全文HTML
--> --> --> 图 1 基于硫化物电解质的固态锂电池界面面临的主要挑战[24-26]
图 1 基于硫化物电解质的固态锂电池界面面临的主要挑战[24-26]Figure1. Illustration of the interfacial issues in all-solid-state lithium batteries with sulfide solid electrolytes[24-26].
硫化物电解质材料在空气中的稳定性差, 对水分十分敏感, 易与之发生反应产生H2S气体, 尤其是含P元素的硫化物电解质. 由于P-S键的键能远低于P-O键, 从而使得P-S结构容易发生氧化或者脱硫. 当硫化物固态电解质材料与空气中的氧气、水蒸气发生不可逆的化学反应时材料会发生结构变化导致离子电导率降低, 因此硫化物电解质材料的合成及电池组装过程均需在惰性气氛保护下进行. 得益于硫化物固态电解质高的离子电导率, 用其组装的固态电池欧姆电阻得到显著下降, 而由固态电解质和固体电极之间固/固界面引起的高界面阻抗问题显得十分突出. 因此, 电池组装及循环过程中电极/固态电解质界面的形成及演化行为对固态电池的性能有着至关重要的影响, 是固态电池商业化要解决的关键问题之一. 而电极/固态电解质界面的有效设计与调控需要深入理解界面的形成及动态演化机制, 这些都有赖于清晰、准确的界面表征手段, 除了传统的电化学测试技术之外, 近年各种先进的表征技术正逐步被应用到固态电池的固/固界面研究之中, 在深入理解和揭示各种界面现象中发挥了非常重要的作用.
本文主要对近年来硫化物固态电解质材料的空气稳定性以及对金属锂负极和正极材料的界面稳定性问题的研究进展进行了回顾和总结, 详细讨论了空气不稳定性的起源、电极/固态电解质界面形成机制、机械应力导致的界面失效问题, 以及针对以上问题的界面优化改性策略, 同时对电极/固态电解质界面形成机制及动态演化过程的先进表征技术进行了总结与评述.


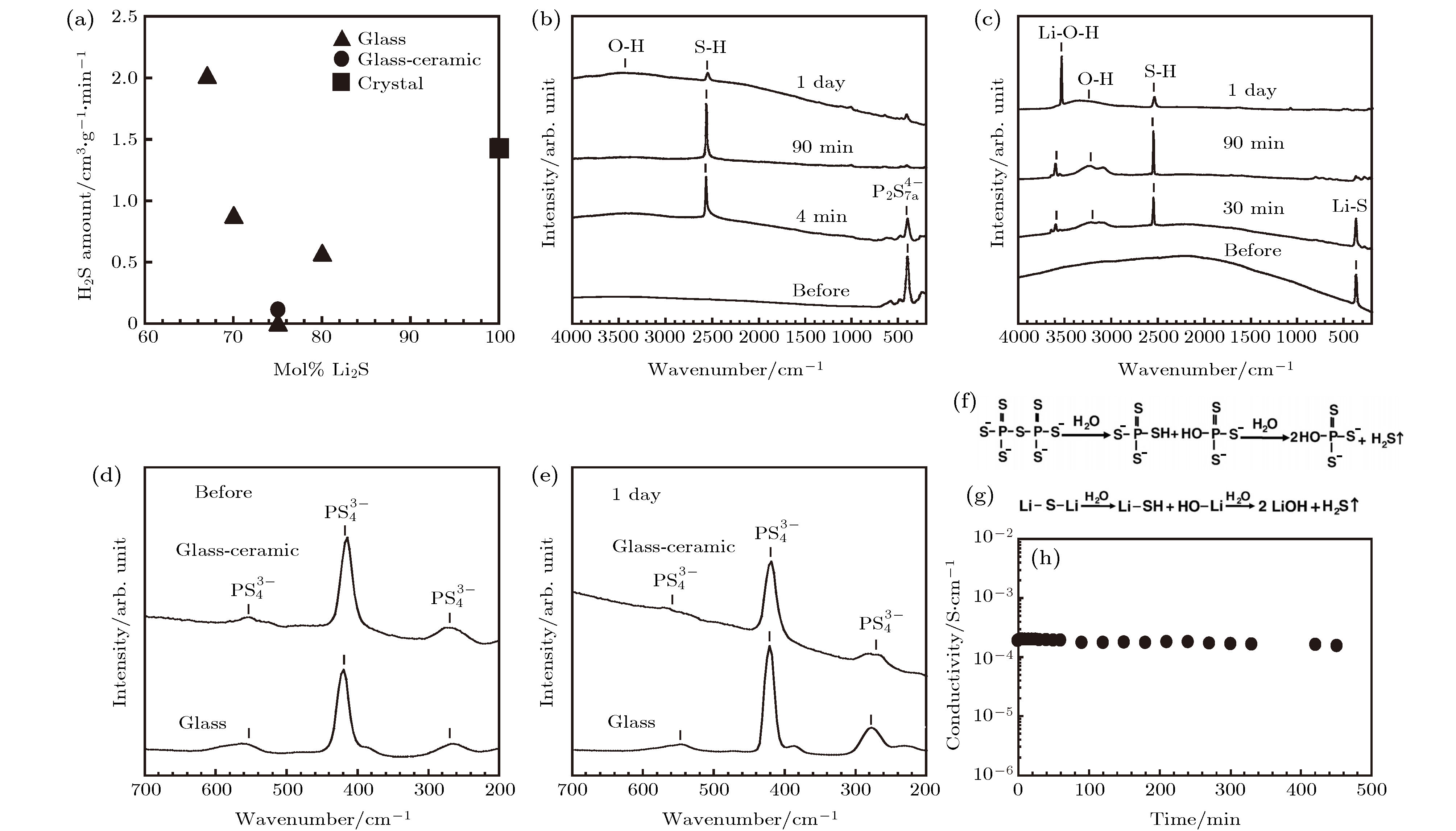 图 2 (a) 不同Li2S含量的Li2S-P2S5玻璃硫化物电解质生成的H2S量; (b), (c) 67Li2S·33P2S5 和Li2S在空气中暴露前和暴露4 min, 90 min以及一天后的拉曼光谱; (d) 75Li2S·25P2S5玻璃和 (e) 玻璃陶瓷硫化物电解质在空气中暴露前和暴露一天后的拉曼光谱; (f) 67Li2S·33P2S5和 (g) Li2S玻璃暴露于空气中的结构变化; (h) 75Li2S·25P2S5玻璃陶瓷硫化物电解质暴露于空气中电导率随着暴露时间变化图[28]
图 2 (a) 不同Li2S含量的Li2S-P2S5玻璃硫化物电解质生成的H2S量; (b), (c) 67Li2S·33P2S5 和Li2S在空气中暴露前和暴露4 min, 90 min以及一天后的拉曼光谱; (d) 75Li2S·25P2S5玻璃和 (e) 玻璃陶瓷硫化物电解质在空气中暴露前和暴露一天后的拉曼光谱; (f) 67Li2S·33P2S5和 (g) Li2S玻璃暴露于空气中的结构变化; (h) 75Li2S·25P2S5玻璃陶瓷硫化物电解质暴露于空气中电导率随着暴露时间变化图[28]Figure2. (a) Amounts of H2S generated from pelletized Li2S–P2S5 glasses with different Li2S contents; (b) Raman spectra of the 67Li2S·33P2S5 glass before exposure to the atmosphere and after exposure for 4 min, 90 min and 1 day; (c) Raman spectra of the Li2S crystal before exposure to the atmosphere and after exposure for 4 min, 90 min and 1 day; Raman spectra of the 75Li2S·25P2S5 glass and glass-ceramic (d) before exposure to the atmosphere and, (e) after exposure for 1 day; structural changes of the (f) 67Li2S·33P2S5 glass and (g) Li2S crystal in air; (h) conductivity of the pelletized 75Li2S·25P2S5 glass-ceramic as a function of exposure time to air at room temperature[28].
为了更深层次地理解硫化物固态电解质在空气中分解过程的潜在机制, Kim等[29]使用密度泛函理论 (DFT) 对β-Li3PS4玻璃陶瓷电解质表面结构和电子性质进行模拟来深入研究其空气不稳定性. 基于模拟结果, 他们构建了由 (100), (010), (001), (111), (110) 和 (101) 六个晶面组成的平衡晶体形状的β-Li3PS4模型 (图3(a)和图3(b)). 其中具有低表面能 (Φ) 的 (001), (111), (110) 和 (101) 四个晶面是主要面, 计算结果表明其稳定性很高, 而具有高Φ的 (100) 和 (010) 晶面的稳定性很差. 因此, 通过在电解质合成过程中合理的控制电解质暴露的晶面, 从而有望改善硫化物固体电解质在空气中的稳定性. 此外, 采用软硬酸碱 (HSAB)理论也能对硫化物晶态电解质在空气中的分解机制进行分析, 基于HSAB理论选择硫化物电解质中的中心原子, 从而设计得到空气稳定性更高的固态电解质[30,31]. 为了进一步设计具有更好空气稳定性的硫化物固态电解质, 系统性地研究材料的耐潮湿稳定性与材料化学组分的关系至关重要. Zhu和Mo[32]采用第一性原理计算材料数据库的热力学分析, 系统地研究了含锂硫化物的水解反应和耐潮湿稳定性. 研究发现含锂硫化物潮湿稳定性普遍较差, 但依然存在一些能够提高其耐潮湿稳定性的阳离子. 他们首先计算了简单二元化合物Li2S的耐潮湿稳定性 (图3(c)). 通过计算水解反应能, 他们发现Li2S在不同化学势下的稳定相主要为氢氧化物和氧化物. 进一步, 他们考察了含锂硫化物的耐潮湿稳定性 (图3(d)), 与实验报道一致, 含有P5+的硫化物耐潮湿稳定性较差. 含有第四、五、六周期的准金属离子的化合物, 如Ga3+, Ge4+, Sn4+, Sb5+, Pb4+, Bi3+, 则具有较好的耐潮湿稳定性, 这与传统的软硬酸碱理论是一致的. 含有前过渡金属离子的材料, 如Zr4+, Hf4+, Ta5+, Nb5+, Cr6+, W6+也具有较差的耐潮湿稳定性, 而Y3+和Cr3+则相对较好. 镧系金属离子的特征较为一致, 对潮湿都较为敏感. 大多数碱金属和碱土金属离子具有较好的耐潮湿稳定性. 后过渡金属离子, 如Zn2+, Cd2+, Cu2+, Ag+, 则表现出最好的耐潮湿稳定性. 通过进一步比较二元硫化物M-S和三元硫化物Li-M-S的稳定性趋势, 发现三元化合物Li-M-S的稳定性介于M-S和Li2S之间, 这是由于三元硫化物的化学组分介于M-S和Li2S之间, 因此其耐潮湿稳定性也介于两者之间. 最后他们探讨了不同化学组分对锂金属还原的电化学稳定性和耐潮湿稳定性两方面的影响 (图3(e)). 计算结果表明, Sc3+, Y3+, In3+ 的掺杂兼具较好的电化学稳定性和耐潮湿稳定性, 是材料设计中可用于掺杂或替换的理想阳离子. 因此, 全面系统地理解化学组分对耐潮湿稳定性的影响, 有利于指导和设计具有良好耐潮湿稳定性和电化学稳定性的硫化物固态电解质材料.
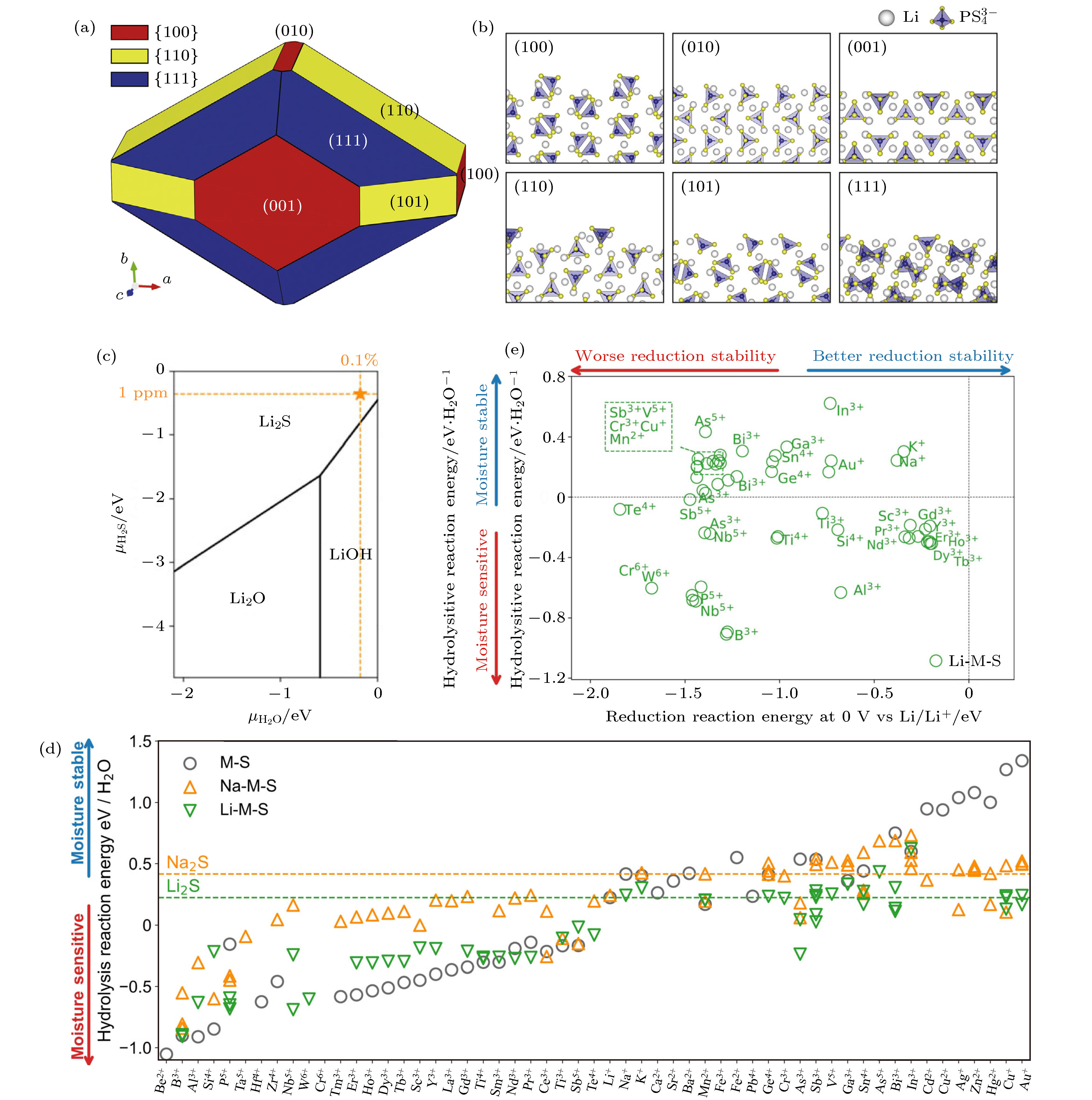 图 3 (a) 基于理论模拟预测构建的由 (100), (010), (001), (111), (110) 和(101)六个晶面组成的平衡晶体形状的β-Li3PS4模型, {100}, {110} 和 {111} 平面分别由红色、黄色和蓝色表示; (b) 平衡晶体形状中六个低指数面的原子结构图, 灰色和蓝色的四面体分别表示锂离子和
图 3 (a) 基于理论模拟预测构建的由 (100), (010), (001), (111), (110) 和(101)六个晶面组成的平衡晶体形状的β-Li3PS4模型, {100}, {110} 和 {111} 平面分别由红色、黄色和蓝色表示; (b) 平衡晶体形状中六个低指数面的原子结构图, 灰色和蓝色的四面体分别表示锂离子和
Figure3. (a) The predicted ECS of the β-Li3PS4 structure. The red, yellow, and blue colors represent the {100}, {110}, and {111} planes; (b) atomic images of six low indexed surfaces in ECS. The grey sphere and blue tetrahedron represent the Li-ion and

为了提高硫化物固态电解质的空气稳定性, 近年来研究工作者进行了大量的探索工作, 已取得一些进展[27]. 其提高硫化物电解质空气稳定性的方法可以归纳为: 1) 利用氧原子或氧化物掺杂, 抑制硫化氢的产生, 提高电解质的空气稳定性[27,32]. Ohtomo等[33]研究表明, 用氧化锂中的氧来部分取代硫化物电解质中硫可以抑制其在空气中产生H2S气体. 他们的实验结果证实, 在xLi2O·(75–x)Li2S·25P2S5体系中, 暴露在空气中的电解质产生H2S气体含量随着x值的增加而逐渐降低 (图4(a)). 这可归因于随着O原子的掺入, 体系形成了非桥键O, 非桥键O相比于桥键S更加稳定, 从而提高了电解质的空气稳定性. 但是, Ohtomo等同时发现氧元素的引入会导致电解质的导电率降低 (图4(b)). 在xLi2O·(75–x)Li2S·25P2S5体系中, 当x = 7时, 7Li2O·68Li2S·25P2S5表现出高的空气稳定性的同时保持了较高的电导率 (2.5 × 10–4 S·cm–1). 因此, 通过合理调控替换氧化物的含量可以抑制硫化物电解质在空气中产生H2S气体的同时维持较高的导电率. Liu等[34]通过对Li3PS4玻璃陶瓷电解质进行ZnO掺杂, 首次成功合成了一系列新型的Li3+3xP1–xZnxS4–xOx固体电解质(x = 0.01, 0.02, 0.03, 0.04, 0.05, 0.06). X-射线衍射(XRD) 结果显示 (图4(c)), 掺杂后的新型硫化物电解质只有β-Li3PS4的特征峰, 而没有ZnO特征峰, 这证实了ZnO不是与Li3PS4的简单物理混合而是掺杂到其晶体结构中. 理论计算优化的结果也表明ZnO掺杂后的β-Li3PS4晶体骨架具有与β-Li3PS4相同的结构, 由排列成锯齿形的孤立的PS4 四面体组成, 具有较高的稳定性 (图4(d)). 他们的理论和实验结果证明, 当x = 0.02时, 所合成的Li3.06P0.98Zn0.02S3.98O0.02可获得最高的离子导电率, 且将其暴露在空气中3 h后产生的H2S的浓度仅为0.0175 cm3·g–1, 几乎不与空气中的H2O反应, 表现出了非常好的空气稳定性 (图4(e)). 因此, 通过在硫化物电解质中掺入一定量的ZnO能显著改善其空气稳定性. 此外, Hayashi等[35]发现, 通过在硫化物电解质 (如Li3PS4) 中添加金属氧化物 (如Fe3O4, ZnO或Bi2O3), 可以作为H2S吸收剂, 从而减少电解质暴露在空气中产生的H2S气体含量, 提高电解质的空气稳定性. 如图4(f)所示, Hayashi等将三种与H2S反应吉布斯能量变化很负的纳米尺寸的金属氧化物Fe2O3, ZnO和Bi2O3简单的物理添加到Li3PS4玻璃态电解质中可以显著降低电解质在空气中产生的H2S含量. 实验结果发现, 添加氧化物后的90Li3PS4·10MxOy (MxOy: ZnO, Fe2O3和Bi2O3)复合材料暴露在空气中产生H2S含量的顺序与金属氧化物吉布斯能量变化的顺序相对应. 对添加ZnO后得到的90Li3PS4·10ZnO复合材料和无添加的Li3PS4玻璃电解质在空气中暴露一天后进行XRD表征可以看出 (图4(g)), 添加ZnO的复合电解质显示出ZnO, ZnS以及一些从Li3PS4玻璃析出的未知相的特征峰. ZnO特征峰的存在证实其是通过简单的物理方式掺杂到Li3PS4电解质中, 而ZnS的形成则表明ZnO作为吸收剂与硫化物电解质中产生的H2S反应, 除去了电解质中产生的H2S气体[36]. 因此, 通过添加具有很大的负的吉布斯能量变化且颗粒尺寸较小的金属氧化物到硫化物固态电解质中也可以显著提升其空气稳定性.
 图 4 xLi2O·(75–x)Li2S·25P2S5 (x = 0, 4, 7, 11, 17) 在空气中暴露后产生的 (a) H2S气体的含量及 (b) 室温下的电导率[33]; (c) Li3+3xP1–xZnxS4–xOx (x = 0, 0.02, 0.06) 和0.98Li3PS4·0.02ZnO的XRD图; (d) 第一性原理计算得出的Li3+3xP1–xZnxS4–xOx (x = 0.02)结构模型图; (e) Li3.06P0.98Zn0.02S3.98O0.02暴露于潮湿空气中产生硫化氢气体量随暴露时间的变化图[34]; (f) 90Li3PS4·10MxOy (MxOy: ZnO, Fe2O3和Bi2O3)复合材料和Li3PS4玻璃在空气中暴露后产生的H2S量随暴露时间的变化图; (g) 90Li3PS4·10ZnO复合材料和Li3PS4玻璃在空气中放置一天后的XRD图谱[35]
图 4 xLi2O·(75–x)Li2S·25P2S5 (x = 0, 4, 7, 11, 17) 在空气中暴露后产生的 (a) H2S气体的含量及 (b) 室温下的电导率[33]; (c) Li3+3xP1–xZnxS4–xOx (x = 0, 0.02, 0.06) 和0.98Li3PS4·0.02ZnO的XRD图; (d) 第一性原理计算得出的Li3+3xP1–xZnxS4–xOx (x = 0.02)结构模型图; (e) Li3.06P0.98Zn0.02S3.98O0.02暴露于潮湿空气中产生硫化氢气体量随暴露时间的变化图[34]; (f) 90Li3PS4·10MxOy (MxOy: ZnO, Fe2O3和Bi2O3)复合材料和Li3PS4玻璃在空气中暴露后产生的H2S量随暴露时间的变化图; (g) 90Li3PS4·10ZnO复合材料和Li3PS4玻璃在空气中放置一天后的XRD图谱[35]Figure4. (a) Amounts of H2S gas generated from the xLi2O·(75–x)Li2S·25P2S5 (x = 0, 4, 7, 11, and 17) glass powders prepared using mechanical milling; (b) electrical conductivities of the pelletized xLi2O·(75–x) Li2S·25P2S5 (x = 0, 4, 7, 11, and 17) glass powders prepared using mechanical milling at room temperature[33]; (c) XRD patterns of Li3+3xP1–xZnxS4–xOx (x = 0, 0.02, 0.06) and 0.98Li3PS4·0.02ZnO; (d) structure of Li3+3xP1–xZnxS4–xOx (x = 0.02) from DFT calculations; (e) the amount of H2S generated from Li3.06P0.98Zn0.02S3.98O0.02 when exposed in the humid air with different duration time[34]; (f) H2S amount as a function of exposure time to air for the 90Li3PS4·10MxOy (MxOy: ZnO, Fe2O3 and Bi2O3) composites and the Li3PS4 glass; (g) XRD patterns of the 90Li3PS4·10ZnO composite and the Li3PS4 glass after exposure to air for 1 day[35].
2) 基于经验性的软硬酸碱理论 (HSAB), 使用较“软”的阳离子进行掺杂或替代: 如Sn4+, Ge4+, As5+, Sb5+等, 改善硫化物电解质的空气稳定性[27,32]. HSAB的原理是硬酸优先与硬碱反应, 软酸更易于与软碱反应. 基于HSAB理论, 相比于P, Sn是一种更软的酸, 与软碱S结合会更加稳定, 因此锡基硫化物电解质 (如Li2SnS3和Li4SnS4-LiI等) 表现出更高的空气稳定性[27,37,38]. Kimura等[39]选择与P处于同一主族, 但比P更软的酸Sb作为电解质的中心原子, 通过机械化学方法合成了Li3SbS4玻璃态和玻璃陶瓷态电解质. 他们的研究结果表明 (图5(a)), 相比于Li3PS4玻璃电解质和Li4SnS4样品, Li3SbS4玻璃电解质暴露于潮湿空气中产生的H2S气体量很少, 且测试后, Li3PS4玻璃变为了液态, 而Li3SbS4玻璃和Li4SnS4研磨样品仍然保持固态. 这说明制备的Li3SbS4显示出了很好的空气稳定性. 此外, Li3SbS4玻璃与玻璃陶瓷电解质的拉曼测试结果都检测出了相同的SbS4单元 (图5(b)), 说明即使暴露在潮湿条件下, SbS4单元中的Sb-S键也是稳定的, 进一步证实了制备得到的Li3SbS4玻璃态电解质表现出了优异的空气稳定性. 因此, 选择Sb作为硫化物电解质的中心原子是设计具有高离子电导率和良好空气稳定性的硫化物固态电解质的有效策略[40]. Zhao等[41]将Li6PS5I (LPSI) 电解质PS4四面体结构中的P用更软的酸Sn部分替代, 成功制备了具有优异空气稳定性的LPSI-20Sn电解质. DFT计算结果得出, LPSI和LPSI-20Sn电解质的氧置换反应能量(ΔE)分别为2.667 eV和9.298 eV, 也就是说用当S被O替换时, PS4四面体中P-S的键比 (P/Sn)S4四面体中 (P/Sn)-S的键能弱得多, 即晶格中取代的Sn倾向于与S而不是O结合, 因此LPSI-20Sn电解质具有稳定的晶体结构 (图5(c)), 显示出更高的空气稳定性. Wang等[42]选择相对更软的酸Cu(I)和Ge(IV)作为硫化物电解质的中心原子, 成功合成了一种离子电导率高达0.9 × 10–4 S·cm–1的硫化物超级导体Li4Cu8Ge3S12 (图5(d)). 这种化合物具有牢固的共价键和刚性稳定的Cu-Ge-S骨架, 强而刚的共价骨架对空气和水中的氧气具有较低的亲和力. 将其暴露于15%湿度的空气和2 M的氢氧化锂溶液中, 其晶体结构未发生任何变化, 显示出了电解质优异的空气或水溶液稳定性 (图5(e)). 阻抗测试结果表明 (图5(f)), 未处理的Li4Cu8Ge3S12电解质, 暴露于15%湿度的空气和2 M的氢氧化锂溶液后, 室温离子电导率分别为0.9 × 10–4, 1.2 × 10–4和1.3 × 10–4 S·cm–1, 离子电导率仅发生了很小的变化, 说明其晶体结构和电化学性能在潮湿的空气或水溶液中未发生较大变化, 显示出了高的稳定性. 上述系列研究结果表明, 基于HSAB理论, 通过选择合适的软酸原子作为电解质的中心原子, 有望设计和合成出高空气稳定性的硫化物固态电解质.
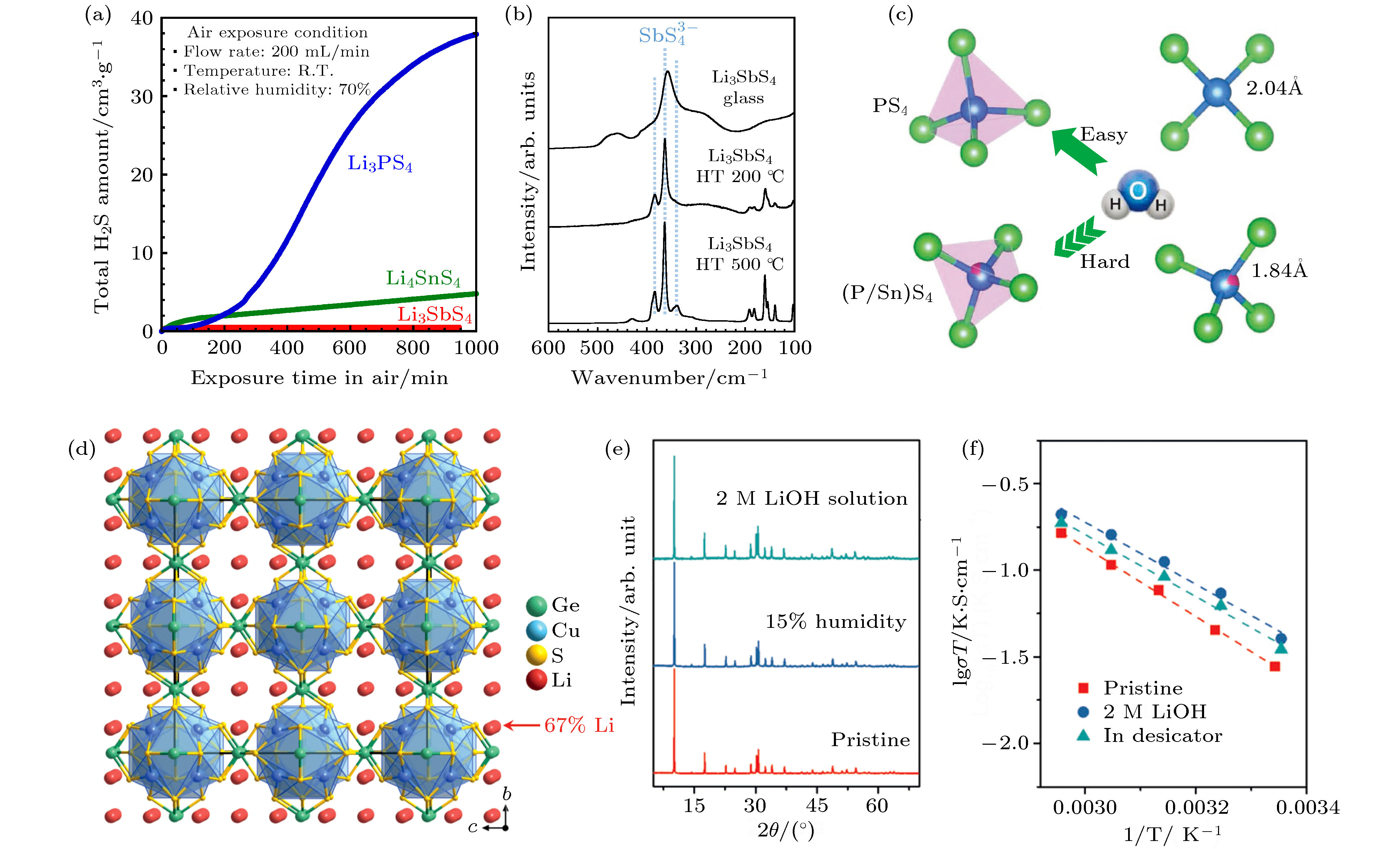 图 5 (a) 由Li3SbS4, Li3PS4和Li4SnS4粉末放出的H2S气体的量; (b) 在200和500 ℃加热条件下, 合成的Li3SbS4和其玻璃陶瓷的拉曼光谱图[39]; (c) 基于第一性原理计算的PS4和 (P/Sn)S4四面体很难被H2O氧化程度的示意图[41]; (d) Li4Cu8Ge3S12的晶体结构; (e) Li4Cu8Ge3S12固态电解质暴露于15%湿度的空气和2 M的氢氧化锂溶液前后的XRD谱图; (f) Li4Cu8Ge3S12固态电解质暴露于15%湿度的空气和2 M的氢氧化锂溶液前后的对比阿伦尼乌斯图[42]
图 5 (a) 由Li3SbS4, Li3PS4和Li4SnS4粉末放出的H2S气体的量; (b) 在200和500 ℃加热条件下, 合成的Li3SbS4和其玻璃陶瓷的拉曼光谱图[39]; (c) 基于第一性原理计算的PS4和 (P/Sn)S4四面体很难被H2O氧化程度的示意图[41]; (d) Li4Cu8Ge3S12的晶体结构; (e) Li4Cu8Ge3S12固态电解质暴露于15%湿度的空气和2 M的氢氧化锂溶液前后的XRD谱图; (f) Li4Cu8Ge3S12固态电解质暴露于15%湿度的空气和2 M的氢氧化锂溶液前后的对比阿伦尼乌斯图[42]Figure5. (a) Amounts of H2S gas generated from the Li3SbS4 glass, Li3PS4 glass, and Li4SnS4 milled powders; (b) Raman spectra of the prepared Li3SbS4 glass and the glass-ceramic heated at 200 and 500 ℃[39]; (c) schematic diagram of the difficult degree of being oxidized by H2O of PS4 and (P/Sn)S4 tetrahedrons based on the DFT calculation[41]; (d) crystal structure of Li4Cu8Ge3S12 viewed along the [100] direction; (e) XRD patterns of Li4Cu8Ge3S12 before and after exposure in 15% moist air and a 2 M LiOH aqueous solution; (f) comparative Arrhenius plots show a minor change in conductivity and activation energy before and after exposure[42].
除了上述提到的可以改善硫化物固态电解质空气稳定性的方法, 其他方法如与疏水的聚合物复合形成硫化物复合固态电解质也可以有效改善其空气稳定性. Tan等[43]通过粉浆浇注法制备了Li7P3S11与疏水的聚合物[polystyrene-block-polyethylene-ranbutylene-block-polystyrene (SEBS)] 复合制备了复合硫化物固态电解质 (图6(a)). 研究发现, 疏水的SEBS的引入能显著改善Li7P3S11的空气稳定性. 他们将100 mg未复合和复合后的Li7P3S11硫化物固态电解质放入一个充入空气 (空气潮湿度50%—55%) 的密闭容器中 (图6(b)), 通过检测硫化物固态电解质在空气中产生H2S气体的量来评价其耐潮湿稳定性. 如图6(c)所示, 相比于未复合的Li7P3S11硫化物固态电解质, 引入5%质量分数的疏水性SEBS形成的复合硫化物固态电解质暴露于一定体积潮湿空气中后能显著降低其产生的H2S气体的量. 此外, 将去离子水分别加入装有100 mg两者的容器中 (图6(d)), 可以明显地发现, 未复合的纯样品很快就在水中分解了. 然而, 复合后的固态电解质即使完全浸泡在水中也未发生明显改变, 显示了很好的耐潮湿稳定性. 此项研究证实, 将硫化物固态电解质与合适的一定质量比的疏水性聚合物复合形成复合硫化物固态电解质也能显著改善其空气稳定性.
 图 6 (a) 95%质量分数Li7P3S11硫化物固态电解质和5%质量分数的SEBS聚合物复合固态电解质的侧面扫描电子显微镜 (SEM) 图; (b) 100 mgLi7P3S11硫化物固态电解质在潮湿空气水解产生H2S气体量的检测装置; (c) 100 mg未复合和复合后的Li7P3S11硫化物固态电解质在定量体积潮湿空气中产生的H2S气体的量随暴露时间的变化图; (d) 100 mg未复合和复合后的Li7P3S11硫化物固态电解质随着去离子水中的涌入后的稳定性照片图[43]
图 6 (a) 95%质量分数Li7P3S11硫化物固态电解质和5%质量分数的SEBS聚合物复合固态电解质的侧面扫描电子显微镜 (SEM) 图; (b) 100 mgLi7P3S11硫化物固态电解质在潮湿空气水解产生H2S气体量的检测装置; (c) 100 mg未复合和复合后的Li7P3S11硫化物固态电解质在定量体积潮湿空气中产生的H2S气体的量随暴露时间的变化图; (d) 100 mg未复合和复合后的Li7P3S11硫化物固态电解质随着去离子水中的涌入后的稳定性照片图[43]Figure6. (a) Cross-section SEM image of composite electrolyte film fabricated with 95 wt % Li7P3S11 and 5 wt % SEBS; (b) setup used for H2S gas measurements from 100 mg of Li7P3S11 hydrolysis in air; (c) H2S amount released vs time for fixed volume air exposed to 100 mg of bare Li7P3S11 and 100 mg of composite with hydrophobic SEBS polymer; (d) figures of bare Li7P3S11 and composite electrolyte film before and after flooding in water[43].
要实现固态电池的高能量密度, 对于固态电解质除了要求有高的离子电导率外, 还要具有宽的电化学稳定窗口. 正极或负极的电势超出电解质的稳定窗口时, 电解质会被氧化或还原而分解, 形成电极/电解质相间界面层. 界面层的组成和结构对界面阻抗和固态电池的性能有着非常重要的影响. 目前, 理论计算和实验结果均表明 (图7), 硫化物固态电解质的电化学稳定窗口十分有限, 约为1.6—2.3 V vs Li+/Li[44,45]. 因此, 几乎所有常用电极材料都无法与硫化物固态电解质形成热力学稳定的界面. 为实现固态电池的高性能稳定循环, 要求形成的相间界面层具有钝化特性的同时拥有高的离子电导率. 固态电解质的类型和电极材料决定了界面层的组成、结构以及物理和化学性质[24,48,54]. 如果形成的界面相是电子绝缘的, 将有利于钝化硫化物固态电解质, 从而抑制电解质的进一步分解, 维持电池的稳定循环. 界面阻抗主要取决于相间界面层的离子电导率. 另一方面, 如果形成的界面相是混合导体相, 电子和离子的同时传输将导致热力学上有利的电解质分解反应持续发生. 因此, 混合导体中间相无法在界面形成必要的钝化层. 持续的分解反应形成厚而不稳定的界面相, 导致界面阻抗不断增大和电池性能的持续劣化. 为抑制电极材料和硫化物固态电解质间的副反应, 获得稳定电极/电解质界面, 许多界面修饰改性策略被开发出来, 最常见的是在电极和固态电解质之间使用缓冲层. 研究表明, 电子绝缘而离子导电的缓冲层可降低电极和硫化物之间的界面阻抗, 改善电池高倍率性能, 并提高固态电池的循环寿命[12,55-60]. 尽管对于硫化物固态电解质需要使用界面缓冲层已经非常明确, 但是其内在的物理和化学机制仍有待于更深入的研究, 这对于开发新型界面缓冲层以进一步提高固态电池性能至关重要. 值得注意的是, 界面缓冲层的引入消除了硫化物固态电解质和电极材料原始的两相接触界面, 同时引入了两个新的界面, 即固体电解质/缓冲层界面和缓冲层/电极材料界面. 因此, 需要从物理、化学及机械力学等方面对这两个新引入界面的特性及功能进行仔细的表征分析, 揭示其作用机制和动态演化行为, 从而为高效界面缓冲层的理性设计提供新的解决方案和实现途径.
 图 7 (a) LGPS固态电解质锂化和去锂化时的电势分布和相平衡的第一原理计算结果[45]; (b) Li/LGPS/LGPS-Pt/Pt半阻塞电极的循环伏安图, 扫描速率为0.1 mV·s–1, 电压范围为0?2.0和1.0?3.5 V[45]; (c) 原始LSPS及 (LSPS + 乙炔黑 (AB))复合材料在充电至4.2 V前、后的X-射线光电子能谱 (XPS) 谱图[53]
图 7 (a) LGPS固态电解质锂化和去锂化时的电势分布和相平衡的第一原理计算结果[45]; (b) Li/LGPS/LGPS-Pt/Pt半阻塞电极的循环伏安图, 扫描速率为0.1 mV·s–1, 电压范围为0?2.0和1.0?3.5 V[45]; (c) 原始LSPS及 (LSPS + 乙炔黑 (AB))复合材料在充电至4.2 V前、后的X-射线光电子能谱 (XPS) 谱图[53]Figure7. (a) The first principles calculation results of the voltage profile and phase equilibria of LGPS solid electrolyte upon lithiation and delithiation[45]; (b) cyclic voltammetry of Li/LGPS/LGPS‐Pt/Pt semiblocking electrode at a scan rate of 0.1 mV·s–1 in the voltage range of 0?2.0 and 1.0?3.5 V[45]; (c) X-ray photoelectron spectra of the pristine LSPS, (LSPS+Acetylene black (AB)) composite before and after charged to 4.2 V[53].
通过调控硫化物固态电解质的组成及结构以提高其离子电导率, 并拓宽电化学稳定窗口, 是解决硫化物固态电解质与正负极界面相容性问题的重要途径. Wu等[61]和Fitzhugh等[62]基于机械电化学效应, 通过控制硫化物固态电解质的合成参数和相应的核壳微观结构组成, 显著拓宽了硫化物电解质的电化学稳定窗口 (图8). 通过控制合成条件制备了核/壳结构硫化物电解质, 其中单晶Li9.54Si1.74P1.44S11.7Cl0.3 (LSPSC) 核被非晶壳包围, 核和壳部分具有不同的原子组成, 具体取决于合成温度. 随着退火温度的升高 (从450到500 ℃), 壳中的Si含量从 ≈40%降低至10%, 而S的含量从≈40%升高至 ≈80%. 较低的温度下处理的硫化物电解质 (较高的Si含量) 电化学稳定窗口拓宽到了0.7—3.1 V, 远高于之前报道的1.7—1 V. 利用第一性原理模拟, 从结构的机械可压缩性角度解释了这种电压窗口的优化. LSPSC的机械模量倾向于随着Si含量的增加而增加, 而随着S含量的增加而降低. 因为核分解产物的体积大于原始物质, 而刚性壳抑制了膨胀, 因此高机械模量壳对核体积膨胀的约束, 抑制了电解质的分解反应, 从而拓宽电化学稳定窗口 (图8(c)). 在此基础上, Fitzhugh等提出了广义的应变稳定模型 (图8(b)), 该模型表明在哪些电压下, 应变诱发的稳定作用可以导致陶瓷-硫化物相的亚稳定性. 显示了在LGPS随着分解而膨胀的情况下, 机械约束如何使得材料在显著拓宽的电位窗口范围内处于亚稳态而不发生分解. 该模型表明, 界面钝化层通过将锂离子维持在机械约束的颗粒内, 最大化反应应变, 从而显著改善了电化学稳定性. 这为实现基于硫化物电解质固态电池在高电压下工作获得高比能量密度开辟了新的广阔空间.
 图 8 (a) 450, 460, 480和500 ℃退火的LSPS-Cl粉末的形貌和组成的微观结构, SEM和透射电子显微镜 (TEM) 图像显示退火的LSPS-Cl电解质具有典型的核壳结构, 扫描透射电子显微镜结合能谱分析 (STEM EDS) 分析表明, 随着退火温度的升高, 样品的壳中Si浓度不断降低, S浓度不断提高[61]; (b) 应变对LGPS电解质分解的影响机制示意图, 其中xD为LGPS分解的分数. 橙色虚线表示当施加的压力可以忽略时, 原始LGPS和任意一组分解产物(D) 二元组合的吉布斯自由能 (G0(xD)). 绿色实线表示将机械约束应用于LGPS时的吉布斯自由能曲线. 由于LGPS在分解时倾向于发生体积膨胀, 因此当施加这种机械约束时, 应变吉布斯自由能 (Gstrain) 增加. 在某个转折点 (表示为xf) 处, 系统的吉布斯自由能量超过了破坏机械约束所需的能量 (蓝色虚线). 突出显示的灰色路径为机械约束LGPS系统的基态. 如果?xDG′ > 0, 则区域xD < xf是亚稳的[62]; (c) 在不同温度下退火的LSPS-Cl样品的循环伏安 (C-V) 曲线[61]
图 8 (a) 450, 460, 480和500 ℃退火的LSPS-Cl粉末的形貌和组成的微观结构, SEM和透射电子显微镜 (TEM) 图像显示退火的LSPS-Cl电解质具有典型的核壳结构, 扫描透射电子显微镜结合能谱分析 (STEM EDS) 分析表明, 随着退火温度的升高, 样品的壳中Si浓度不断降低, S浓度不断提高[61]; (b) 应变对LGPS电解质分解的影响机制示意图, 其中xD为LGPS分解的分数. 橙色虚线表示当施加的压力可以忽略时, 原始LGPS和任意一组分解产物(D) 二元组合的吉布斯自由能 (G0(xD)). 绿色实线表示将机械约束应用于LGPS时的吉布斯自由能曲线. 由于LGPS在分解时倾向于发生体积膨胀, 因此当施加这种机械约束时, 应变吉布斯自由能 (Gstrain) 增加. 在某个转折点 (表示为xf) 处, 系统的吉布斯自由能量超过了破坏机械约束所需的能量 (蓝色虚线). 突出显示的灰色路径为机械约束LGPS系统的基态. 如果?xDG′ > 0, 则区域xD < xf是亚稳的[62]; (c) 在不同温度下退火的LSPS-Cl样品的循环伏安 (C-V) 曲线[61]Figure8. (a) Microstructural study on the morphology and composition of LSPS-Cl powders annealed at 450, 460, 480, and 500 ℃. SEM and TEM images show their typical core-shell structures. STEM EDS analyses of these samples show a continuously decreasing Si concentration and increasing S concentration in the shell of these samples versus the increasing annealing temperature[63]; (b) illustration of the impact of strain on LGPS decomposition, where xD is the fraction of LGPS that has decomposed. The dashed-orange line represents the Gibbs energy (G0(xD)) of a binary combination of pristine LGPS and an arbitrary set of decay products (D) when negligible pressure is applied. The solid-green line shows the Gibbs when a mechanical constraint is applied to the LGPS. Since LGPS tends to expand upon decomposition, the strain Gibbs (Gstrain) increases when such a mechanical constraint is applied. At some fracture point, denoted xf, the Gibbs energy of the system exceeds the energy needed to fracture the mechanical constraints (the dashed-blue line). The highlighted gray path is the suggested ground state for a mechanically constrained LGPS system. The region xD < xf is metastable if ?xDG′ > 0[62]; (c) cyclic voltammetry (C-V) test and analyses of the LSPS-Cl samples annealed at various temperatures[63].
2
3.1.锂金属负极界面
金属锂由于其高的理论比容量 (3840 mAh·g–1) 和低的电化学势 (相对于标准氢电极为–3.04 V) 而被认为是锂电池的理想负极. 然而, 在传统液态锂离子电池中, 由于锂枝晶导致电池内短路带来的严重安全隐患, 制约了金属锂负极的应用. 得益于使用固态电解质替代易燃液态电解质带来的安全性能提升, 人们希望在固态电池中使用金属锂负极以进一步提升电池能量密度. 然而, 研究表明, 金属锂负极应用于硫化物电解质的固态电池仍面临着锂枝晶导致短路及界面稳定性差等问题. 与传统的液态锂电池类似, 在固态锂电池中同样面临锂金属不均匀沉积-溶出导致的锂枝晶生长问题, 锂枝晶穿透固体电解质导致电池性能下降甚至短路[64-66]. 在典型的硫化物电解质, 如LGPS[67,68], Li3PS4(玻璃或陶瓷)[69-71]和Li6PS5Cl (LPSCl)[72,73], 均观察到了锂枝晶的形成. 虽然机理仍不明确, 实际上发现尽管固态电解质的机械强度比锂金属要高得多, 硫化物固态电解质中的锂枝晶生长比液态电解质中更容易. 通常认为硫化物固态电解质内部的空隙和晶界以及锂负极和固态电解质之间界面的不稳定性是导致硫化物电解质中锂枝晶形成的两个主要原因. 由于其较窄的电化学稳定窗口, 硫化物固态电解质对金属锂负极都不稳定, 和锂金属接触时会被还原形成界面层 (图9)[24,48,74-76]. 其中, 含有Ge, Cl, I等元素的硫化物电解质与金属锂接触后会形成含有Li-Ge, LiCl和LiI等的界面层; Li7P3S11在2.28 V时会被还原成Li3PS4, 在1.71 V时被进一步还原形成包含Li3P和Li2S的界面层[44]. 当硫化物电解质被还原形成的界面层由Li2S, LiCl, Li3P, LiI等电子绝缘产物构成时, 可以阻止电解质和金属锂的进一步反应, 形成稳定的界面, 如Li3PS4和Li7P3S11电解质 (图9(d)). 在硫银锗矿型电解质Li6PS5X (X = Cl, Br, I)中, Li6PS5I表现出异常高的界面阻抗增长, 具体机制目前仍不清楚 (图9(e)和图9(f)). 当电解质中含有Ge, Sn和Si等元素时, 如Li10GePS12和Li10SnPS12 (LSPS), 被金属锂还原后形成含有Li-Ge, Li-Sn合金的电子-离子混合导体界面层, 此时界面副反应则会持续进行, 界面层不断增厚, 导致电池性能不断下降甚至失效 (图9(b)和图9(c)). 为此, Yao等[77]在锂负极与LGPS电解质之间引入75Li2S·24P2S5·1P2O5组成双层电解质结构稳定负极界面. 双层电解质中LGPS提供高的锂离子电导率, 而75Li2S·24P2S5·1P2O5可与金属锂负极形成稳定的界面层.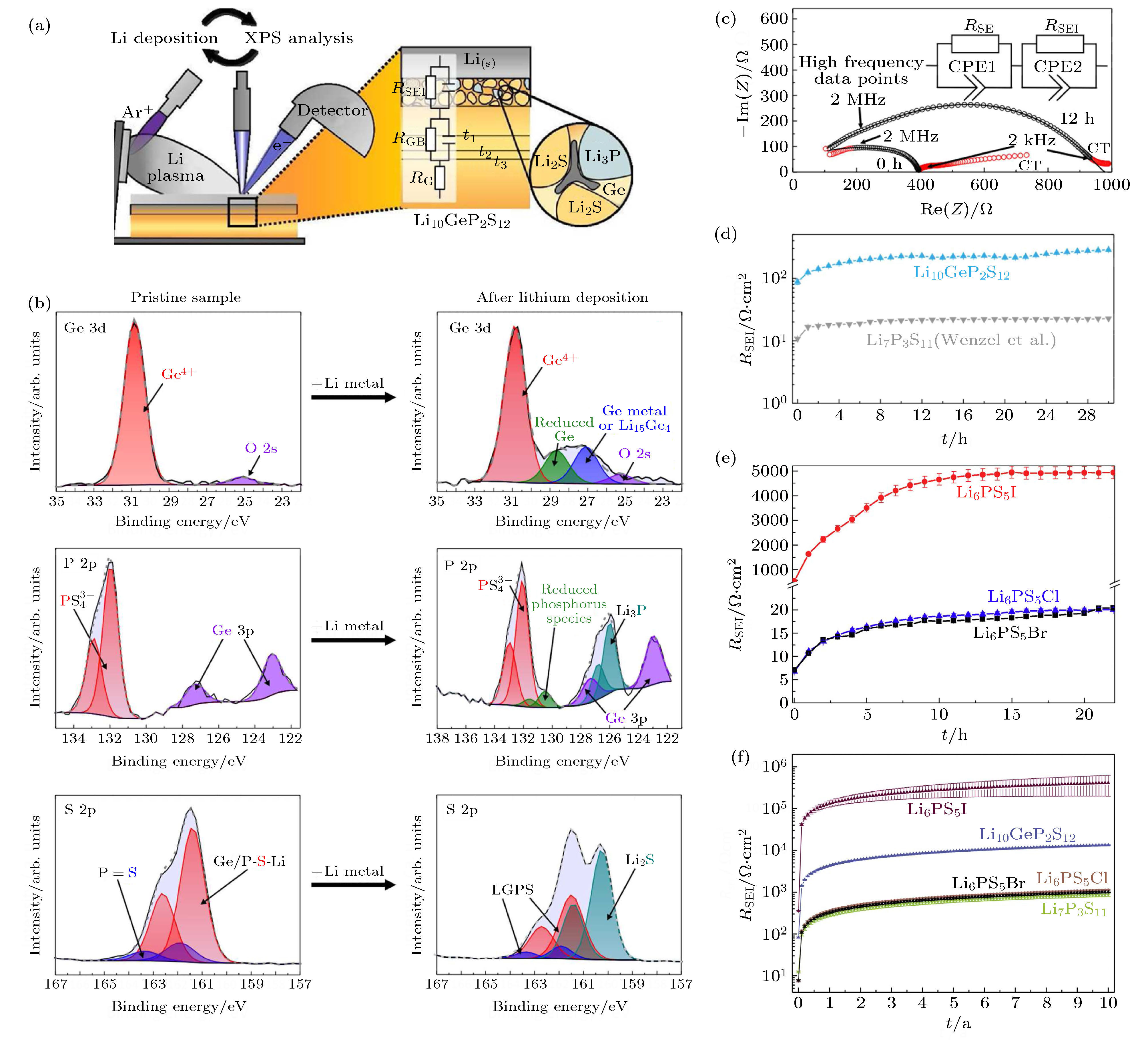 图 9 (a) 用于监测硫化物电解质与金属锂之间反应的原位XPS实验装置; (b) 原始LGPS样品以及沉积31 nm的锂金属后S 2p, Ge 3d和P 2p的XPS谱图和拟合结果. 拟合鉴定的物种以不同的颜色标记. 微弱的氧信号是由XPS腔室气氛中的微量的氧气引起的; (c) Li/LGPS/Li电池组装后 (0 h) 和组装后12 h的阻抗. 插图为用于拟合的等效电路, 拟合过程去除了红色数据点; (d) LGPS/Li与Li7P3S11/Li界面电阻增加的比较. 拟合的不确定性绘制为误差线, 通常明显小于5 Ω·cm2; (e) Li/Li6PS5X/Li电池组装后 (0 h) 和20 h后的界面电阻均增加情况. 在初始的快速增长后, 界面电阻的增加表现出渐近行为; (f) Li7P3S11, Li10GeP2S12, Li6PS5Cl, Li6PS5Br和Li6PS5I电解质10年内界面电阻的模拟结果. 误差线由拟合误差得出[24,76]
图 9 (a) 用于监测硫化物电解质与金属锂之间反应的原位XPS实验装置; (b) 原始LGPS样品以及沉积31 nm的锂金属后S 2p, Ge 3d和P 2p的XPS谱图和拟合结果. 拟合鉴定的物种以不同的颜色标记. 微弱的氧信号是由XPS腔室气氛中的微量的氧气引起的; (c) Li/LGPS/Li电池组装后 (0 h) 和组装后12 h的阻抗. 插图为用于拟合的等效电路, 拟合过程去除了红色数据点; (d) LGPS/Li与Li7P3S11/Li界面电阻增加的比较. 拟合的不确定性绘制为误差线, 通常明显小于5 Ω·cm2; (e) Li/Li6PS5X/Li电池组装后 (0 h) 和20 h后的界面电阻均增加情况. 在初始的快速增长后, 界面电阻的增加表现出渐近行为; (f) Li7P3S11, Li10GeP2S12, Li6PS5Cl, Li6PS5Br和Li6PS5I电解质10年内界面电阻的模拟结果. 误差线由拟合误差得出[24,76]Figure9. (a) Experimental setup of the in situ XPS experiment to monitor the reaction between sulfide and Li; (b) S 2p, Ge 3d, and P 2p XPS spectra and model fits for the pristine LGPS sample and after deposition of 31 nm Li metal. The identified species are marked and labeled in different colors. The small oxygen signal is caused by tiny fractions of oxygen in the atmosphere of the XPS chamber; (c) impedance of a Li/Li10GeP2S12/Li cell directly after assembly (0 h) and after 12 h. The equivalent circuit used as fit model is shown in the inset. Red data points were removed for the fitting process; (d) increase of the Li10GeP2S12/Li interphase resistance, in comparison to Li7P3S11/Li. The uncertainty of the fit is drawn as error bars, which are generally significantly smaller than 5 Ω·cm2; (e) the SEI resistance increase for Li/Li6PS5X/Li cells directly after assembly (0 h) and after 20 h. The increasing resistance appears to show asymptotic behavior after initial growth; (f) simulation of the SEI resistance over 10 years for Li7P3S11, Li10GeP2S12, Li6PS5Cl, Li6PS5Br and Li6PS5I. Error bars were derived from the fit errors[24,76].
通过锂负极/硫化物固态电解质界面修饰提高界面稳定性, 被认为是抑制固态电解质中锂枝晶生长的重要途径. 虽然具体机理仍不明确, 在硫化物固态电解质中引入LiI, LiF, ZnO, P2O5等被证明在改善金属锂/电解质界面稳定性的同时可有效地抑制锂枝晶的生长[71,78-80]. 对锂负极/固态电解质界面进行修饰, 提高界面稳定性, 也是抑制锂枝晶生长的有效策略. 为抑制锂枝晶体的生长, 界面层应具有高的界面能. 通过在硫化物电解质与锂负极处引入富含LiF的界面修饰层可有效改善界面兼容性, 抑制锂枝晶生长[57,67,81]. Fan等[57]利用LiFSI与Li的反应在锂负极与Li3PS4电解质界面处原位形成一层富含LiF的界面层, 有效地抑制了锂枝晶的生长. 他们认为, 富含LiF的界面层能够抑制锂枝晶向固态电解质内部的渗透, 而其低电子电导和高机械强度能够提高金属锂与Li3PS4的界面稳定性. Xu等[81]将高含氟的HFE溶剂渗透至Li7P3S11电解质内部与金属锂原位反应形成富含LiF的界面修饰层, 实现了0.5 mA·cm–2电流密度、0.1 mAh·cm–2截止容量下稳定循环超过200 h. Zheng等[58]通过在金属锂负极/LSPS电解质界面引入极少量的锂盐/离子液体, 在金属锂表面上原位形成一层只传导锂离子的界面层, 很好地抑制LSPS与Li金属之间的副反应, 从而显著改善了界面稳定性. 对比不同锂盐/离子液体修饰Li/LSPS界面的结果发现, 相比于LiTFSI锂盐, LiFSI锂盐体系会在锂金属表面形成LiF富集的SEI层, 导致电池循环过程中阻抗不断增大 (图10). Zhao等[80]对LPSCl(LPSCl)电解质进行氟 (F) 取代, 制备了氟化的硫化物电解质LPSCl1–xFx, 显著提升了其对金属锂负极的界面稳定性 (图11). LPSCl0.3F0.7电解质在Li-Li对称电池中表现出优异的锂沉积-溶出稳定性, 可在6.37 mA·cm–2电流密度、5 mAh·cm–2截止容量下稳定循环超过250 h. 其优异的性能主要得益于金属锂与LPSCl0.3F0.7电解质原位形成的高度氟化、致密的界面层, 不仅可以有效地阻止界面副反应还可以抑制锂枝晶的生长. Liang等[82]基于溶液反应在金属锂表面原位沉积一层LixSiSy中间层, 经过修饰的锂金属负极对空气和对Li3PS4电解质均表现出优异的稳定性, 可有效抑制锂枝晶的生长. Zhang等[68]通过将H3PO4溶解于THF中, 并与锂金属反应, 在锂金属表面原位形成LiH2PO4保护层. LiH2PO4界面层在保持循环过程中金属锂与LGPS电解质界面紧密接触的同时可有效地抑制界面副反应发生, 从而改善了电池的循环性能. Wang等[83]利用分子层沉积技术在锂金属负极和LSPS硫化物电解质之间沉积一层有机无机混合界面修饰层, 有效避免了LSPS和金属锂之间副反应, 同时抑制了锂枝晶的生长. 此外, 原子层沉积技术 (ALD) 等方法也被应用来制备界面缓冲层 (如Al2O3) 以改善硫化物电解质与金属锂负极的界面稳定性[84-86]. Wang等[87]将固态塑晶电解质 (PCE) 用作LGPS电解质和锂负极的界面修饰层, 有效改善了界面兼容性, 并抑制了枝晶生长.
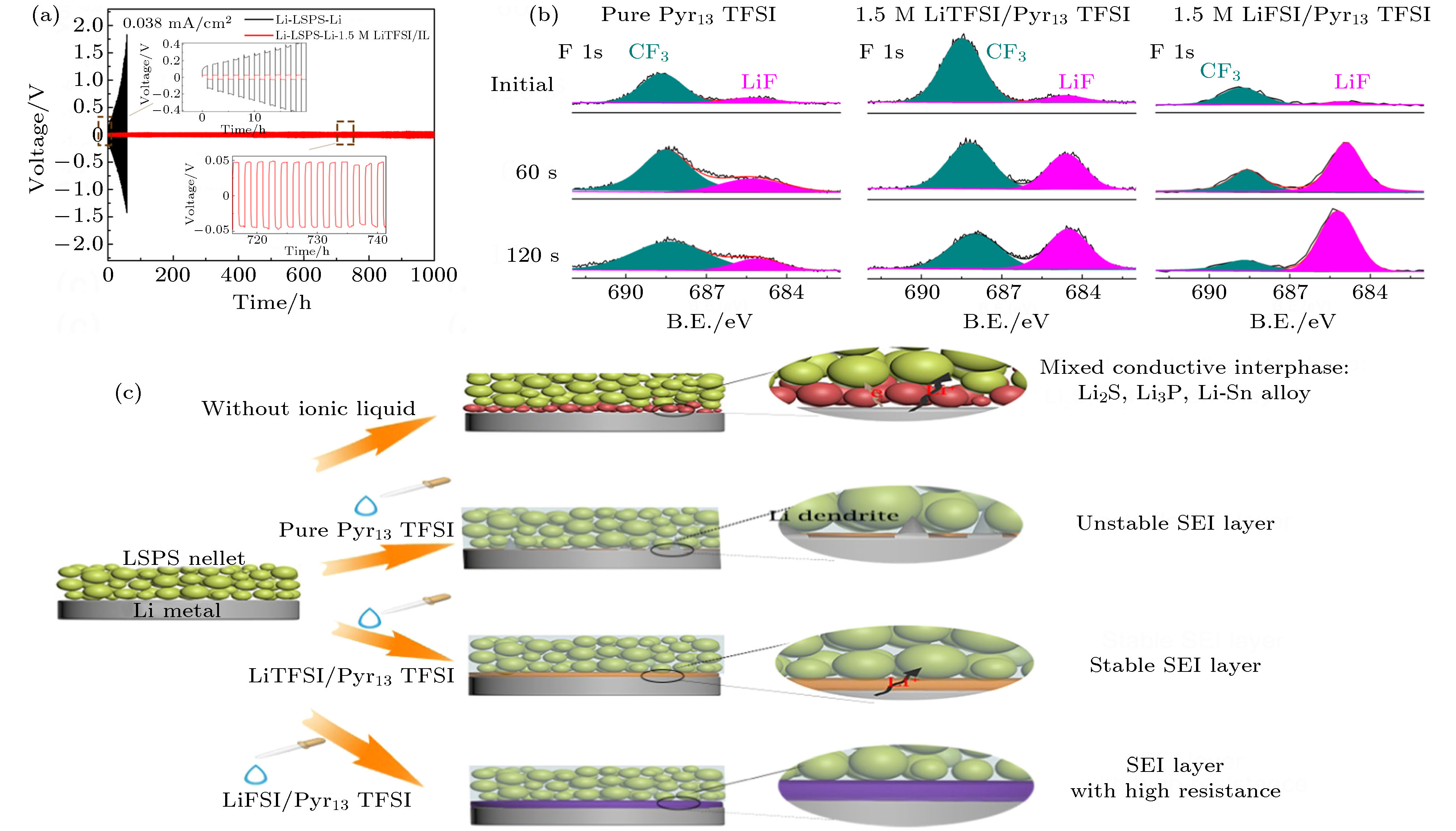 图 10 (a) 1.5 M LiTFSI/Pyr13TFSI离子液体修饰前后Li/LSPS/Li对称电池在0.038 mA·cm–2电流密度下的锂沉积-溶出曲线; (b) 加纯Pyr13TFSI离子液体修饰、1.5 M LiTFSI/Pyr13TFSI离子液体修饰和1.5 M LiTFSI/Pyr13TFSI离子液体修饰的Li/LSPS/Li对称电池循环140 h后Li金属不同刻蚀时间下的F 1s XPS谱图; (c)不同锂盐/Pyr13TFSI修饰界面的机理示意图[58]
图 10 (a) 1.5 M LiTFSI/Pyr13TFSI离子液体修饰前后Li/LSPS/Li对称电池在0.038 mA·cm–2电流密度下的锂沉积-溶出曲线; (b) 加纯Pyr13TFSI离子液体修饰、1.5 M LiTFSI/Pyr13TFSI离子液体修饰和1.5 M LiTFSI/Pyr13TFSI离子液体修饰的Li/LSPS/Li对称电池循环140 h后Li金属不同刻蚀时间下的F 1s XPS谱图; (c)不同锂盐/Pyr13TFSI修饰界面的机理示意图[58]Figure10. (a) Li+ stripping/plating curves of Li/LSPS/Li symmetric cells with and without 1.5 M LiTFSI/IL at a current density of 0.038 mA cm–2 and the cycle performance of Li/LSPS/Li symmetric cell with 1.5 M LiTFSI/IL; (b) F 1s XPS spectra of Li metal at different etching times disassembled from (i) Li/(LSPS + pure Pyr13TFSI)/Li, (ii) Li/(LSPS + 1.5 M LiTFSI/Pyr13TFSI)/Li, and (iii) Li/(LSPS + 1.5 M LiFSI/Pyr13TFSI)/Li symmetric cells after cycling for 140 h; (c) schematic illustration of interfacial modification mechanism of different Li salt/Pyr13TFSI[58].
 图 11 (a) Li/LPSCl0.3F0.7/Li对称电池中的锂沉积-溶出曲线; (b) Li金属/ LPSCl0.3F0.7固态电解质界面的XPS深度分析; Li/LPSCl0.3F0.7界面处Li 1s和F 1s XPS光谱随溅射深度的整体变化情况 (上面栏); 从上图提取的代表性Li 1s和F 1s XPS谱图[80]
图 11 (a) Li/LPSCl0.3F0.7/Li对称电池中的锂沉积-溶出曲线; (b) Li金属/ LPSCl0.3F0.7固态电解质界面的XPS深度分析; Li/LPSCl0.3F0.7界面处Li 1s和F 1s XPS光谱随溅射深度的整体变化情况 (上面栏); 从上图提取的代表性Li 1s和F 1s XPS谱图[80]Figure11. (a) Li plating and stripping in Li/LPSCl0.3F0.7/Li symmetric cells; (b) XPS depth profiling analysis of the Li metal/SSE interface. Overall evolution of the Li 1s and F 1s spectra with sputtering depth at the Li/LPSCl0.3F0.7 interface. Representative Li 1s and F 1s XPS spectra extracted from the upper panel[80].
Lee[88]等设计开发了一种银-碳 (Ag-C) 复合材料作为金属锂负极和硫化物固态电解质的界面缓冲层, 有效地抑制了锂枝晶生长, 改善了负极界面稳定性. 以高镍三元层状氧化物 (LiNi0.9Co0.05Mn0.05O2, NCM955) 为正极, LPSCl为固态电解质制备了软包的全固态电池, 获得了高的能量密度和优异的循环稳定性. 电池组装过程负极不含锂 (Ag-C缓冲层涂覆在负极集流体上), 以充电过程沉积的锂为负极。虽然具体机理不明确, 他们发现Ag-C过渡层能够调节锂的沉积-溶出行为, 促进循环过程中锂的均匀沉积-溶出, 从而抑制锂枝晶生长.
2
3.2.正极/硫化物固态电解质界面
由于硫化物固态电解质较低的电化学稳定窗口, 层状氧化物等高电压正极材料 (如LiCoO2, LiNi(1–x–y)CoxMnyO2等)与硫化物电解质由于界面相容性差而产生高的界面阻抗. 硫化物固态电解质与正极的界面问题主要包括: 界面空间电荷层、高电压下硫化物电解质的氧化分解、界面化学反应 (元素互扩散) 以及充放电过程中产生的界面应力. 空间电荷层的形成是由于Li+在正极和硫化物电解质中化学势的较大差异, 导致Li+从电解质向正极材料扩散, 从而在界面的电解质侧形成高电阻的Li+缺陷层[56,89,90]. 由于氧化物正极材料为离子-电子混合导体, 界面处氧化物侧Li+的浓度梯度可以通过电子传导来平衡, 这使得更多的Li+从电解质向正极扩散以平衡界面两侧化学势差, 形成更厚的空间电荷层, 从而产生高的界面阻抗. 此外, 硫化物电解质通常在2.1 V (vs Li+/Li) 以上会发生氧化分解 (对应于S(0)/S(–2)氧化还原对), 而在界面处生成硫 (S) 和五硫化二磷 (P2S5) 等具有低离子电导率的副产物[44,91]. 因此, 硫化物电解质与氧化物正极直接接触, 特别是在充电过程中处于高电位时, 会被氧化分解而形成高阻抗界面层 (图12). 界面化学反应是影响硫化物电解质固态电池电化学性能的另一个重要因素. 由于界面反应, 硫化物电解质与氧化物正极 (如LiCoO2, LiNi(1–x–y)CoxMnyO2, LiNi0.85Co0.1Al0.05O2和LiMn2O4) 之间的界面上会生成硫酸盐、磷酸盐、单质硫、多硫化锂和硫化物等反应产物[92-95]. Richards等[47]的计算结果表明, 由于氧化物电极材料的高电压和高氧化学势, 通常与硫化物电解质之间具有较大的反应能, 如硫化物中的PS4基团可与氧化物正极反应生成PO4基团和金属硫化物. 硫化物固体电解质与正极材料界面的机械应力对其界面稳定性同样有着不可忽视的重要影响 (图11(b))[52,96]. Zhang等[93]结合原位阻抗谱与原位拉曼等表征手段研究了LPSCl电解质与LiNi0.8Co0.1Mn0.1O2 (NCM811) 正极界面的演化行为, 间接揭示了空间电荷层的演变. 研究表明, 在电池循环初期, 空间电荷层是影响电池界面阻抗变化的主要因素, 而且由于空间电荷层引起的界面处锂离子迁移与重新分布, 同时导致了界面处LPSCl内P—S键状态的改变. 随着电池循环次数增加, 空间电荷层、界面反应、界面机械失效、锂枝晶生长等共同作用导致界面的进一步劣化和电池性能的衰退. Jung等[51]研究了高镍三元氧化物正极材料与LPSCl电解质之间化学反应对固态电池性能劣化的影响. 研究表明, 在荷电状态(SOC) 为0时, 界面化学反应就已经开始, 生成了NiS, LiPxCly和LiCl等产物, 严重恶化电池性能. 化学反应产物与通常在电化学循环后观察到的分解产物不同, 并且它们降低了晶界处的离子传导. 同时化学反应过程会导致正极材料与硫化物电解质之间的物理接触劣化或裂纹形成. 化学 (电化学) 反应诱导的体积变化在界面处产生巨大的应力会破坏界面的机械完整性, 导致界面阻抗的增加和电池性能衰退. 界面化学反应及其诱导的机械性能劣化都对电池性能产生非常不利的影响, 增大界面阻抗, 降低初始放电容量和能量效率. 对于大多数电极材料而言, 在脱锂/锂化过程中不可避免地会出现不同程度的体积变化. 不同于传统液态锂离子电池, 可以通过液态电解质的自由流动来缓冲/吸收这种体积变化. 电极材料充放电过程的体积变化会在硫化物电解质与氧化物正极的固-固界面处产生巨大的应力/应变.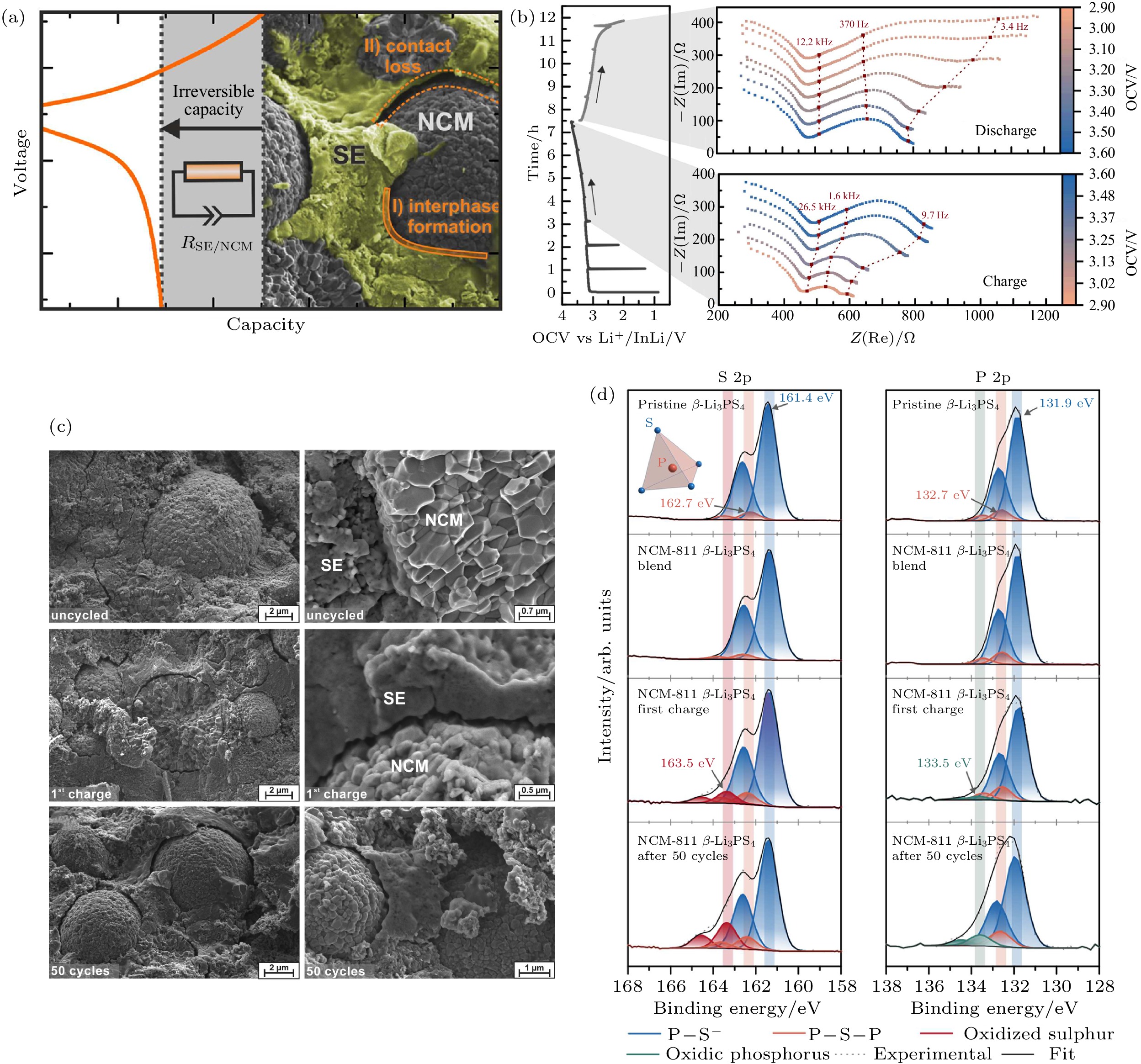 图 12 (a) LiNi0.8Co0.1Mn0.1O2 (NCM811)正极和β-Li3PS4固态电解质相间界面形成和化学机械过程的示意图; (b) Li–In|β-Li3PS4|NCM811/β-Li3PS4固态电池恒流充放电过程阻抗变化. 左图: 0.1 C下首次充放电曲线显示出与阻抗测量周期相对应的电流中断. 右: 充电期间和放电期间的阻抗谱, 分别在充电或放电1 h后进行测量, 红色点表示阻抗谱的选定频率; (c) Li–In|β-Li3PS4|NCM811/β-Li3PS4 固态电池复合正极中NCM811和β-Li3PS4的SEM图. 电池组装后未施加电流或电势的SEM图;电池在0.1 C下首次充电至4.3 V vs Li+/Li后的SEM图; 给定电池经过50个循环后, 在放电状态下的SEM图. NCM-811颗粒在脱锂 (充电) 过程中会收缩并失去与SE的接触. NCM形态在电解质上的残留痕迹表明, 活性材料和电解质之间最初紧密接触; (d) NCM811正极和β-Li3PS4固态电解质界面XPS表征. 原始β-Li3PS4; NCM811和β-Li3PS4的原始混合物, 不施加任何电流或电势; 首次充电后(0.1 C) 正极NCM811和β-Li3PS4复合残留; 50个循环后正极复合材料的S 2p和P 2p XPS谱图拟合结果. 蓝色峰归因于Pδ+—Sδ-键, 橙色峰与
图 12 (a) LiNi0.8Co0.1Mn0.1O2 (NCM811)正极和β-Li3PS4固态电解质相间界面形成和化学机械过程的示意图; (b) Li–In|β-Li3PS4|NCM811/β-Li3PS4固态电池恒流充放电过程阻抗变化. 左图: 0.1 C下首次充放电曲线显示出与阻抗测量周期相对应的电流中断. 右: 充电期间和放电期间的阻抗谱, 分别在充电或放电1 h后进行测量, 红色点表示阻抗谱的选定频率; (c) Li–In|β-Li3PS4|NCM811/β-Li3PS4 固态电池复合正极中NCM811和β-Li3PS4的SEM图. 电池组装后未施加电流或电势的SEM图;电池在0.1 C下首次充电至4.3 V vs Li+/Li后的SEM图; 给定电池经过50个循环后, 在放电状态下的SEM图. NCM-811颗粒在脱锂 (充电) 过程中会收缩并失去与SE的接触. NCM形态在电解质上的残留痕迹表明, 活性材料和电解质之间最初紧密接触; (d) NCM811正极和β-Li3PS4固态电解质界面XPS表征. 原始β-Li3PS4; NCM811和β-Li3PS4的原始混合物, 不施加任何电流或电势; 首次充电后(0.1 C) 正极NCM811和β-Li3PS4复合残留; 50个循环后正极复合材料的S 2p和P 2p XPS谱图拟合结果. 蓝色峰归因于Pδ+—Sδ-键, 橙色峰与
Figure12. (a) Schematic illustration of interphase formation and chemomechanical processes in LiNi0.8Co0.1Mn0.1O2 cathode and β-Li3PS4 solid electrolytes; (b) impedance spectra recorded intermittently during galvanostatic battery cycling. Left: First cycle charge and discharge profile of a Li–In|β-Li3PS4|NCM811/β-Li3PS4 cell at 0.1 C showing current interruption corresponding to the periods of impedance measurement. Right: Impedance spectra during charge and discharge periods. Measurements were conducted after 1 h of charging or discharging, respectively. Red points indicate selected frequencies in the spectra; (c) scanning electron micrographs of the cathode composite of NCM811 and β-Li3PS4. As prepared in a solid-state cell but without the application of current or potential; SEM images of a Li–In | β-Li3PS4| NCM-811 /β-Li3PS4 cell after single charging to 4.3 V vs Li+/Li at 0.1 C; SEM images of a given cell after 50 full battery cycles in the discharged state. The NCM-811 particles shrink during delithiation (charge) and lose contact with the SE. The remaining imprint of the NCM morphology on the electrolyte shows the initially intimate contact between the active material and the electrolyte; (d) peak deconvolution of the S 2 p and P 2 p spectra of pristine β-Li3PS4 for the pristine mixture of NCM811 and β-Li3PS4 without any application of current or potential, the cathode composite of NCM811 and β-Li3PS4 after the first charge (0.1 C), and the cathode composite of NCM811 and β-Li3PS4 after 50 cycles. Blue peaks are attributed to equivalent Pδ+–Sδ– bonds. Orange peaks were correlated to bridged P–S–P in

因此, 对硫化物固态电解质与氧化物正极界面进行修饰, 以消除空间电荷层, 阻止硫化物电解质高电位下氧化分解, 抑制界面副反应, 以及吸收界面应力稳定界面机械结构, 对于固态电池的稳定循环显得十分关键. 由于硫化物电解质对空气不稳定, 其表面修饰改性受到很大限制. 目前, 界面修饰工作主要集中在对氧化物正极材料进行包覆改性上. LiNbO3 (LNO)[22,55,97-99], Li4Ti5O12 (LTO)[56,100], LiTaO3[101], Li2SiO3[102,103], Li3PO4[104], ZrO2[105]和Al2O3[106]等均被应用于正极材料表面包覆. 这些材料通常具有宽的电化学稳定性窗口, 以及与氧化物正极和硫化物电解质良好的化学稳定性. 其中应用最广泛的是LiNO, 其室温离子电导率为10–6 S·cm–1, 且可以通过溶液法方便地制备包覆层.
Jung等[107]通过水溶液方法在LiCoO2 (LCO)表面包覆Li3BO3–Li2CO3 (LBCO), 显著改善了LiCO|LPSCl|Li-In固态电池的电化学性能. 他们发现利用Li3BO3水溶液包覆技术, 在高温煅烧过程中可以将LCO表面的Li2CO3杂质 (锂离子不良导体) 转化为具有高离子电导率的LBCO (在30 °C下最高离子电导率为6.0 × 10–7 S·cm–1). 因此, 与纯的Li3BO3相比, LBCO包覆材料表现出更好的效果. 在0.2和2 C下循环, LBCO包覆的LCO材料的放电容量分别达到142和94 mAh·g–1; 而 未包覆LCO材料的放电容量仅为107和18 mAh·g–1. XPS等分析结果表明, LBCO包覆层可避免含有Co3S4等混合离子-电子导体界面相的形成, 同时在界面处生成磷酸盐有效钝化界面. Zhang等[108]利用介电谱测试技术结合多种表征手段, 研究了WS2和LiBr双重掺杂对Li7P3S11电解质离子电导率和与LCO正极界面稳定性的影响. 基于LCR数字电桥的介电谱测试测试结果表明, WS2和LiBr双重掺杂后Li7P3S11固体电解质与LCO正极之间的界面电容减小, 这表明掺杂抑制了Li7P3S11电解质与LCO正极界面空间电荷层效应, 改善了界面相容性. Wang等[109]通过表面双层包覆调控其与硫化物电解质界面之间的纳米结构, 首先利用ALD技术在LCO表面包覆了一层LNO作为内壳层, 然后通过湿化学分散法将具有高离子导电率的LGPS电解质包覆在核壳结构的LNO@LCO表面, 得到了双壳层结构的LGPS@LNO@LCO. LNO内壳层可以稳定LCO与硫化物电解质的界面, 而外壳LGPS可以实现LCO与LGPS之间的良好固-固接触. 得益于双壳层的界面结构设计, LCO电极的极化显著降低, 电化学性能明显改善. 在0.1 C下, 表现出125.8 mAh·g–1的首次放电比容量和90.4%的高库伦效率, 1 C下放电比容量为87.7 mAh·g–1. Banerjee等[110]结合多种先进的表征技术和第一性原理计算研究了LPSCl电解质与LiNi0.85Co0.1Al0.05O2 (NCA)正极的界面问题. 通过控制材料的荷电状态, 以研究NCA与LPSCl之间的化学反应以及LPSCl的电化学氧化分解对界面劣化的贡献. 研究表明, NCA与LPSCl本质上是不稳定的, 并且这种不稳定性在更高荷电状态下会进一步加剧. XRD, X-射线吸收谱 (XAS), XPS, Raman和冷冻TEM (cryo-TEM) 分析表明, NCA和LPSCl接触后会发生自发化学反应, 生成Ni3S4, LiCl, Li3PO4和氧化的LPSCl(oLPSCl)等界面产物. 第一性原理计算和分子动力学模拟也支持以上结论. 引入LNO包覆层后, NCA和LPSCl之间的热力学稳定性得到改善. 但包覆层并不能阻止高电位下LPSCl自身的电化学氧化分解. LPSCl的电化学分解仅发生在首圈循环过程中, 同时其分解产物可形成自钝化界面层, 从而使得电池可以实现长期稳定循环.
为了进一步提高电池的能量密度, 近年来研究者们越来越关注具有高比容量的高镍正极材料在固态锂电池中的应用. 不同于LCO材料, 高镍正极通常为多晶结构, 由较小的一次颗粒团聚而成的微米级二次颗粒组成. 高镍材料循环过程中相变引起的各向异性体积变化会诱导二次颗粒内部微裂纹的产生, 破坏正极颗粒的机械完整性, 导致正极材料二次颗粒内部以及正极和硫化物电解质界面严重的接触损失, 从而加速了电池性能的劣化. 因此, 对于多晶高镍材料, 在稳定材料表面与硫化物电解质的接触界面的同时, 抑制由于体积变化导致的正极材料二次颗粒内部晶界及正极和硫化物电解质界面机械失效也显得十分重要.
Jung等[111]对比了不同微观形貌的Li (Ni0.798 Co0.160Al0.042)O2(NCA80), Li(Ni0.751Co0.101Mn0.148) O2 (FCG75) 材料在硫化物电解质固态电池中的电化学性能, 研究了电化学循环过程中机械应力对高镍正极材料电化学性能的影响. NCA80材料中一次颗粒随机取向堆积, 而FCG75材料的一次颗粒则呈规律的放射状堆积更有利于锂离子传输以及缓冲体积变化. 研究表明, 具有特殊结构的FCG75材料表现出更高的容量以及更稳定的循环性能, 且颗粒在循环过程中更易保持完整的结构. 同时由于FCG75材料的镍含量呈梯度分布, 表面镍含量低, 因此界面处形成的岩盐相更薄, 电解质分解更少. Deng等[112]利用ALD技术对高镍NMC811材料晶界和表面进行了Li3PO4(LPO)双功能分级包覆, 以解决NMC811材料面临的双重挑战. 首先将10 nm厚的LPO包覆层注入NMC811颗粒的内部, 然后在其表面沉积LPO包覆层, 获得了双功能分级包覆的HLPO@NMC811. 并利用XANES, XPS和TEM等表征技术研究了表面化学和结构演变. 结果表明, LPO修饰不仅显著地抑制了硫化物电解质与NMC811的界面副反应, 同时有助于缓解充放电过程中应力导致的机械失效. 考虑到与其他氧化正极材料相比, LCO与硫化物电解质具有较好的界面相容性, Li等[63]利用LCO和LNO对NMC811材料进行双重修饰, 制备了NCM @LCO @ LNO材料. 首先, 利用共沉淀方法在NCM表面涂覆LCO, 形成表面贫镍的核-壳结构NCM@LCO, 其中富Ni的NCM核可以提供较高的比容量, 贫Ni的表面稳定与硫化物电解质的界面. 然后, 在NCM@LCO二次颗粒的表面包覆少量的LNO, 以进一步优化界面. 得益于双重修饰对界面的稳定, Li-In|LGPS|NCM@LCO@LNO固态电池显示出高的放电容量, 优异的倍率性能和循环和循环稳定性. 35 ℃下, 0.1 C循环首圈放电比容量达182 mAh·g–1, 3 C倍率下首圈容量为103 mAh·g–1, 0.3 C下循环585圈后容量保持率为80%. Wang等[113]通过控制LiNb0.5Ta0.5O3包覆层退火温度来调控界面包覆层的离子电导率, 研究了其对Li-In|LGPS|LiNi0.5Mn0.3Co0.2O2固态电池电化学性能的影响. 研究表明, 包覆层的离子电导率决定了界面Li+的传输动力学, 提高界面包覆层的离子电导率可以显著提高界面Li+传输, 从而改善电池性能. Li等[114]在NCA表面构建双缓冲层 (电化学活性的厚缓冲层+惰性的薄缓冲层), 显著改善了其在硫化物电解质固态电池中的电化学性能 (图13). 他们首先将NCA材料设计成具有核壳结构的CS-NCA材料, 该结构由富Ni的LiNi0.85Co0.15O2核和富Al的LiNi0.6Co0.15Al0.25O2壳组成. 然后, 在CS-NCA材料表面包覆上一层薄的LNO惰性缓冲层, 制备了具有双缓冲层结构的CS-NCA@ LNO材料. 双缓冲层结构可以更好地抑制空间电荷层效应, Li-In|LGPS|CS-NCA@LNO固态电池表现出优异的电化学性能. 60 ℃ 下, 0.06 C时显示出184.1 mAh·g–1的高比容量和90.2%的库伦效率, 4.2 C放电比容量达130 mAh·g–1, 0.3 C下循环400圈后容量保持率为89.4%.
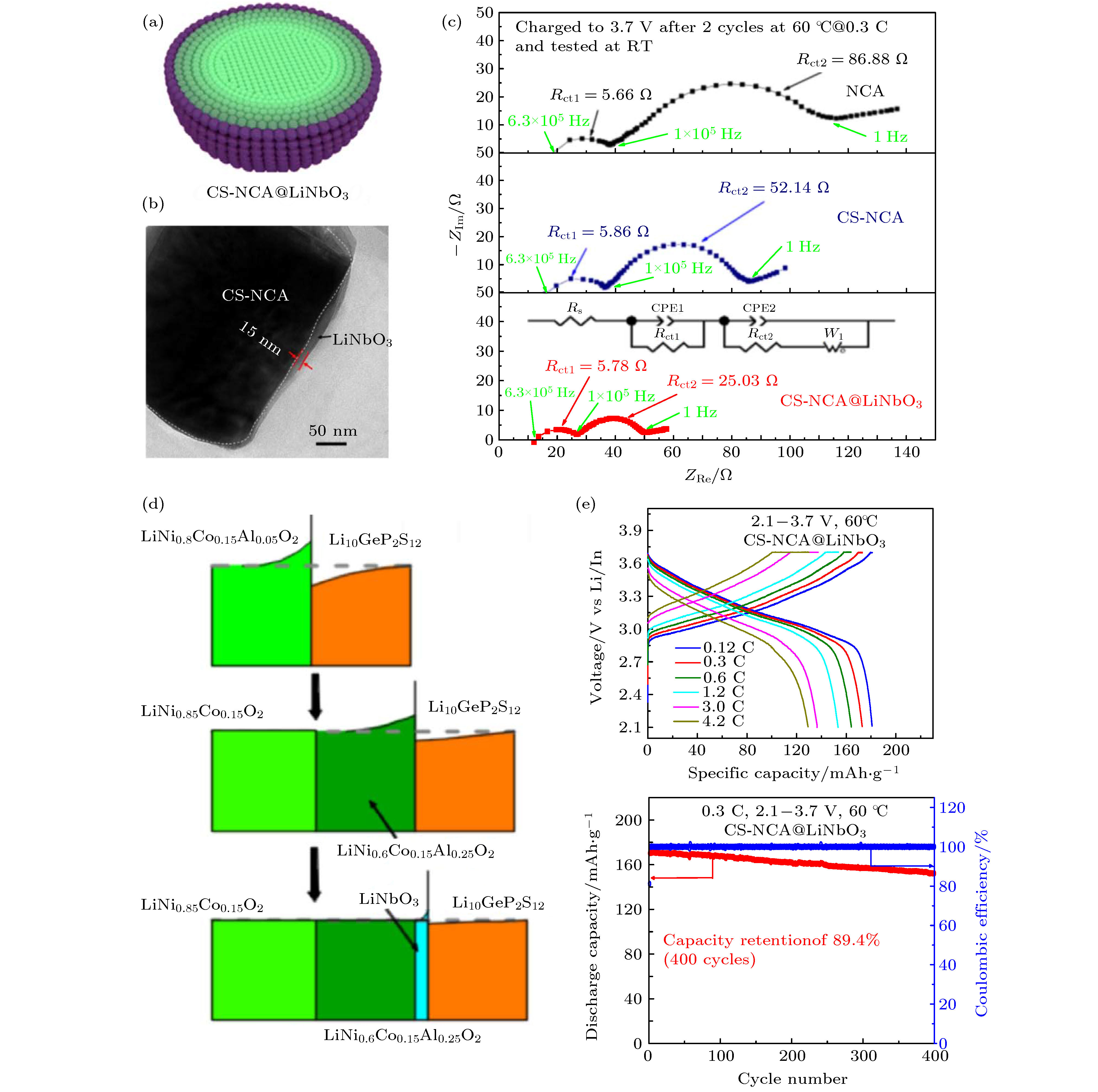 图 13 (a) CS-NCA@LiNbO3材料的结构示意图 (以LiNi0.85Co0.15O2为核心, LiNi0.6Co0.15Al0.25O2作为第一缓冲层, LiNbO3作为第二缓冲层); (b) CS-NCA@LiNbO3材料的高分辨透射电镜图像; (c) 在60 °C下循环2周后, 使用NCA, CS-NCA和CS-NCA@LiNbO3正极的全固态锂电池充电态的阻抗谱图. 阻抗测试在RT进行; (d) 使用NCA, CS-NCA和CS-NCA@LiNbO3正极的全固态锂电池中正极/电解质界面Li浓度分布的示意图; (e) 在60 °C下, 基于CS-NCA@LiNbO3正极的全固态锂电池的倍率性能和循环性能曲线. 惰性LiNbO3缓冲层的包覆量为1.37 wt%. 1 C = 200 mA·g–1[114]
图 13 (a) CS-NCA@LiNbO3材料的结构示意图 (以LiNi0.85Co0.15O2为核心, LiNi0.6Co0.15Al0.25O2作为第一缓冲层, LiNbO3作为第二缓冲层); (b) CS-NCA@LiNbO3材料的高分辨透射电镜图像; (c) 在60 °C下循环2周后, 使用NCA, CS-NCA和CS-NCA@LiNbO3正极的全固态锂电池充电态的阻抗谱图. 阻抗测试在RT进行; (d) 使用NCA, CS-NCA和CS-NCA@LiNbO3正极的全固态锂电池中正极/电解质界面Li浓度分布的示意图; (e) 在60 °C下, 基于CS-NCA@LiNbO3正极的全固态锂电池的倍率性能和循环性能曲线. 惰性LiNbO3缓冲层的包覆量为1.37 wt%. 1 C = 200 mA·g–1[114]Figure13. (a) Illustration of the structure of CS-NCA@LiNbO3 (LiNi0.85Co0.15O2 as the core, LiNi0.6Co0.15Al0.25O2 as the first buffer layer and LiNbO3 as the second buffer layer); (b) HRTEM images of CS-NCA@LiNbO3 materials; (c) Nyquist plots of charged ASSLB using NCA, CS-NCA and CS-NCA@LiNbO3 cathodes after 2 cycles at 60 °C. EIS tests are conducted at RT; (d) schematic illustrations of the interfacial Li concentration distribution in ASSLB using NCA, CS-NCA and CS-NCA@LiNbO3 cathodes; (e) rate performance and cycle performance curves of CS-NCA@LiNbO3 cathodes in ASSLB at 60 °C. Coating amount of inactive LiNbO3 buffer layer is 1.37 wt.%. 1 C=200 mA·g–1[114].
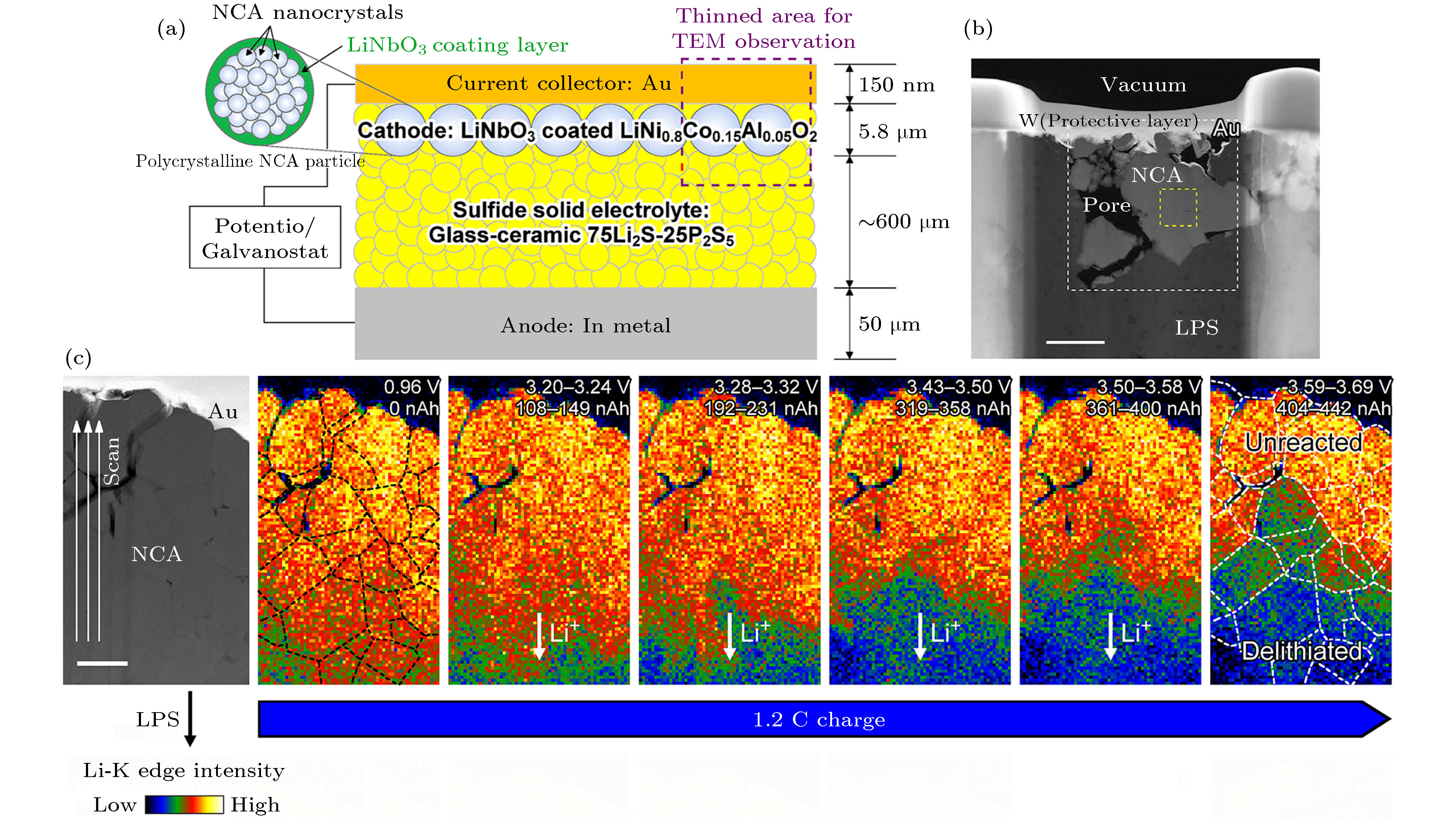 图 14 (a) 块体全固态电池示意图, 微米尺寸的多晶NCA正极是有很多堆积在一起的纳米晶组成, 紫色虚线矩形出显示用于TEM观察的减薄的区域; (b) NCA正极部分的ADF-STEM图像, 标尺是1 μm; (c) ADF-STEM图像和1.2 C充电下一系列不同状态下的NCA正极中的锂浓度分布图, 标尺是500 nm[119]
图 14 (a) 块体全固态电池示意图, 微米尺寸的多晶NCA正极是有很多堆积在一起的纳米晶组成, 紫色虚线矩形出显示用于TEM观察的减薄的区域; (b) NCA正极部分的ADF-STEM图像, 标尺是1 μm; (c) ADF-STEM图像和1.2 C充电下一系列不同状态下的NCA正极中的锂浓度分布图, 标尺是500 nm[119]Figure14. (a) Schematic diagram of the bulk-type SSLIB. NCA forms micrometer-sized polycrystalline particles composed of aggregated nanocrystals (top-left inset). The purple dashed rectangle shows the thinned area for TEM observation; (b) ADFSTEM image around the NCA cathodes. Scale bar: 1 μm; (c) ADF-STEM image and series of corresponding Liconcentration maps of NCA acquired during 1.2 C charging. Scale bar: 500 nm[119].
原位近边结构X射线吸收光谱法 (in situ XANES) 能在电化学和化学反应期间连续监测界面组分结构的动态变化, 从而获得工况下化学/电化学界面结构和化学信息[117]. Wang等[120]和Li等[121]利用in situ XANES表征手段研究了不同正极和硫化物电解质间的界面反应机理. 例如, 他们用in situ XANES技术动态观测了LiNbOx包覆和未包覆高镍正极LiNi0.8Mn0.1Co0.1O2 (NMC811)与硫化物固态电解质Li10GeP2S12 (LGPS) 间的界面演化行为 (图15(a)和图15(b))[121]. 从图15(c)和图15(d) S元素的K近边吸收谱及相应的mapping图可以看出, LGPS与没有包覆的NMC811的界面在充放电过程中处于不稳定的状态. LGPS在最初的充电过程中变成了亚稳的中间态, 其中部分LGPS在随后的充电过程中分解形成了硫化锂杂质相, 最终导致了NMC811表面结构重组. 相比之下, LiNbOx包覆的NCM与LGPS的界面在相同的充放电过程中的S元素的K近边吸收谱及相应的mapping图未发生明显变化(图15(f)和图15(g)), 说明包覆层能够提升正极和硫化物电解质界面的化学/电化学稳定性. 然而, 从图15(e)和/图15(h)可以看出, LiNbOx包覆和未包覆NMC811正极中Ni元素的K近边吸收谱在长时间循环后都向更高能量的方向偏移, 表明了NMC811正极在固态电池中Ni元素不可逆的化学态变化. 采用非原位STEM结合EELS表征手段对循环后的样品进行分析, 他们进一步证实了未包覆的NMC811与LGPS界面的副反应可以诱导NCM811表面晶格演变. 相比之下, 包覆后的NMC811的表面相没有明显变化. 这项研究证实, 通过对高镍正极表面进行合理的修饰和改性, 可以有效地抑制与硫化物固态电解质的界面互扩散, 从而保持固/固界面在循环过程中的稳定性.
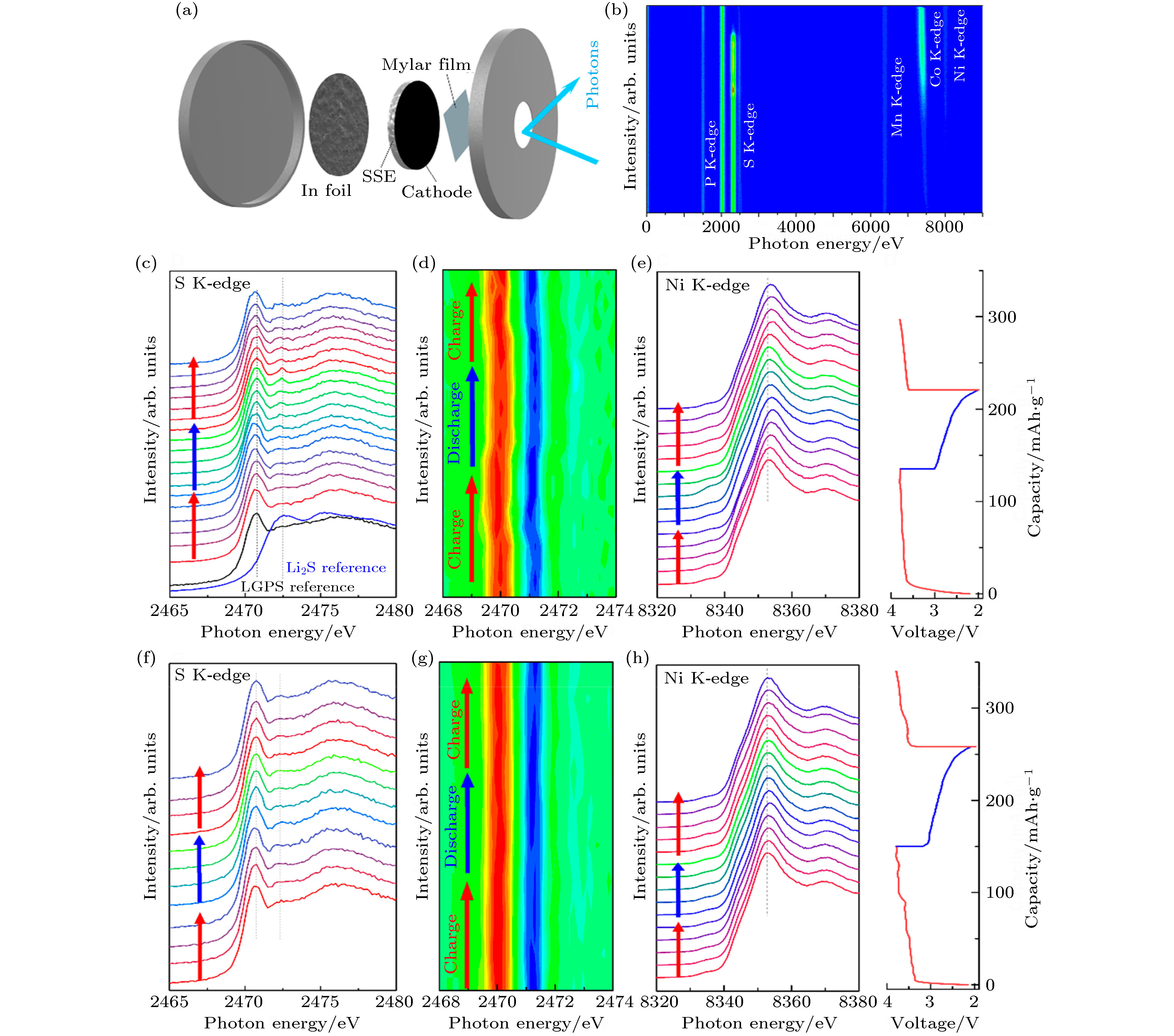 图 15 (a) 原位近边结构X射线吸收光谱法的基于硫化物固态电解质的全电池实验装置图; (b) 在大光子能量区域获得布拉格光谱图; (c) 未包覆的NCA811的原位S元素的K近边吸收谱及 (d) 相应的mapping图; (e) Ni元素的K近边吸收谱和充放电曲线, LiNbOx包覆的NMC811正极的原位; (f) S元素的K近边吸收谱及(g)相应的mapping图; (h) Ni元素的K近边吸收谱和充放电曲线[121]
图 15 (a) 原位近边结构X射线吸收光谱法的基于硫化物固态电解质的全电池实验装置图; (b) 在大光子能量区域获得布拉格光谱图; (c) 未包覆的NCA811的原位S元素的K近边吸收谱及 (d) 相应的mapping图; (e) Ni元素的K近边吸收谱和充放电曲线, LiNbOx包覆的NMC811正极的原位; (f) S元素的K近边吸收谱及(g)相应的mapping图; (h) Ni元素的K近边吸收谱和充放电曲线[121]Figure15. Operando XANES study of SSLIBs with bare and LNO-coated NMC811 cathodes during cycling: (a) Schematic diagram of theoperando cell; (b) Bruker spectrum in a large photon energy region. Operando S K-edge spectra with first derivative mapping, Ni K-edge spectra, and charge/discharge profiles of (c)?(e) bare NMC811-LGPS and (f)?(h) LNO NMC811-LGPS SSLIBs[121].
拉曼 (Raman) 效应起源于材料对单色光的非弹性散射, 利用拉曼效应可以得出材料的拉曼光谱, 它与具有拉曼活性的分子的振动模式相关. 因此, 所有影响分子振动模式的因素, 比如晶体结构、应力和化学状态, 都会影响拉曼光谱. 也就说, 利用原位拉曼光谱 (in situ Raman) 的变化可以动态地分析电极/电解质界面处结构和化学状态发生的改变, 这将有助于揭示充放电过程中的动态界面反应机理. Zhang等[93]搭建了原位拉曼检查装置, 动态观测了LiNi0.8Co0.1Mn0.1O2 (NCM)与硫化物固态电解质Li6PS5Cl界面处的拉曼光谱变化 (图16). 他们首先通过对比不同位置的拉曼变化, 确定了界面处电解质的位置. 结合循环伏安法, 来观察每个电位下界面处拉曼峰的原位变化. 他们的结果发现, 和之前的研究一样, 425 cm–1位置的拉曼峰可归结于Li6PS5Cl结构中的





 图 16 (a) 正负电压扫描区间的NCM/Li6PS5Cl界面的原位拉曼光谱; (b), (c) 来自NCM/Li6PS5Cl界面在两个电压 (第一次正电压扫描时为3.6和3.7 V) 的归一化原位拉曼光谱的拟合结果; (d) 在正电压和负电压扫描期过程中, 两个拉曼峰在418和425 cm–1处的归一化峰面积百分比演变; (e), (f) 基于NCM/Li6PS5Cl界面
图 16 (a) 正负电压扫描区间的NCM/Li6PS5Cl界面的原位拉曼光谱; (b), (c) 来自NCM/Li6PS5Cl界面在两个电压 (第一次正电压扫描时为3.6和3.7 V) 的归一化原位拉曼光谱的拟合结果; (d) 在正电压和负电压扫描期过程中, 两个拉曼峰在418和425 cm–1处的归一化峰面积百分比演变; (e), (f) 基于NCM/Li6PS5Cl界面
Figure16. (a) In situ Raman spectra at NCM/Li6PS5Cl interface during the positive and negative potential sweeps; (b), (c) examples of normalized peak fitting results from in situ Raman spectra at NCM/Li6PS5Cl interface at two voltages (3.6 and 3.7 V in the first positive potential sweep); (d) normalized peak area percentages evolutions of two Raman peaks at 418 and 425 cm–1 during the positive and negative potential sweeps; (e)?(h) schematic illustrations of interfacial evolution during the charge–discharge processes based on different vibration states of P-S bond in

中子散射相关技术具有与X-射线互补的散射截面, 特别是轻元素 (例如H, Li和O核) 对于中子具有大的散射截面, 相比于X-射线技术, 中子散射技术在探测轻元素方面显示出独特的优势. 中子散射技术在电池充放电过程中电极材料晶体结构演变、表/界面结构及形态变化以及监测锂的发布和传输路径等方面都有着非常重要的应用. 中子与原子核相互作用, 因此在低中子能量的情况下可实现对样品的无损分析. 同时其弱的相互作用可获得高穿透深度, 有利于对样品的整体探测和电池的原位研究. Han等[64]利用时间分辨原位中子深度剖析技术研究了典型固体电解质中锂枝晶形成机理 (图17). 对锂沉积过程中Li浓度分布动态演变的原位监测表明, Li3PS4电解质高的电子电导率是导致锂枝晶生长的重要原因. 因此, 他们认为降低固体电解质的电子电导率是抑制锂枝晶生长要考虑的一个重要方向.
 图 17 (a) 电化学原位中子深度分析实验装置示意图; (b) Li/Li3PS4/Pt电池结构的示意图, Li沉积在Pt上, 在锂沉积过程中从电池的上表面开始进行深度剖析; (c) 在100 ℃下测试的Li/Li3PS4/Pt电池的时间分辨锂浓度曲线, 灰色箭头表示Li的连续沉积; (d) 在100 ℃下, Li/Li3PS4/Pt电池随着放电电流密度阶跃增大的电压-时间曲线; (e) 在100 ℃下锂沉积过程中, 不同时间Li3PS4中的锂浓度分布曲线[64]
图 17 (a) 电化学原位中子深度分析实验装置示意图; (b) Li/Li3PS4/Pt电池结构的示意图, Li沉积在Pt上, 在锂沉积过程中从电池的上表面开始进行深度剖析; (c) 在100 ℃下测试的Li/Li3PS4/Pt电池的时间分辨锂浓度曲线, 灰色箭头表示Li的连续沉积; (d) 在100 ℃下, Li/Li3PS4/Pt电池随着放电电流密度阶跃增大的电压-时间曲线; (e) 在100 ℃下锂沉积过程中, 不同时间Li3PS4中的锂浓度分布曲线[64]Figure17. (a) Schematic diagram of the experimental set-up for operando NDP; (b) schematic structures of Li/Li3PS4/Pt cells. Li is plated on Pt, and the depth profiles are measured from the top surfaces of the cells during plating; (c) time-resolved lithium concentration profiles for Li/Li3PS4/Pt cells tested at 100 ℃. The grey arrows indicate continuous plating of Li; (d) voltage–time curves of the Li/Li3PS4/Pt cells discharged with the magnitude of the current density increased step-wise at 100 ℃; (e) lithium concentration profiles in Li3PS4-100 ℃ at different times during lithium plating[64].
除了上述提到的原位表征, 其他的原位表征包括可以表征全固态电池中界面处电势的分布情况的原位电子全息技术 (quantitative electron holography, EH) 与电子能量损失谱 (electron energy loss spectroscopy, EELS) 的联用[122]或直接采用原位开尔文探针力显微镜 (in situ Kelvin probe force microscopy, KPFM) 技术[123]. 分析界面处副产物的化学价态的原位XPS技术[124]. 实现无损监测全固态电池界面和体相处Li的分布情况的原位三维7Li核磁共振成像 (three-dimensional 7Li magnetic resonance imaging, MRI)[125], 以及监测循环过程中界面组分的结构变化的原位中子衍射技术 (ND)[126]等. 随着各种先进表征技术在固态电池界面研究中的不断应用的发展, 将为硫化物电解质各种界面问题的解决提供理论基础和设计依据, 推动界面设计、优化与调控策略的发展. 但是目前单一的原位表征技术仍然在动态研究、定量分析和检测分辨率等方面面临困难和挑战. 虽然, 固体核磁技术 (固体核磁共振及其成像技术) 具有无损、原位定量分析的特有优点, 但受制于谱仪有限的梯度场强度, 以及固体材料中探测核较宽的谱峰等限制, 目前在空间分辨率和灵敏度上还难以满足固态电解质/电极界面表征的需求 (< 1 μm). 未来需要进一步提升固体核磁共振谱仪硬件/软件水平, 以及设计新型成像脉冲, 不断提高电化学原位核磁共振 (EC-NMR) 及其成像 (MRI) 技术的分辨率和灵敏度, 以实现对固态电池中各界面特性及其动态演化行为的高精度定量分析. 未来原位动态电子显微学的方法研究固/固界面问题的发展应注重如下几个方面: 1) 发展低加速电压下的球差消除技术, 大大地降低电子束诱导的副反应, 进一步提高分辨率, 实现单个空位、表面台阶等固/固界面演变过程的动态观察及其对电学传输性能影响的原位定量测量; 2) 提高时间分辨率, 实现动态观察固/固界面处原子结构变化过程, 并确定固/固界面处原子及外场 (热、力、电等) 对界面结构演变的影响; 3) 发展新型光路系统, 能够同时观察固/固界面结构和整体形貌, 并提高电子能量损失谱分析能力, 最终实现原子尺度下观察及分析固/固界面演化过程 (如界面反应位置, 反应产物成分), 并深入分析不同电化学条件对界面结构及性能的影响. 而随着具有更高亮度及相干度的新一代同步辐射光源的建设和快速发展, 基于同步辐射的各种电化学原位表征技术 (如X-射线衍射、X-射线吸收及X-射线衍射成像等) 可以实现更高的时间、空间及能量分辨率, 在固态电池各种界面问题研究中将发挥重要作用. 可以实现在多时间和空间尺度下, 对处于平衡和非平衡条件下硫化物固态电解质与电极界面反应及其动力学特性的高精度实时原位监测. 总而言之, 通过综合应用多种原位和非原位表征手段, 并结合理论计算和模拟方法, 才能对硫化物电解质与电极固/固界面的构效关系有清晰准确的认识. 需要不断地发展更高时间/空间分辨、能量分辨以及更加精确的定量分析表征手段以及各类实验技术的综合集成, 以满足固态锂电池中各界面问题研究的广泛需求.
虽然, 大量研究工作表明, 通过界面缓冲层的构建 (如锂负极表面修饰、氧化物正极材料表面包覆等) 可以显著地降低电极/硫化物电解质的界面接触阻抗, 改善固态电池倍率性能, 提高循环寿命. 然而, 在界面修饰层特性和修饰方法选择等方面仍有很大的改进空间. 深入研究修饰层的作用机理, 明确修饰层的功能特性, 对于开发新型修饰层以进一步提高电池性能至关重要. 为消除空间电荷层效应, 抑制硫化物电解质分解, 正极包覆层首要特性是要能传导锂离子而阻挡电子, 即理想的包覆材料应该是具有高氧化稳定性的固体电解质材料. 在包覆层有效地实现正极材料和硫化物电解质电子绝缘的同时, 需要在正极材料颗粒之间构建高效的电子导电网络, 显然完整的电子绝缘包覆层对于这一点是不利的. 如何实现这两者之间的平衡还需在微观电子和离子输运层面开展更深入的研究工作. 同时, 作为正极和电解质的界面缓冲层, 包覆层除了有宽的电化学稳定性窗口外, 还应同时与正极材料和硫化物电解质具有良好的化学兼容性. 此外, 为防止充放电过程中电极材料体积膨胀和收缩产生的应力对界面的破坏, 包覆层最好能具有一定柔性 (机械性能), 以通过弹性和/或塑性变形缓冲 (吸收) 充放电过程的界面应力/应变. 单一包覆材料往往难以满足以上所有要求, 发展多组分的复合功能界面修饰层是一种可行的解决方案.
