相及以上)模型, 包括当前主流的Carter模型、Steinbach模型和Chen模型. 下述讨论中, 假定体系存在$n$ 个相, $m$ 个组分, ${f_\alpha }$ 为$\alpha $ 相的自由能密度. 有时为了更细致地区分一个相(通常是固相)的不同状态, 需要用多个相场变量去描述该相, 例如晶粒的不同取向. 此时可将该相的不同状态视为若干个更精细的“亚相”, 每个“亚相”引入一个相场变量, 意味着一个相(普通意义)可以用若干个相场变量描述. 为了表述上的方便, 下述讨论中, 每个相(“亚相”意义)引入一个相场变量描述体系的相态.2.2.1.Carter模型 -->2.2.1.Carter模型 在Carter模型中, 相场变量被解释为相分数, 满足归一化条件, 即$\displaystyle\sum\nolimits_\alpha {{\phi _\alpha }} = 1$ ; 总浓度以相同的值分配到各个相, 即组分$i$ 在每个相中的相浓度都相同, 等于总浓度. 该模型使用如下的自由能密度[33 ] 进行描述:${W_{\alpha \beta }}$ 为势垒高度, ${\varepsilon _{\alpha \beta }}$ 和${\kappa _{ij}}$ 分别为相场和浓度的梯度能系数, 它们均为模型的唯象参数. 方程(7 )表达的自由能密度具有非常明晰的物理意义: 右边前两项只与场变量有关, 称为均匀自由能密度, 它包含两部分: 各相自由能密度的加权平均$\bigg( \displaystyle\sum\limits_{\alpha = 1}^n {{\phi _\alpha }{f_\alpha }(\{ {c_i}\} )}\bigg)$ 、相间势垒$\bigg( \displaystyle\sum\limits_{\beta > \alpha = 1}^n {{W_{\alpha \beta }}{\phi _\alpha }{\phi _\beta }}\bigg)$ . 后两项与场变量的梯度相关, 称为非均匀自由能密度, 也包含两部分: 相场梯度的贡献$\bigg( \displaystyle\sum\limits_{\alpha, \beta = 1}^{n - 1} \dfrac{{{\varepsilon _{\alpha \beta }}}}{2}\times \nabla {\phi _\alpha } \cdot \nabla {\phi _\beta } \bigg)$ 和浓度梯度的贡献$\bigg( \displaystyle\sum\limits_{i, j = 1}^{m - 1} {\frac{{{\kappa _{ij}}}}{2}\nabla {c_i} \cdot \nabla {c_j}} \bigg)$ .8 )是方程(1 )的特殊情形, 它满足自由能最小化原理. 该模型的演化方程形式简单, 计算复杂度较低, 编程求解时比较方便. 但是相场方程与相场变量的归一化条件不相容, 这意味着独立相场变量的不同选取将影响模拟结果, 然而真实材料体系的微结构演化是一个确定的过程, 它应该与对体系的描述方式无关. 在实际求解中需要数值截断和重整化来确保每一个相场变量都在区间[0, 1]中, 尽管这些处理对模拟效率影响甚微, 但却丧失了数学上的合理性和严谨性.2.2.2.Steinbach模型 -->2.2.2.Steinbach模型 同Carter模型一样, Steinbach模型的相场变量也被解释为相分数, 并区分了相浓度$y_i^\alpha $ 和总浓度${c_i}$ , 用相浓度来构造相能量密度. 其自由能密度为[31 ] ${h_{\rm{S}}}({\phi _\alpha }) \!=\! \dfrac{1}{{\text{π}}}[2(2{\phi _\alpha } \!-\! 1)\sqrt {{\phi _\alpha }(1 \!-\! {\phi _\alpha })} + \arcsin (2{\phi _\alpha } - 1)] + \dfrac{1}{2}$ 为相自由能密度的权重因子(以下简称相权重), 它满足${h_{\rm{S}}}(0) = 0$ , ${h_{\rm{S}}}(1) = 1$ , ${h'_{\rm{S}}}(0) = {h'_{\rm{S}}}(1) = 0$ , 满足此条件的相权重函数称为恰当相权重, 图2 显示了${h_{\rm{S}}}({\phi _\alpha })$ 和${\phi _\alpha }$ 的差异. 该模型不使用相场变量作为相权重是为了消除体相内的驱动力, 确保相变只发生于相边界处. 对于二相系, 相权重是归一的, 即${h_{\rm{S}}}({\phi _\alpha }) + {h_{\rm{S}}}({\phi _\beta }) = 1$ . 但对于三相及以上体系, 一般$\displaystyle\sum\nolimits_\alpha {{h_{\rm{S}}}({\phi _\alpha })} \ne 1$ . 该模型为了确保相平权(相权重具有相同的函数形式)和模拟结果的合理性而放弃相权重的归一化. 和Carter模型相比, Steinbach模型中相场梯度对非均匀自由能密度的表达式也有差异, 它只考虑相场梯度交叉项($\nabla {\phi _\alpha } \cdot \nabla {\phi _\beta }$ )的贡献, 未考虑相场梯度对角项($\nabla {\phi _\alpha } \cdot \nabla {\phi _\alpha }$ )的贡献. 相比方程(7 ), 方程(10 )略去了浓度梯度对自由能密度的贡献.图 2 相场变量与Steinbach模型、Chen模型中的相权重函数的比较Figure2. Comparison of phase field and phase-weight functions in Steinbach and Chen models.[31 ] $N$ 是局域相数, 与上面自由能密度表达式中$n$ (整体相数)是不同的. 该模型的相场方程不仅满足自由能最小化原理, 而且还与相场变量的归一化条件相容, 这意味着该模型的模拟结果与独立相场变量的选取方式是无关的. 为了便于耦合热力学数据库, 该模型第一次构造了相浓度的演化方程. 在Steinbach模型之前, 解决热力学数据库和总浓度方程对接的方法就是依据等化学势原则将总浓度分解为相浓度, 这个思想由KKS模型首先给出[30 ] . 随着组元和相的增加, 这种处理方法的计算量增加很快, 而且等化学势原则对平衡、弱非平衡体系是适用的, 但不适用于强非平衡体系. 而Steinbach模型依据演化方程来确定相浓度, 不仅大大降低了处理多元多相系的复杂性, 而且也适用于强非平衡过程, 如快速凝固. 虽然该模型克服了Carter模型的与相场变量的归一化条件不相容的缺陷, 但它也引入了其他问题. 方程(11 )包含了不连续的量—局域相数, 这导致模拟结果违背近距作用, 即两相即便不接触也可能出现相互作用. 方程(11 )是对体系相演化的局域描述(在${\phi _\alpha } = 0$ 时, ${\phi _\alpha }$ 的演化方程没有意义), 在实际应用中, 需要根据一定的准则判断相对于$\alpha $ 相的邻近界面区(包含: 界面和接近界面的体相)与非邻近界面区. 对多相系而言, 相对于$\alpha $ 相的相区判断需要大量的计算, 这会降低模拟效率.2.2.3.Chen模型 -->2.2.3.Chen模型 与Carter模型和Steinbach模型不同, Chen模型中的相场变量被解释为序参量, 它们只是相的标度, 没有明确的物理意义. 该模型用总浓度${c_i}$ 描述组分演化, 用相浓度$y_i^\alpha $ 构建相自由能密度, 它用KKS模型的处理实现总浓度方程与体系的自由能密度的对接, 即按照一定的原则将总浓度分解为相浓度. 其自由能密度为[19 ,34 ] ${h_{\rm{C}}}({\phi _\alpha }) = \phi _\alpha ^3(6\phi _\alpha ^2 - 15{\phi _\alpha } + 10)$ 是Chen模型的相权重, 与Steinbach模型中的${h_{\rm{S}}}$ 一样, 该模型的${h_{\rm{C}}}$ 也是恰当相权重, 图2 展示了${h_{\rm{C}}}({\phi _\alpha })$ 和${\phi _\alpha }$ 的差异. 与Steinbach模型相比, 该模型的相间势垒是相场变量的四次多项式, 非均匀自由能密度只考虑了相场梯度和浓度梯度的对角项, 没有考虑交叉项. 可以看出, 该模型中, 多相系中的其中一项被选为参考相, 其余相为独立相, 引入相场变量${\phi _\alpha }$ 来描述独立相$\alpha $ , 参考相无需引入相场变量描述, 即该模型的$n$ 相系需要$n - 1$ 个相场变量来描述, 这些相场变量彼此独立. ${\phi _1} = 0,\cdots,$ ${\phi _\alpha } = 1,\cdots,$ ${\phi _{n - 1}} = 0$ 表示纯$\alpha $ 相; ${\phi _1} \!=\! 0,\cdots,$ ${\phi _\alpha } \!=\! 0,\cdots,$ ${\phi _{n - 1}} \!=\! 0$ 表示纯参考相. 相比Steinbach模型, Chen模型正好相反, 为了保持相权重的归一化放弃了相平权. 从方程(13 )中容易看出, 独立相彼此平权, 独立相和参考相不平权.13 )中的相间势垒项在${\phi _\alpha } = 0$ 和${\phi _\alpha } = 1$ 时有最小值, 该模型的相场方程可以自动将相场变量约束在区间[0, 1]内, 即使数值求解不做截断处理, 也可以得到符合物理事实的结果. 和Steinbach模型相比, 该模型的相场方程是对体系演化的整体描述, 数值求解时无需相区判断, 故模拟效率比较高. 由于该模型舍弃了相平权, 所以选取$\alpha $ 为参考相与选取$\beta $ 为参考相的模拟结果在理论上存在差异. 事实上, 真实材料体系的微结构演化是一个确定的过程, 它应该与对体系的描述方式无关.3.电化学储能材料的已有相场模拟结果简介 电化学储能系统是一个十分复杂的体系, 充电和放电对应金属阳离子在阳极和阴极之间的反复迁移, 这往往涉及多个组分的扩散和固态相变[35 ] , 因而带来了许多问题, 如各向异性输运和相分离, 弹性和塑性变形, 裂纹扩展和断裂, 以及枝晶生长等等. 相场模型是解决这些问题的强有力的工具之一. 将相场方法应用于电化学储能系统的最初尝试是研究电极材料(如LiFePO4 )在充放电过程中的相分离. Carcia等[36 ] 将Cahn-Hilliard方程与Maxwell方程耦合, 阐明了电磁活性体系的变分原理; Guyer等[37 ,38 ] 提出了模拟电化学界面的双层和Butler-Volmer动力学的相场模型; Han等[39 ] 利用Cahn-Hilliard方程模拟了锂离子在LiFePO4 中的扩散行为. 以上工作被公认为相场模拟应用于电化学储能材料的奠基工作, 启发了科研工作者利用相场模型研究电化学储能材料的各种微结构演化过程. 利用相场模拟研究电化学储能材料微结构演化的一般步骤如图3 所示.图 3 相场模型中研究电化学储能材料微结构演化的步骤Figure3. Steps for investigating microstructure evolution of electrochemical energy storage materials in phase-field model.3.1.电化学相场模拟中的化学反应、电场、弹性 3.1.电化学相场模拟中的化学反应、电场、弹性 由于电化学反应的存在, 相场和浓度的演化方程中考虑源项${n_{\alpha, i}}$ (单位体积和时间内组分$i$ 生成的物质的量), 方程(1 )和方程(2 )相应修改为${n_{\alpha, i}}$ 通常是电流密度、相场和浓度的函数.$\varPhi $ 来描述电化学储能材料体系的电场, 则电场能密度${f^{{\rm{elec}}}}$ 可以表达为${\rho ^{{\rm{elec}}}}$ 为电荷密度; ${F_0}$ 为法拉第常数; ${z_i}$ 为组分$i$ 的化合价; ${V_{{\rm{mol}}}}$ 是系统的摩尔体积. 电势场是描述电化学储能材料体系的基本场变量, 其演化方程由电荷守恒定律给出:${\sigma ^{{\rm{elec}}}}$ 为电化学储能材料的电导率.${{\varepsilon }}$ 描述电化学储能材料体系的形变, 则${f^{{\rm{elas}}}}$ 可以表示为${{{\varepsilon }}_\alpha }$ 和${{\varepsilon }}_\alpha ^0$ 分别为$\alpha $ 相的总应变张量和本征应变张量, ${{{C}}_\alpha }$ 为$\alpha $ 相的弹性张量. 由于应力的弛豫时间远远小于相变和扩散的弛豫时间, 一般认为应力始终平衡, 故应变张量${{{\varepsilon }}_\alpha }$ 的演化方程为3.2.相场模型在电化学储能材料中的应用 -->3.2.相场模型在电化学储能材料中的应用 33.2.1.离子输运和相分离 -->3.2.1.离子输运和相分离 在可充电电池中, 离子输运对于正极材料电极容量、充放电效率和电池体系的循环性能等关键性质具有很大的影响. 例如, 锂离子电池充电过程中, 在电池正负极施加足够大的电流, 若锂离子在电池内部的扩散速度不够快, 就会出现浓度梯度, 可能会导致电极材料晶体结构内锂离子的重组, 进而引起相分离. 电化学储能系统中通过描述离子输运特性刻画微观物理图像至关重要. 实验研究发现锂在FePO4 晶体中的扩散具有很强的各向异性, Lix 4 具有分离成富锂相和贫锂相的强烈倾向. Lix 4 完美晶体中的Li输运局限于y 方向的一维通道, 横向扩散的可能性很小(图4(a) ), 这一观点也从相场角度得到了验证[40 -43 ] . Hong等[44 ] 运用相场模型为锂离子扩散各向异性和相边界迁移机制提供了重要的理解(图4(b) ). Chueh?等[45 ] 通过相场模拟研究了Lix 4 纳米颗粒的相干非平衡亚表面相形态. Fleck等[46 ] 发展了一个考虑各向异性(正交)和非均匀弹性效应的连续相场模型, 并模拟了FePO4 纳米粒子中嵌入Li时的FePO4 相和LiFePO4 相之间的转变过程. 目前, 通过相场模拟方法可较清晰刻画电化学储能材料中的离子输运物理图像.图 4 (a) 锂离子嵌入到FePO4 时活跃面(010)快速扩散(蓝色箭头)和相分离示意图[43 ] ; (b) LiFePO4 相变过程中的三种可能的扩散路径: 体扩散、表面扩散和电解液扩散[45 ] Figure4. (a) Schematic diagram of diffusion of active surface (010) (blue arrows) and phase separation when Li-ion intercalates FePO4 [43 ] ; (b) three potential migration paths in phase transition of LiFePO4 : bulk, surface, and electrolyte diffusions[45 ] .[6 ] , 如何将相场模拟与其他计算方法相结合, 通过多尺度探究离子输运特性等物理图像来指导电池的设计, 也是未来需要研究的问题.3.2.2.电极材料中的力学行为 -->3.2.2.电极材料中的力学行为 金属阳离子嵌入和脱出电极的过程, 会导致电极晶体结构膨胀和收缩从而产生应力[47 ,48 ] . 阳离子在固态电极中的扩散会使电极成分偏离其化学计量状态, 同样会导致体积变化并产生应力[47 ,49 ] . 应力的产生通常会造成电极的弹性和塑性变形(图5 )[50 ] . Chen等[51 ] 将相场模型与弹塑性变形相结合以研究硅电极嵌锂过程中内部的相、形貌和应力的变化. 该模型中引入总变形梯度乘法分解$F = {F^{\rm{e}}}{F^{\rm{p}}}{F^{\rm{s}}}$ , 其中${F^{\rm{e}}}$ , ${F^{\rm{p}}}$ 和${F^{\rm{s}}}$ 分别是弹性、塑性和锂离子的嵌入/脱出对变形梯度的贡献. 可以看出, 总变形可以看作是一个非弹性变形和一个弹性变形的累积[52 ] , 这一理论适用于需要考虑间隙原子扩散和弹塑性变形耦合效应的情况. 随后, Zhao等[53 ,54 ] 和Bower等[55 ] 设计了模拟扩散诱导塑性变形的计算框架. Walk等[56 ] 分别采用小变形和大变形耦合Cahn-Hilliard方程分析应力演化. Gao和Hong[50 ] 进一步发展了弹塑性相场模型, 该模型将体积和偏非弹性变形(deviatoric inelastic deformation)都视为锂化的直接结果.图 5 硅颗粒的弹性和塑性变形[50 -52 ] Figure5. Elastic and plastic deformations of Si particles[50 -52 ] .[48 ] . 2009年, Hakim 和Karma[26 ] 以及Aranson等[57 ] 发展了一个研究裂纹扩展的相场模型. 之后, Marconi和Jagla[58 ] , Spatschek等[59 ] 和Miehe等[60 -62 ] 利用该模型进行了大量的研究. Bhandakkar等建立了裂纹扩展的内聚模型, 探讨了裂纹扩展的临界特征维数[63 ,64 ] . Woodford等[65 ] , Zhu等[66 ] , Gao和Zhou [67 ] 以及Klinsmann[68 ] 等分别利用改进的相场模型研究了扩散诱导应力、锂诱导软化、实际的电化学加载条件和惯性对锂电池断裂的影响等. Zhao等[54 ,69 ] 和Zuo与Zhao[48 ] 利用相场模型研究了Si粒子中裂纹的萌生条件和裂纹扩展过程. Huttin和Kamlah[70 ] 以及Liang和Chen[71 ] 分别研究了Lix 2 O4 和LiFePO4 颗粒的应力产生和裂纹演化. Miehe等[72 ] 和Zhao等[73 ] 采用相场模型研究了化学反应对断口表面组分与微结构的影响 (图6 ), 发现最初的Li分布浓度是均匀的, 外层失去Li后出现两相分布和相偏析, 从而导致了裂纹的扩展.图 6 锂化过程中的裂纹扩展 (a)初始状态; (b)相偏析导致裂纹扩展; (c)相界面赶上裂纹尖端; (d)裂纹尖端在相界面处开始分叉; (e)相界面离开裂纹尖端向中心移动; (f)反应停止[73 ] Figure6. Crack propagation during lithiation process: (a) Initial state; (b) phase segregation generates crack propagation; (c) the phase interface catches up with the crack tip; (d) the crack tip starts to branch at the phase interphase; (e) phase interface leaves crack tip and moves towards center; (f) the reaction stops [73 ] .[74 ] 通过第一性原理计算与力学-电化学耦合方程相结合的方法探讨了锂在铜、铝等集流体中的扩散机理, 结果表明锂能在具有一定量空位的集流体中以单空位机制扩散, 扩散带来的体积膨胀可以显著促进离子在“无负极”电池材料中的扩散, 这也是将来相场模拟需要考虑的问题之一. 锂离子电池电极的断裂会破坏电极结构的完整性, 进而对电池安全性及电化学性能造成严重的影响. Lu等[75 ] 综述了电极断裂的三种典型类型: 活性层断裂、界面脱层和金属箔断裂, 并揭示了复合电极和薄膜电极中的不同断裂现象及其相应的机理. 文中提到, 目前关于电极断裂的研究主要是基于实验观察与典型机械损伤理论模型, 有关电化学性能和电极断裂之间的关系的定量研究还很缺乏. 为了推进力学与电化学交叉学科的发展, 从多尺度、多物理的角度认识断裂是十分必要的. 此外, 对于有裂纹的电极, 裂纹扩展后暴露出新的表面, 并在与电解液接触的区域发生电化学反应, 导致富锂相成核. 裂纹尖端附近的应力更容易引起电极变形, 这种变形对成核的影响还需要进一步研究. 为了加快电池材料的设计和优化, 往往需要结合DFT, MD, MC以及相场模拟等方法进行多尺度模拟, 以获得对材料体系的整体性理解. Hong和Viswanathan[76 ] 总结了目前具有相场模拟功能的开源软件包, 强调了建立完全开源的多尺度模拟框架的重要性, 并提出了“完全开源多尺度仿真框架”的概念.3.2.3.枝晶生长 -->3.2.3.枝晶生长 锂枝晶的形成是锂离子电池广泛应用的主要障碍之一, 它会导致可逆容量的降低和内部短路[76 ] . 因此, 了解这种复杂的非平衡体系的基本物理机制对于提高锂离子电池的性能至关重要. 在过去的十几年里, 研究者们从热力学、动力学和电化学结合的角度出发, 提出了几种相场模型来理解锂的沉积和溶解行为[77 -82 ] .[83 ] 首次建立了电化学枝晶生长模型, 该模型是锂聚合物电池中锂枝晶尖端高度和生长速度演化的综合数学模型. 2004年, Guyer等[37 ,38 ] 建立了一维相场模型, 研究了电沉积过程中的界面电荷分离和动力学行为. 随后Shibuta等[84 ] 将Cahn-Hilliard方程与Butler-Volmer方程耦合, 模拟二维电沉积过程. 围绕电极反应过程中电极/电解质界面动力学与界面形貌展开研究, 并考察了界面形貌与外加电压和反应系数的关系, 证实了超电势是影响电沉积速率的因素, 并发现电沉积尖端半径与枝晶生长速度的平方根成反比, 这与凝固过程中的枝晶生长理论一致[85 ] . 然而, 以上模型都以线性电化学反应动力学为基础, 当体系远离平衡时, 该反应动力学将不再适用. 针对这一情况, Miehe等[72 ] 和Liang等[86 ] 提出了一个非线性相场模型, 该模型考虑了Butler-Volmer反应动力学, 可以在不考虑SEI层效应的情况下模拟和预测锂离子电池充电过程中的锂沉积行为. 在此基础上, Chen等[87 ] 建立了热力学一致的相场模型来研究锂枝晶结构, 图7 为序参量、Li+ 浓度和电势分布的演变. 在充电过程中, 相场的演化与锂离子浓度和电势有关, 这是由于电极和/或镀层附近存在浓度梯度和电位梯度. 由于尖端效应, 沉积锂的尖端有较大的Li+ 浓度和电势梯度, 从而产生较大的过电位加速其生长最终形成枝晶结构.图 7 相场模型模拟的Li枝晶生长 (a) 序参量, (b) Li+ 浓度和(c)电势的演化[87 ] Figure7. Dendrite growth of Li simulated by phase-field model: evolution of (a) order parameter, (b) concentration of Li+ , and (c) electric potential [87 ] .[88 ] 采用拉格朗日描述法对各向异性电解质溶液中的枝晶生长模型进行了改进. Yurkiv等[89 ,90 ] 改进了Tan等的模型以研究应力和SEI对锂电沉积的影响, 重现了实验中经常观察到的Li的丝状结构. Yan等[91 ] 通过相场方法研究了热效应对锂枝晶生长的影响, 并定义了垂直和水平方向的枝晶长度比用以描述枝晶形貌的变化. Hong和Viswanathan[92 ,93 ] 在开源软件MOOSE上建立了一个基于大电位的非线性相场模型, 揭示了枝晶生长与离子输运和电化学反应之间的密切竞争关系, 定义出“各向异性生长因子”(AGF)来量化尖端和谷底区域界面生长的不均匀性. 为研究电池自生热抑制枝晶的现象, Hong和Viswanathan[94 ] 结合能量平衡方程提出了一个完全热耦合的相场模型. 将离子输运和电化学反应速率之间的相互作用视为温度的函数, 探索了利用自生热诱导抑制树枝晶的可能性. 研究发现, 根据电化学反应势垒和离子扩散势垒的不同, 自加热可以加速(较大的反应势垒)或减慢(较大的扩散势垒)枝晶的形成. Jana等[79 ] 使用相场方法评估了电流密度对锂枝晶生长的影响, 研究表明, 在低电流密度下, 锂枝晶生长的驱动力来源于尖端控制, 但形态通过塑性流动形成, 使得金属锂表面生长较平整. 在高电流密度下, 由于弹性能的局域性, 电沉积速率主导应力弛豫动力学, 有利于枝晶的分支. 对于非常高的电流密度, 质量快速积聚导致尖端应力弛豫很慢, 因此仍局限于在低级的分支. 由于尖端处应力局域化与浓度梯度的作用, 形成了树枝状形貌. 这种非均相电沉积形成小的表面扰动, 形成了细长并高度分支的锂枝晶. Mu等[95 ] 通过相场模拟发现致密的SEI结构可以更好地抑制锂枝晶的生长. Zhang等[96 ] 采用相场模型描述了不同导电结构的Li枝晶生长, 发现枝晶的形态与不同的电子导电结构有关.4.总结与展望 24.1.理想的新相场模型及其应有的性质 4.1.理想的新相场模型及其应有的性质 目前电化学储能材料的相场模拟已从二相系迈向多相系, 从二元系迈向多元系, 从无外场体系迈向一个或多个外场的体系, 其微结构演化模拟需要一个适用于多元多相系的更严谨更自洽的相场模型. 理想的多元多相系的新相场模型应具以下性质:$\displaystyle\sum\nolimits_\alpha {{\phi _\alpha }} = 1$ 相容, 相浓度方程需要与$\displaystyle\sum\nolimits_i {y_i^\alpha } = 1$ ($y_i^\alpha $ 为$\alpha $ 相中$i$ 组分的摩尔分数)相容.$\beta $ 相逐渐消失, 则它对$\alpha $ 相演化的贡献要逐渐变为0; 如果$j$ 组分逐渐消失, 则它对$i$ 组分的扩散通量贡献也要逐渐变为0.$\alpha $ 相的区域还是在无$\alpha $ 相的区域, 实际模拟中的${\phi _\alpha }$ 都可以通过在整个模拟区域求解其演化方程而得到.4.2.相场模型在电化学储能材料领域的应用展望 -->4.2.相场模型在电化学储能材料领域的应用展望 目前, 可充电电池中大多数的相场模拟仍然处于定性、半定量和定量研究混合的层次. 可充电电池相场模拟向定量化发展需要体系的精确热力学和动力学数据库. 毕竟, 相场模型的有效性和合理性在很大程度上依赖于体系的物理、化学和力学参数以及准确的热力学和动力学数据库. 此外, 实际的可充电电池是十分复杂的体系, 电池中发生的物理过程跨越多个空间和时间尺度, 这使得电池的直接建模非常困难, 目前仍然缺乏将不同尺度的建模工作进行整合的统一多尺度计算模型. 因此, 进一步的相场模型需要结合DFT, MD等计算方法发展成多尺度模型, 消除可调参数, 通过理论计算或实验测量得到模拟中的所有参数. 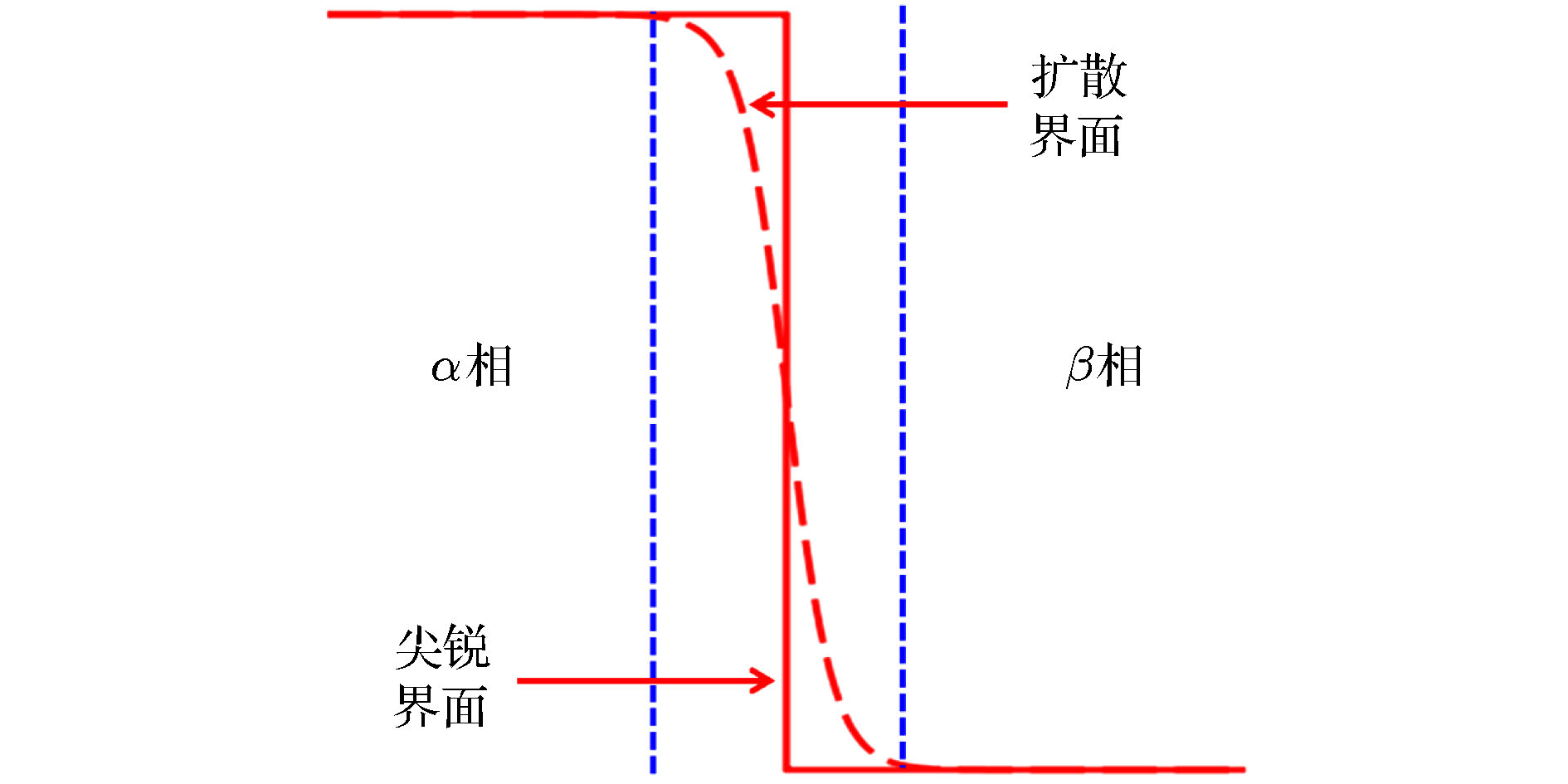 图 1 相变界问题的尖锐界面和扩散界面的对比
图 1 相变界问题的尖锐界面和扩散界面的对比





































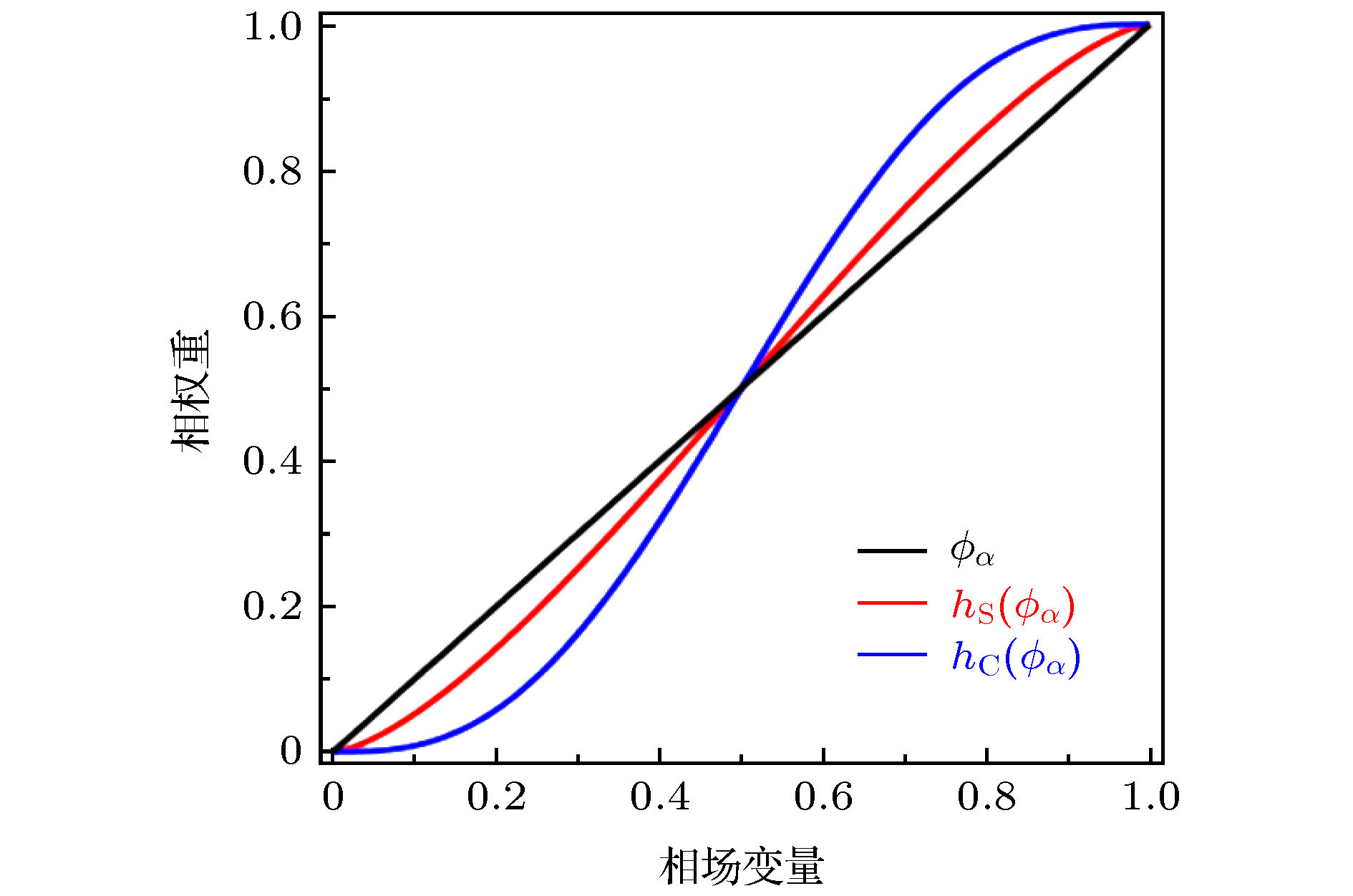 图 2 相场变量与Steinbach模型、Chen模型中的相权重函数的比较
图 2 相场变量与Steinbach模型、Chen模型中的相权重函数的比较



























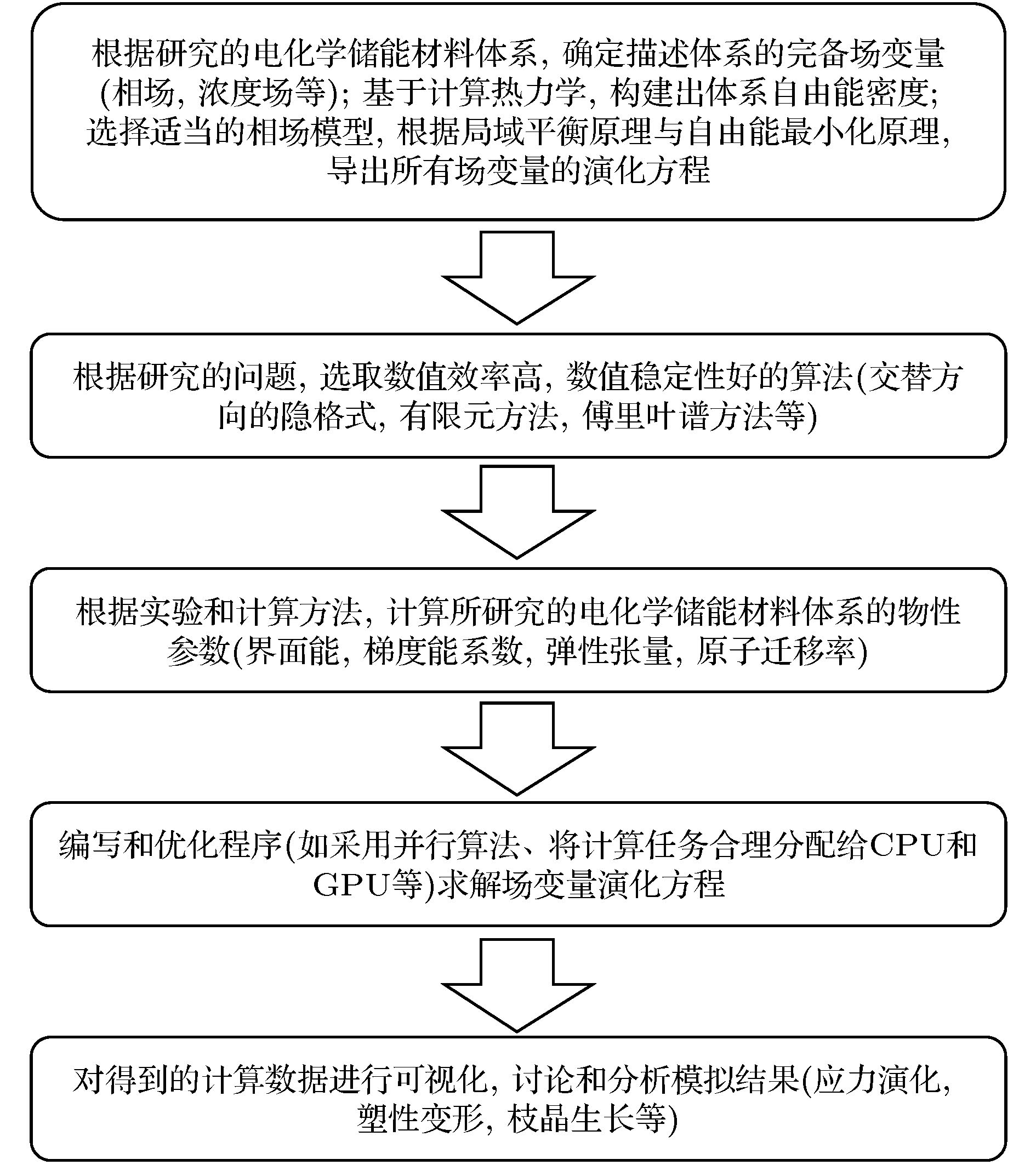 图 3 相场模型中研究电化学储能材料微结构演化的步骤
图 3 相场模型中研究电化学储能材料微结构演化的步骤


















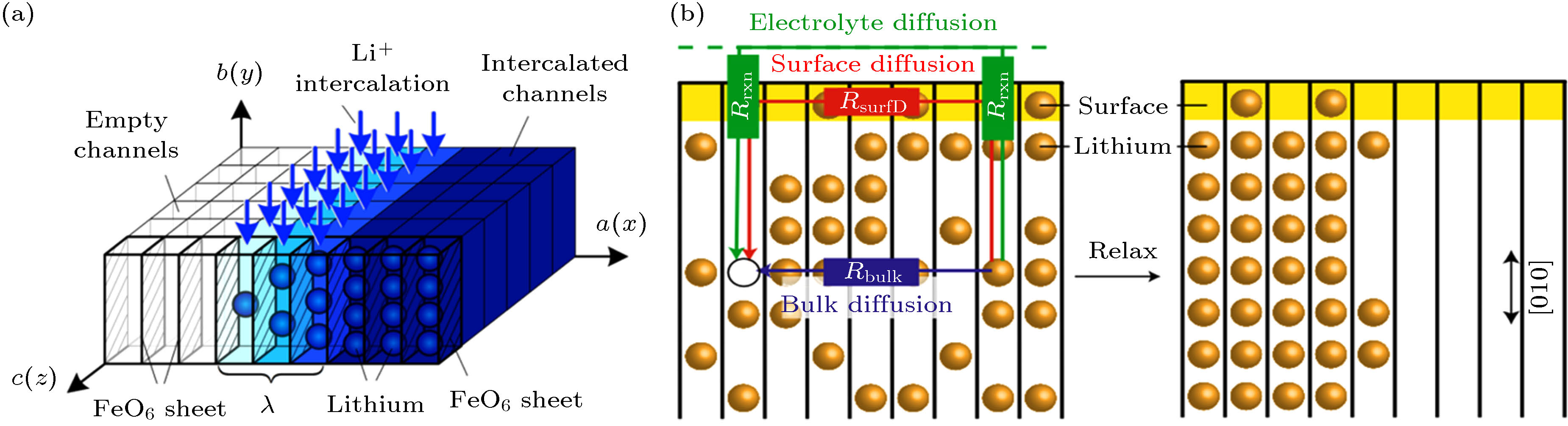 图 4 (a) 锂离子嵌入到FePO4时活跃面(010)快速扩散(蓝色箭头)和相分离示意图[43]; (b) LiFePO4相变过程中的三种可能的扩散路径: 体扩散、表面扩散和电解液扩散[45]
图 4 (a) 锂离子嵌入到FePO4时活跃面(010)快速扩散(蓝色箭头)和相分离示意图[43]; (b) LiFePO4相变过程中的三种可能的扩散路径: 体扩散、表面扩散和电解液扩散[45]



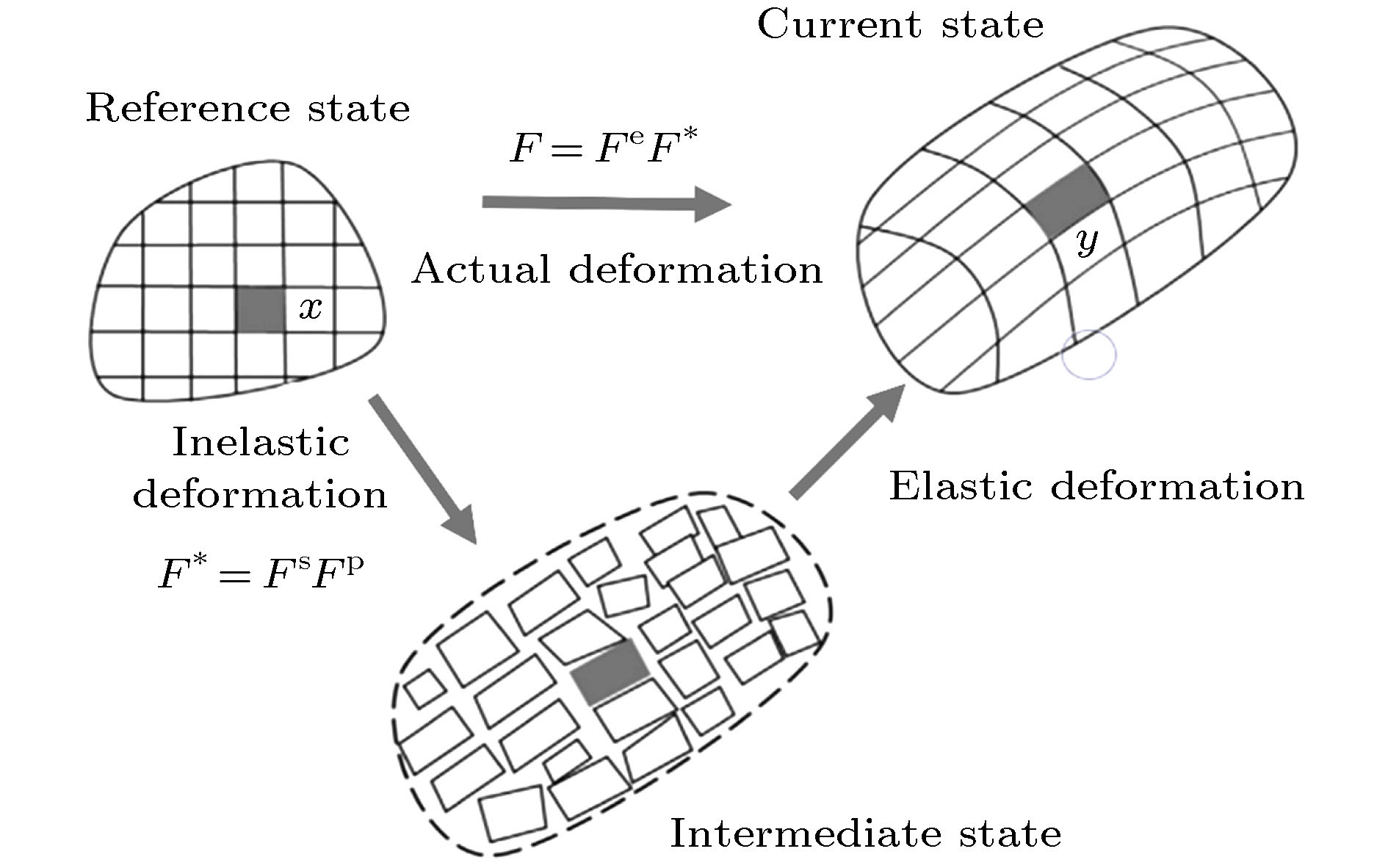 图 5 硅颗粒的弹性和塑性变形[50-52]
图 5 硅颗粒的弹性和塑性变形[50-52]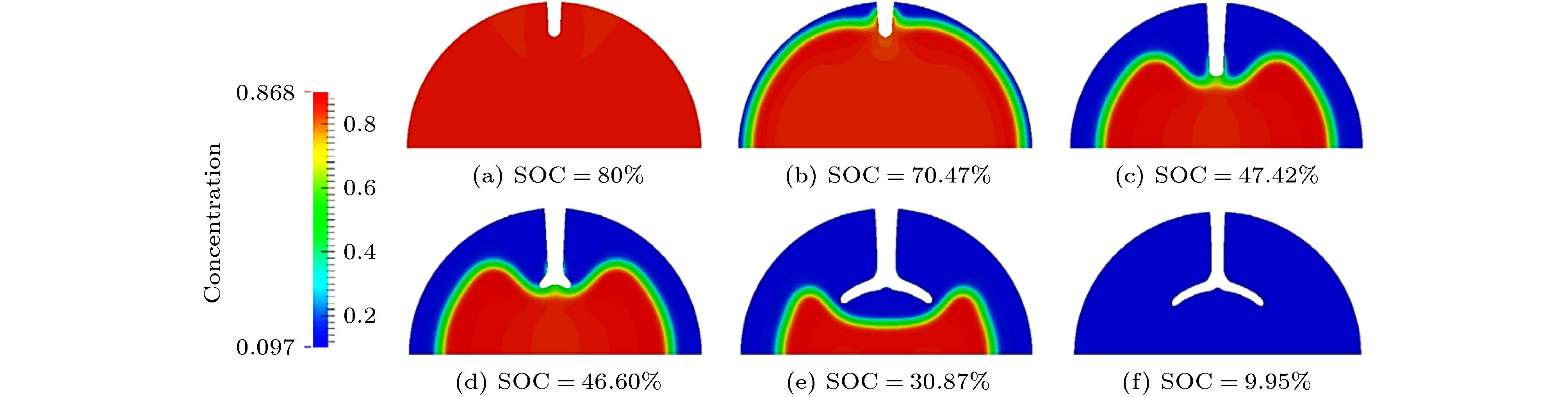 图 6 锂化过程中的裂纹扩展 (a)初始状态; (b)相偏析导致裂纹扩展; (c)相界面赶上裂纹尖端; (d)裂纹尖端在相界面处开始分叉; (e)相界面离开裂纹尖端向中心移动; (f)反应停止[73]
图 6 锂化过程中的裂纹扩展 (a)初始状态; (b)相偏析导致裂纹扩展; (c)相界面赶上裂纹尖端; (d)裂纹尖端在相界面处开始分叉; (e)相界面离开裂纹尖端向中心移动; (f)反应停止[73] 图 7 相场模型模拟的Li枝晶生长 (a) 序参量, (b) Li+浓度和(c)电势的演化[87]
图 7 相场模型模拟的Li枝晶生长 (a) 序参量, (b) Li+浓度和(c)电势的演化[87]











